

Abstract
A striking heterogeneous distribution of topographic and cellular huntingtin immunoreactivity was observed within the human neostriatum using three distinct huntingtin antibodies. Patchy areas of low huntingtin immunoreactivity were present in both the caudate nucleus and putamen, surrounded by an intervening area of greater immunoreactivity. Comparison of huntingtin immunoreactivity with contiguous serial sections stained for enkephalin and calbindin D28k immunoreactivities showed that the topographic heterogeneity of huntingtin immunostaining corresponded to the patch (striosome) and matrix compartments within the striatum. Huntingtin immunoreactivity was confined primarily to neurons and neuropil within the matrix compartment, whereas little or no neuronal or neuropil huntingtin immunostaining was observed within the patch compartment. There was marked variability in the intensity of huntingtin immunolabel among medium-sized striatal neurons, whereas a majority of large striatal neurons were only faintly positive or without any immunoreactivity. Combined techniques for NADPH-diaphorase enzyme histochemistry and huntingtin immunocytochemistry, as well as double immunofluorescence for either nitric oxide synthase or calbindin D28k in comparison with huntingtin expression, revealed a striking correspondence between calbindin D28k and huntingtin immunoreactivities, with little or no colocalization between NADPH-diaphorase or nitric oxide synthase neurons and huntingtin expression. These observations suggest that the selective vulnerability of spiny striatal neurons and the matrix compartment observed in Huntington’s disease is associated with higher levels of huntingtin expression, whereas the relative resistance of large and medium-sized aspiny neurons and the patch compartments to degeneration is associated with low levels of huntingtin expression.
- Huntington’s disease
- huntingtin
- neostriatum
- immunofluorescence
- NADPH-diaphorase
- nitric oxide synthase
- calbindin D28k
Huntington’s disease (HD) is a progressive, fatal neurological disorder characterized by prominent striatal degeneration, chorea, and dementia (Bruyn, 1968; Young, 1994). The HD gene defect is an expanded, unstable DNA segment containing a polymorphic trinucleotide CAG repeat in the coding sequence of the IT15 gene on chromosome 4, which encodes the highly conserved protein huntingtin (Huntington’s Disease Collaborative Research Group, 1993; Gusella and MacDonald, 1995). Although the normal function of huntingtin is unknown, expression is observed throughout the nervous system as well as in non-neural tissues.
Although both mRNA and immunohistochemical studies have yielded generally consistent results, differences in both the cellular and topographic distribution of huntingtin have been reported. An analysis of the localization of huntingtin mRNA suggests that it is present in all neurons (Li et al., 1993; Strong et al., 1993; Landwehrmeyer et al., 1995). Large neurons contain higher levels of message, but, when corrected for size, there is no difference from other neurons (Landwehrmeyer et al., 1995). Although several different huntingtin antibodies have been used in immunohistochemical studies, the topographic distribution and cellular localization of huntingtin in the brain have not yet been characterized fully. One of the first reports, using an antipeptide polyclonal antibody, noted that huntingtin was found in both the nucleus and cytoplasm of neurons and did not describe any regional variations (Hoogeveen et al., 1993). Subsequent studies have not detected nuclear huntingtin and suggest that huntingtin is a cytoplasmic protein widely distributed in neurons throughout the brain (DiFiglia et al., 1995; Gutekunst et al., 1995; Persichetti et al., 1995; Sharp et al., 1995; Trottier et al., 1995; Bhide et al., 1996). Within the striatum it has been reported that neuropil labeling is light, with little or no immunoreactivity within striatal neurons (Sharp et al., 1995; Trottier et al., 1995). Other studies suggest that, although most neurons are immunopositive for huntingtin throughout the brain, there is some variability in neuronal expression and that large striatal neurons have a greater signal than other striatal neurons (Gutekunst et al., 1995; Bhide et al., 1996). Although a patch/matrix pattern of huntingtin immunoreactivity has not been observed in the mouse or nonhuman primate striatum (Gutekunst et al., 1995; Sharp et al., 1995; Bhide et al., 1996), a heterogeneous patch-like staining was reported within the human striatum (Gutekunst et al., 1995).
These studies have been widely interpreted as demonstrating that huntingtin is expressed ubiquitously within the brain and that neurons are labeled without any regional qualitative differences in brain areas susceptible to degeneration in HD, suggesting that some factor other than the expression of the HD gene product underlies the selective regional and neuronal vulnerability observed in HD. It is unclear, however, whether this conclusion is correct, because there is no strong consensus on the topographic distribution and cellular localization of huntingtin within the brain. In the present study we used enzyme histochemical, immunocytochemical, and immunofluorescent techniques to characterize the heterogeneity of huntingtin expression within the normal human striatum.
MATERIALS AND METHODS
Postmortem striatal tissue from 12 patients with no evidence of neurological disease (mean age, 68.5 years; range, 58–79 years) was dissected fresh and placed in cold (4°C) 2% paraformaldehyde–lysine–periodate solution for 24–36 hr. The postmortem intervals did not exceed 12 hr (mean time, 9.7 hr; range, 4–12 hr). Tissue blocks were rinsed in 0.1 m sodium phosphate buffer and placed in cold cryoprotectant in increasing concentrations of 10 and 20% glycerol/2% DMSO solution over 36 hr. Frozen serial sections of the entire striatal tissue block were cut at 50 μm intervals in the coronal plane. The cut sections were stored in 0.1 m sodium phosphate buffer/0.08% sodium azide at 4°C for subsequent immunocytochemistry, enzyme histochemistry, immunofluorescence, and a combination of these techniques.
