

Abstract
Neuronal migration is an essential feature of the developing nervous system, but the intracellular signaling mechanisms that regulate this process are poorly understood. During the formation of the enteric nervous system (ENS) in the moth Manduca sexta, the migration of an identified set of neurons (the EP cells) is regulated in part by the heterotrimeric guanyl-nucleotide binding protein (G protein) Goα. Using an in vivo culture preparation for developing embryos that allows direct access to the ENS, we have shown that EP cell migration is similarly regulated by intracellular Ca2+; treatments that increased intracellular Ca2+inhibited the migratory process, whereas buffering intracellular Ca2+ induced aberrant migration onto inappropriate pathways. Imaging