It is important to note that increased postmortem interval, temperature, fixation, and time interval after tissue sectioning all play a role in the subsequent staining patterns of the topographic and neuronal expression of each of the huntingtin antisera used in these studies. Time course immunostaining studies were performed to detect any differences between staining patterns at intervals directly after tissue sectioning and at 2, 4, and 8 weeks after frozen-sectioning and cold (4°C) storage.
Immunocytochemistry. Three well characterized antibodies against distinct huntingtin epitopes were used in this study: a rabbit polyclonal anti-fusion antibody (HDp549) derived from a segment of human huntingtin (amino acids 549–679; dilution, 3 μg/ml) (Gutekunst et al., 1995), a rabbit polyclonal antiserum (HF1) directed against amino acids 1981–2580 expressed in Escherichia coli as a fusion with glutathione S-transferase (GST; dilution, 1:250) (Persichetti et al., 1995), and a mouse anti-huntingtin monoclonal antibody as a fusion protein from an amino acid huntingtin fragment, 181–810 (dilution, 1:20; Chemicon, Temecula, CA) (Trottier et al., 1995). Immunohistochemical localization of antibodies to choline acetyltransferase (ChAT), a marker for large striatal neurons (dilution, 1:500; polyclonal rabbit antisera, Chemicon); calbindin-D28k, for selective identification of spiny striatal neurons and the striatal matrix compartment (dilution, 1:3000; monoclonal mouse antisera, Swiss Antibodies, Belinzona, Switzerland); met-enkephalin, delineating the striatal patch/matrix compartments (dilution, 1:800; polyclonal rabbit antisera, Incstar, Stillwater, MN); and brain nitric oxide synthase (NOS), which colocalizes with striatal somatostatin, neuropeptide Y/NADPH-diaphorase neurons (dilution 1:500; polyclonal rabbit antisera, Accurate Chemicals, Westbury, NY) was performed with a previously reported conjugated second antibody method (Ferrante et al., 1993). Tissue sections were preincubated in an absolute methanol–0.3% hydrogen peroxide solution for 30 min, washed (3×) in PBS, pH 7.4, for 10 min each, placed in 10% normal goat serum (Life Technologies, Grand Island, NY) for 1 hr, incubated free floating in primary antiserum at room temperature for 12–18 hr (all dilutions of primary antisera above included 0.3% Triton X-100 and 10% normal goat serum), washed (3×) in PBS for 10 min each, placed in periodate-conjugated goat anti-rabbit IgG (1:300 in PBS, Boehringer Mannheim, Indianapolis, IN) or goat anti-mouse IgG (1:300 in PBS, Boehringer Mannheim), washed (3×) in PBS 10 min each, and reacted with 3,3′ diaminobenzidine HCl (1 mg/ml) in Tris-HCl buffer with 0.005% hydrogen peroxide. So that double immunocytochemical and enzyme histochemical methods could be completed, selected striatal tissue sections immunoreacted with huntingtin antisera were not preincubated in absolute methanol–0.3% hydrogen peroxide solution. These huntingtin-labeled sections were wet-mounted with 50% glycerol, coverslipped, photographed at different focal planes of the tissue specimen to ensure that all huntingtin-positive neurons were identified by the use of a Nikon photomicroscope, stored at 4°C for subsequent combined NADPH-diaphorase enzyme histochemistry, and rephotographed.
Specificity for the antisera used in this study was examined in each immunochemical experiment to assist with interpretation of the results. This was accomplished by preabsorption with excess target proteins (e.g., homologous huntingtin fusion proteins) and by omission of the primary antibody to determine the amount of background generated from the detection assay. The HDp549 and HF1 huntingtin antibodies were tested by preadsorption of dilute primary antisera with an excess of appropriate fusion protein (10 μg/ml and 12 μg/ml, respectively) for 6 hr at room temperature before incubation (Fig.1). Both of these huntingtin antibodies were made as a fusion with helminthic GST. Although the polyclonal antibodies are purified over a GST column to remove GST antibodies, each purified aliquot was tested for cross-reactivity to GST, and GST polyclonal antibodies were used as controls to insure lack of cross-reactivity. Fusion protein for preadsorption of the Chemicon huntingtin antisera was unavailable.

Preadsorption studies of huntingtin antisera. Shown are photomicrographs of cellular and neuropil huntingtin immunoreactivity within the caudate nucleus, using HDp549 (A) and HF1 (C) huntingtin antibodies, and their respective preadsorption (B andD), using primary antisera with an excess of fusion protein (see Materials and Methods).
Enzyme histochemistry: nicotinamide adenine dinucleotide phosphate dehydrogenase (NADPH-d) method. Free-floating sections were stained, using a modification of the direct method of Vincent and Johansson (1983) for demonstrating NADPH-d. Tissue sections were incubated at 37°C and monitored intermittently for intensity for 0.5–3 hr in a solution of 10 ml of 0.1 m Tris-HCl buffer, pH 7.4, containing 4 mg of NADPH (Sigma, St. Louis, MO) and 10 mg of nitro blue tetrazolium salt (NBT; Sigma). Increased intensity of reaction product was achieved by the addition of 0.8% Triton X-100 (Sigma). Heat treatment of tissue sections at 60°C for 2 hr or incubating sections with NBT alone served as controls for specificity of enzyme activity.
Fluorescent immunocytochemistry. Immunofluorescence was performed by a previously described method (Ferrante et al., 1987a) by incubating striatal tissue sections in the HDp549 polyclonal rabbit huntingtin antisera (12 μl/ml) and in either a monoclonal mouse brain nitric oxide synthase (bNOS) (dilution, 1:50; Accurate Chemicals) or a monoclonal mouse calbindin-D 28k antisera (dilution, 1:300) in Tris-HCL buffer containing 0.3% Triton X-100 for 24–72 hr at 4°C. Then sections were rinsed in PBS (3 washes for 10 min each) and incubated in the dark with goat anti-rabbit fluorescein isothiocyanate (FITC) conjugate (1:15; Boehringer Mannheim) and with goat anti-mouse tetramethylrhodamine isothiocyanate (TRITC) conjugate (1:10; Boehringer Mannheim) for 2 hr at 20°C. Deletion of huntingtin antisera resulted in an absence of green fluorescence, whereas deletion of either bNOS or calbindin-D 28k antisera resulted in an absence of red fluorescence. Sections were wet-mounted and coverslipped, using 50% glycerol on completion of the technique. Identical microscopic fields were photographed immediately with a Nikon fluorescent microscope, delineating the location of huntingtin and bNOS or huntingtin and calbindin-D 28k immunoreactivities within the same striatal section.
The densities of huntingtin- and calbindin D28k-immunoreactive neurons were determined within the medial portion of the caudate nucleus in 1 mm2 areas (n = 28 and 24, respectively) at the level of the head of the caudate nucleus. The neuronal counts were made by using a ruled graticule eyepiece at 250×. The maximum diameter of huntingtin-, calbindin D28k-, and ChAT-positive neurons also was determined by using an eyepiece graticule at 400×. Results were analyzed by one-way ANOVA, followed by Fisher Probability of Least Significant Difference post hoc test to compare group means. Data are expressed as the mean ± SEM.
RESULTS
Topographic heterogeneity
Pronounced topographic differences in the intensity of huntingtin immunoreactivity were observed throughout the rostral and caudal extent of the normal human neostriatum (Fig. 2). When examined at higher power, the heterogeneity of striatal huntingtin immunostaining was attributable to comparatively reduced immunoreactivity in patchy striatal areas present within both the caudate nucleus and putamen. These patches formed discrete ellipses and circles that were elongate and more irregular basally. The intervening striatal regions had markedly greater cellular and neuropil immunostaining. This heterogeneous expression of huntingtin distribution was most prominent within the medial and ventral caudate nucleus and the nucleus accumbens and was outlined distinctly within the dorsal striatum. Patches of low huntingtin immunoreactivity were less well defined in the putamen. This topographic disposition was present, using each of the three distinct huntingtin antisera, and independently confirmed in the Bedford and Emory laboratories (Fig.3). Although increasing concentrations of the Chemicon antisera were used, striatal huntingtin immunoreactivity was not as intense as with the HDp549 and HF1 antibodies.

Huntingtin (HDp549) immunostaining of the rostral striatum at the head of the caudate nucleus, putamen, and nucleus accumbens (A) and the caudal striatum at the level of the globus pallidus and the body of the caudate nucleus (B). There is a marked heterogeneity of huntingtin immunostaining throughout the neostriatum, with lighter stained patches interspersed on a darker stained background.

Huntingtin-immunostained caudate nucleus with HF1 (A, C) and Chemicon (B,D) immunosera. A and B are at the interface of a lightly stained patch (p) and darker-stained matrix (m), as shown in Figure 2. The darker-stained matrix has greater cellular and neuropil huntingtin immunoreactivity using both HF1 (C) and the Chemicon antisera (D). The Chemicon antisera were characterized by a punctate appearance. Magnification bars: A,B, 500 μm; C, D, 200 μm.
This heterogeneous pattern of huntingtin immunoreactivity was reminiscent of the patch (striosomal) and matrix compartmentalization found within the striatum. When compared with serially cut contiguous striatal tissue sections, the zones of low huntingtin immunoreactivity corresponded with low immunoreactive patches of calbindin D28k and met-enkephalin, respectively (Fig. 4). There were very few differences in matching compartmental areas in adjoining sections processed for these neurochemical substances, with remarkable conformity from case to case.

Photomicrographs of adjacent, serially cut 50 μm frozen sections of the rostral striatum demonstrating the patch and matrix compartments, using antisera against calbindin D28k (A, D, H), huntingtin (HDp549) (B, E,G), and enkephalin (C, F). Areas of low huntingtin immunoreactivity (B,E) correspond to those low immunoreactive areas (patches) in contiguous sections immunostained for calbindin D28k (A, D) and enkephalin (C,F) (arrowheads). The heterogeneity of huntingtin staining was the result of reduced neuronal and neuropil immunoreactivity within the patches. There was markedly greater neuronal and neuropil huntingtin immunoreactivity in the matrix zone (m) in comparison to the patch area (p), as shown in the same adjacent areas immunostained for huntingtin (G) and calbindin D28k (H) immunoreactivity. Magnification inD–H is the same.
Terminal and neuronal huntingtin immunoreactivity were present within both segments of the globus pallidus. Although we have not detected any gross differences in huntingtin immunoreactivity between the internal and external segment of the globus pallidus, a more detailed analysis is underway.
Cellular heterogeneity
Huntingtin immunoreactivity in the striatum was present in medium-sized neurons distributed throughout the caudate nucleus and putamen (Figs. 3, 4, 5). These neurons were confined primarily to the striatal matrix, whereas the patch compartments were devoid of intense neuropil immunoreactivity and labeled cells. Only a few faintly immunoreactive neurons were present within patches (Fig. 4). Immunostaining was confined to the cytoplasm and primary and secondary dendritic arbors of medium-sized striatal neurons within the matrix compartment. Measurements of greatest somal diameters of huntingtin-immunoreactive neurons ranged from 18 to 45 μm in diameter with a mean of 29.7 ± 5.2 μm. Axon fibers and the punctate labeling of axon terminals were not a distinctive characteristic within the striatum.

Huntingtin (HDp549) immunoreactivity in the dorsal (A) and ventral (B) striatal matrix of the caudate nucleus. Huntingtin immunoreactivity is present in medium-sized neurons and is confined to the cytoplasm. Marked variability in neuronal immunoreactive intensity is observed. Neurons are immunolabeled either darkly (arrowheads) or lightly (arrows) for huntingtin (see Fig. 3C,D). The more darkly immunostained neurons are significantly greater in diameter (see Results). Magnification bars in A,B, 200 μm.
The intensity of huntingtin immunoreactivity was markedly variable among labeled neurons, with no obvious regional dorsoventral differences (Fig. 5). These neurons, however, could be classified into two subgroups by the intensity of their immunolabel as either lightly or darkly immunostained. The most intensely immunostained neurons were significantly greater in diameter (range, 28–45 μm; mean, 34.8 ± 4.2 μm; p > 0.01) as compared with the lightly immunostained neurons (range, 18–40 μm; mean, 27.7 ± 4.8 μm).
Qualitative observations suggested that the number of calbindin D28k-positive neurons was much greater than huntingtin-labeled neurons within the striatal matrix compartment. A quantitative analysis comparing these neurochemical subsets of striatal neurons in contiguous stained sections from the same topographic area revealed that the density of calbindin D28k-positive neurons was significantly greater than that of huntingtin-positive neurons (calbindin D28k, 678.3 ± 26.1/mm2; huntingtin, 288.5 ± 18.9/mm2;p > 0.001) (Fig. 6). It is of interest to note that the variability of the intensity of huntingtin label described in these studies is also present within calbindin D28k neurons (Fig. 6).

Comparison of neuronal densities and colocalization of huntingtin (HDp549) (A, C, E) and calbindin D28k (B, D, F) immunoreactivities within the medial caudate nucleus. A greater number of immunopositive calbindin D28k neurons (B, D) are present in the caudate nucleus than huntingtin-positive neurons (A, C). Variability of intensity in calbindin D28k immunoreactivity is present in labeled neurons (D), although not so prominent as that observed in huntingtin-positive neurons (C). Double immunofluorescence for huntingtin (FITC) (E) reveals a striking correspondence with calbindin D28k neurons (F). A moderate number of calbindin D28k neurons have no correspondence with huntingtin neurons within the same tissue section. The blood vessel in the top left corner ofE and F (white circles) acts as a fiduciary mark. Arrowheads delineate some of the corresponding pairs of neurons in E andF. Magnification bars: in A,B, 500 μm; in C–F, 200 μm.
To further characterize any correlation between these neuronal populations, we performed double immunofluorescence for huntingtin (FITC) and calbindin D28k (TRITC) immunoreactivities within the same tissue section. Huntingtin and calbindin D28k were colocalized within striatal neurons. Almost all huntingtin-positive neurons contained calbindin D28k (Fig. 6). There were some instances, albeit few, in which positive immunofluorescent huntingtin neurons did not coexist with calbindin D28k TRITC immunofluorescence. This negative correspondence, or single labeling, may be the result of the lack of absolute coexistence between these two neurochemical substances. A significant number of calbindin D28k-positive neurons, however, in which the presence of huntingtin immunofluorescence could not be detected, confirmed the variance between huntingtin and calbindin D28k neuronal densities. It was not possible to determine whether darkly and lightly immunostained neurons for huntingtin exactly corresponded to similar calbindin D28k-positive neurons in our material.
NADPH-d neurons and huntingtin
Combined NADPH-d enzyme histochemistry and huntingtin immunocytochemistry revealed that NADPH-d-positive aspiny striatal neurons did not contain any demonstrable huntingtin immunoreactivity. Comparison of photomicrographs within the same striatal sections first immunostained for huntingtin and subsequently reacted enzyme histochemically for NADPH-d strongly suggested that NADPH-d neurons contain little or no huntingtin expression (Fig. 7). NADPH-d neurons were present in the tissue section that previously were not detectable by huntingtin immunohistochemistry. In a small number of neurons, however, the formazan end product of the NADPH-d enzyme method reacted with the diaminobenzidine marker, resulting in the deposition of a coarse and blackened precipitate. This was unlike the fine purple-blue punctate formazan reaction product observed in most other neurons in the double-stained section or in sections stained alone for NADPH-d. As a consequence, we were unable to determine whether such neurons were reactive to both huntingtin and NADPH-d. Although we performed serial color photomicrography of different focal planes through the tissue specimens, we could not conclude absolutely whether some weakly huntingtin-positive neurons went undetected and subsequently stained for NADPH-d.

Comparison of huntingtin-positive neurons with NADPH-diaphorase (NADPH-d)-positive and nitric oxide synthase (NOS)-positive neurons in the normal caudate nucleus. Striatal NADPH-d neurons (A) and NOS neurons (B) are morphologically similar and are reported to colocalize (Hope et al., 1991). A striatal caudate section, first immunostained for huntingtin immunoreactivity (C) and subsequently treated for NADPH-d enzyme histochemistry (D), suggests that NADPH-d neurons do not contain huntingtin. Arrows inC and D delineate the same huntingtin-positive neurons within the section. There are no corresponding huntingtin-positive neurons in C where NADPH-d neurons are observed in D(arrowheads). Combined immunofluorescence for huntingtin (FITC) (E) and NOS (TRITC) (F) immunoreactivities in the same section confirm the absence of huntingtin and NADPH-d colocalization found in C andD. NOS-positive neurons in F(arrowheads) do not correspond with any huntingtin neurons in E. The blood vessel in the top left corner of E and F (white circles) acts as a fiduciary mark. Magnification bars inA–F, 100 μm.
It recently has been shown that neuronal NADPH-d and NOS activity are the same (Hope et al., 1991). NOS is localized selectively to medium-sized aspiny striatal interneurons that contain NADPH-d, somatostatin, and neuropeptide Y. To further clarify whether huntingtin and NADPH-d activity were present within the same striatal neurons, we performed double immunofluorescence for huntingtin (FITC) and NOS (TRITC) within the same striatal section. In all four cases examined, FITC/TRITC colocalization of huntingtin with NOS was not observed within striatal neurons. The immunofluorescence of each marker remained distinctly separate, confirming the suggested lack of coexistence with the use of the combined huntingtin immunoreactivity and NADPH-d enzyme histochemistry (Fig. 7).
ChAT neurons and huntingtin
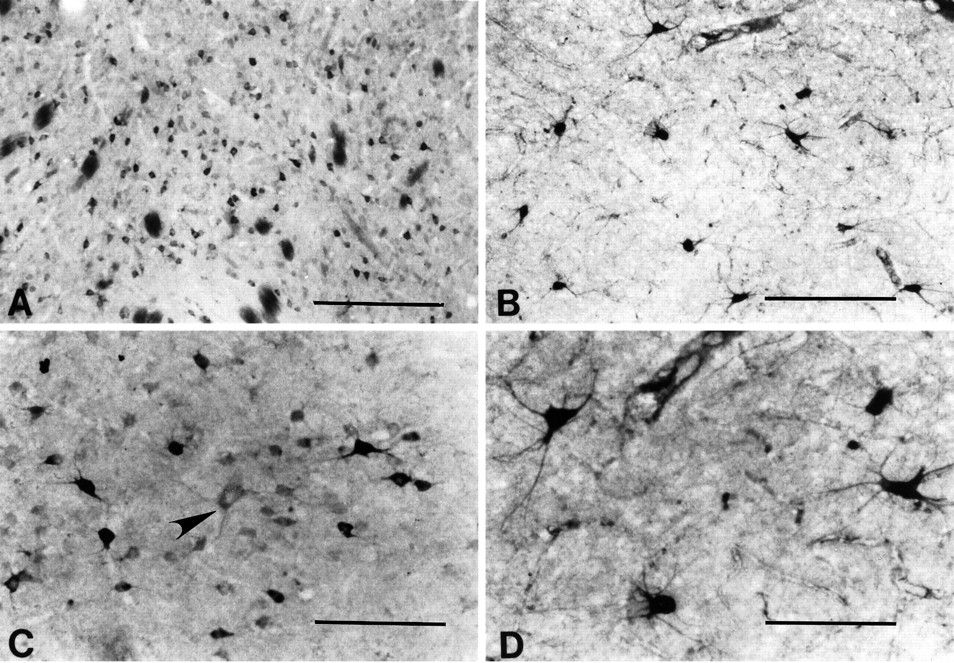
The largest striatal neurons were only lightly immunopositive for huntingtin or did not express huntingtin at all (Fig. 8). Large neurons, moderately immunostained for huntingtin, rarely were observed. ChAT selectively labeled large aspiny striatal neurons. In a comparison between huntingtin neurons and ChAT-positive neurons from contiguous sections, those neurons that were ChAT-positive were significantly larger than any of the huntingtin-positive neurons. These large ChAT-positive neurons were similar in size to weakly or negatively huntingtin-stained large neurons (range, 50–90 μm; mean 68.5 ± 8.7; p> 0.01) (Fig. 8).

Comparison of huntingtin-positive (A, C) and choline acetyltransferase (ChAT)-positive neurons (B, D) in the caudate nucleus. At lower power note the differences in neuronal density and size of each neuronal type. A large neuron, lightly immunostained for huntingtin in C(arrowhead), is morphologically similar to the ChAT neurons observed in D. Magnification bars: inA, B, 500 μm; inC, D, 200 μm.
Postmortem interval and huntingtin
In all cases studied, there was a rapid loss of huntingtin immunoreactivity associated with the number of postmortem hours and the time course in weeks after tissue sectioning. Postmortem specimens fixed after 12 hr from death resulted in relatively poor huntingtin immunostaining within the neostriatum and were not used in these studies. Between the second and fourth week after frozen-sectioning and cold (4°C) storage, the heterogeneous regional and neuronal pattern of huntingtin expression observed after immediate tissue immunostaining was altered. The lightly immunoreactive neurons and huntingtin expression within the neuropil were no longer observed, whereas the more intensely stained medium-sized striatal matrix neurons were still present (Fig. 9). Residual areas of neuronal and neuropil immunostaining resembling patches often were found. These patchy areas of huntingtin immunostaining, however, did not correspond to striatal patch compartments in contiguous stained sections for both calbindin D28k and enkephalin. They were present within the matrix compartment. After 4–8 weeks, the topographic heterogeneity was absent entirely, and most neurons were unstained. The rate of huntingtin immunoreactivity loss was greater than any other neurochemical compound used in these studies.
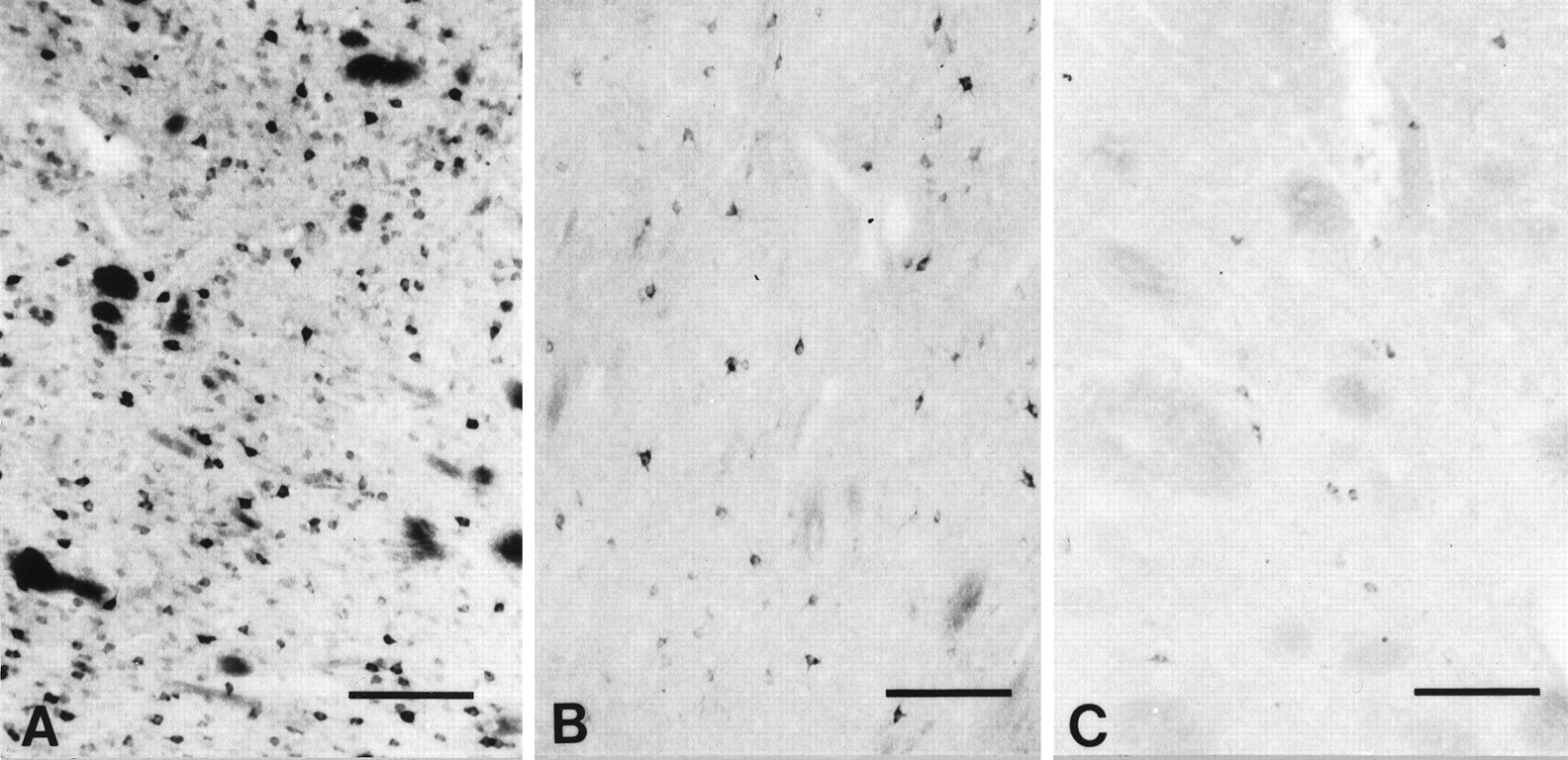
In comparison to tissue sections of the caudate nucleus immunoreacted for Huntingtin (HDp549) directly after tissue sectioning (A), the increased time interval between tissue sectioning and cold storage (4°C) and the subsequent immunostaining with huntingtin antisera resulted in a loss of huntingtin immunoreactivity. Between 2 and 4 weeks (B), there was a significant loss of both neuronal and neuropil immunolabel. After 4 weeks (C), only a few faintly huntingtin-immunostained neurons can be identified. Magnification bars in A–C, 300 μm.
Results in a time course study of the postmortem stability for huntingtin at 0, 24, and 48 hr using immunoblots from unfixed tissue of human cerebral cortex obtained immediately from temporal lobectomy surgery reflected our immunohistochemical findings. A full-length 320 kDa band, clearly visible at 0 hr, was almost undetectable at 24 hr and entirely absent at 48 hr (data not shown). The present results, thus, are dependent on rapid autopsy and immunohistochemistry directly after sectioning of the tissue specimens.
DISCUSSION
Using three distinct anti-fusion huntingtin protein antibodies, we have found a striking heterogeneous organization of huntingtin expression throughout the adult human neostriatum, which conforms to the striatal patch (striosome) and matrix compartments. Huntingtin immunoreactivity was confined primarily to neurons and the neuropil within the matrix area, with low levels of expression in the patch compartments. There was marked variability in the intensity of huntingtin expression among medium-sized striatal neurons. Little or no huntingtin immunoreactivity was present within large striatal neurons. Combined immunofluorescent and enzyme histochemical techniques revealed that huntingtin expression colocalized with calbindin D28k, with little or no coexistence for NADPH-d and NOS activities.
The results of this study show that huntingtin immunoreactivity is associated with striatal spiny neurons and the matrix compartment, both of which have been widely reported to be severely affected in HD. There was little or no localization of huntingtin immunoreactivity to neurons and striatal regions (striosomes) that are relatively spared in this disorder. These observations suggest that the selective neuronal vulnerability to the degenerative process in HD may be dependent on the levels of huntingtin found in affected neurons and provide an explanation for the pattern of striatal neuron loss.
The neuropathological findings in HD have shown that there is a selective pattern of neuronal degeneration within the striatum. Medium-sized spiny striatal neurons, and those neurochemical substances contained within them, are disproportionately affected early and most severely in HD (Marshall et al., 1983; Graveland et al., 1985; Ferrante et al., 1986, 1991; Seto-Oshima et al., 1988; Goto et al., 1989), whereas large and medium-sized aspiny striatal neurons and their chemical components are relatively spared (Dawbarn et al., 1985;Ferrante et al., 1985, 1987a,b; Albin et al., 1990).
Several studies in HD suggest that there is greater pathological involvement within the striatal matrix zone than within the patch compartments (Ferrante et al., 1986, 1987a; Kowall et al., 1987;Seto-Oshima et al., 1988; Richardson, 1990; Ferrante, 1991; Faull et al., 1993). The total area of the matrix compartment, as defined by calbindin and acetylcholinesterase activities, is decreased significantly in HD, whereas the total area of the patch compartments remains within normal limits (Ferrante et al., 1986, 1987a; Kowall et al., 1987; Seto-Oshima et al., 1988). The range in diameter size of striosomes is approximately the same in HD and controls (Ferrante et al., 1987a). Faull and colleagues have observed that patches of GABA–benzodiazepine receptors are relatively spared in HD (Faull et al., 1993). In addition, the homogeneous distribution of glutamate receptors within the normal striatum becomes patchy in HD and conforms to areas of low acetylcholinesterase activity (striosomes) (Olsen et al., 1986). The present findings that huntingtin immunoreactivity is associated primarily with the matrix compartment are consistent with the relative sparing of the patch compartment in HD.
It is of interest that a recent study in HD reports that islands of astrogliosis and neuronal loss corresponding to the patch compartments are present before and during the progressive dorsoventral gradient of striatal degeneration (Hedreen and Folstein, 1995). Patchy gliosis and islands of neuronal loss were not, however, observed in our systematic neuropathological grading of 163 striata from clinically diagnosed HD patients with low, moderate, and severe striatal pathology (Vonsattel et al., 1985). The topographic distribution and the number of neuronal, astroglial, and oligodendroglial cells within the striatum were analyzed carefully in these patients. In addition, patches or islets of preserved neurons in juvenile HD have been reported (Vonsattel et al., 1992). The rapid course of neurodegeneration in juvenile HD may not, however, reflect accurately the neuropathology in adult onset HD. Another quantitative study did not show any significant differences between patch and matrix neuronal loss in HD (Ferrante et al., 1989). The pathological severity may have been too great to detect any differential neuronal degeneration in either striatal compartment.
Indirect evidence also suggests that the patch compartments are spared in HD. Neurons in the striatal matrix compartment primarily project to the substantia nigra reticulata (SNR), whereas patch neurons project to the substantia nigra compacta (Gerfen et al., 1985, 1987). There is a significant reduction in the number of neurons and neuropil area within the SNR of HD patients, whereas the substantia nigra compacta is relatively spared (Ferrante et al., 1989; Richardson, 1990; Ferrante, 1991). The atrophy and neuronal loss within the SNR may reflect the loss of striatal matrix afferents and transneuronal degeneration. This is consistent with findings of SNR neuronal loss in experimental striatal excitotoxic lesions (Saji and Reis, 1987).
The variability of huntingtin expression in striatal spiny neurons may play a role in the differential loss of projection neurons containing enkephalin and substance P in HD. Enkephalinergic neuronal death is reported to precede that of substance P in HD (Albin et al., 1991;Reiner et al., 1988; Sapp et al., 1995). It is possible that these two neurochemically distinct populations of medium spiny neurons do not express equal levels of huntingtin. If greater huntingtin expression correlates with neurodegeneration, substance P neurons may contain less huntingtin immunoreactivity than enkephalinergic neurons.
In addition to NADPH-d/NOS neurons containing little or no huntingtin expression, large ChAT-positive neurons also showed little or no expression. Both of these neuronal populations are relatively spared in HD (Ferrante et al., 1985, 1987; Albin et al., 1990), again suggesting that higher levels of huntingtin may play a role in increased vulnerability. Previous studies reported greater huntingtin immunoreactivity within large striatal neurons with moderate immunostaining in medium-sized neurons (Gutekunst et al., 1995; Bhide et al., 1996). The present observations suggest that the most intense huntingtin-immunoreactive neurons do not coincide with the large ChAT-positive striatal neurons. Species and methodological differences may play roles in this apparent discordance.
In the present study delayed immunostaining resulted in the loss of huntingtin immunoreactivity. Residual areas of patchy neuronal and neuropil immunostaining were present but did not conform to striatal patch compartments. We suggest that these residual circumscribed areas of huntingtin-immunolabeled striatal neurons were attributable to the variable loss of immunoreactivity within the stored tissue sections over time and not representative of in vivo conditions. These results may explain why huntingtin-positive neuronal staining was not identified within the striatum in previous studies (Sharp et al., 1995; Trottier et al., 1995).
In contrast to the present findings, the Emory investigators within our group previously reported a patchy expression of huntingtin immunoreactivity within the normal human striatum (Gutekunst et al., 1995). We are uncertain exactly why this was found in their preliminary work; however, there were differences in tissue preparation and immunostaining. Tissue sections underwent extended storage before staining, and a monoclonal huntingtin antibody was used that may have different sensitivity characteristics than the polyclonal antibodies used in the present study. It is possible that the previous patchy results were attributable to an uneven loss of immunoreactivity, as described in this work.
The mechanism by which the gene defect in huntingtin contributes to neuronal degeneration in HD remains obscure. The targeted disruption of both copies of the HD gene leads to fetal death, suggesting a fundamental role in cellular survival (Duyao et al., 1995; Zeitlin et al., 1995). The loss of one copy, however, has no HD phenotype (Gottfried et al., 1981). It therefore seems that the gene defect causes a gain of function. A number of possible processes by which the HD gene might act at the protein level have been proposed. Proteins with expanded polyglutamine tracts could serve as substrates for transglutaminases and become cross-linked to lysine donors, leading to aggregation of the protein within the cell (Green, 1993). The excessive polyglutamine stretches found in HD may disrupt neuronal function via interactions with other proteins (Albin and Tagle, 1995), such as huntingtin-associated protein 1 (Li et al., 1995). Our results suggest that greater levels of huntingtin correlate with increased vulnerability.
Another hypothesis as to the pathogenesis of HD is that an impairment of energy metabolism may play a critical role by rendering neurons vulnerable to excitotoxicity (Albin and Greenamyre, 1992; Beal, 1992,1994, 1995). There is increasing evidence to suggest that there may be a relationship between the genetic abnormality and a defect in cellular energetics in HD. Consistent with this possibility, it has been reported recently that huntingtin and the dentatorubral–pallidoluysian atrophy gene product (atrophin) both may bind to the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Burke et al., 1996;Roses, 1996). GADPH is a critical enzyme for glycolysis and the production of acetyl-CoA in the Kreb’s cycle. An alteration of GAPDH activity resulting from an interaction with mutant huntingtin therefore could contribute to energy impairment in HD. No matter what the mechanism is by which huntingtin expression leads to neuronal death, the present results suggest that differences in huntingtin expression in striatal neurons may account for selective neuronal vulnerability in HD.
Footnotes
This work was supported by the Huntington’s Disease Society of America (R.J.F.), the Department of Veterans Affairs (R.J.F., N.W.K.), the Emory University Research Committee (S.M.H.), the Huntington’s Disease Foundation (C.A.G.), and National Institutes of Health Grants AG12922 (R.J.F., M.F.B., N.W.K.), 1P30AG13846 (R.J.F., N.W.K.), NS16367 and NS10828 (M.F.B.), NS16367 (J.F.G., F.P., S.M.M., M.E.M.), NS01624 (S.M.H.), and NS35255 (S.M.H., C.A.G., R.J.F.). We thank Karen Smith and Tom Kilgallen for their technical assistance.
Correspondence should be addressed to Dr. Robert J. Ferrante, Geriatric Research Education Clinical Center, Unit 182B, Bedford VA Medical Center, 200 Springs Road, Bedford, MA 01730.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}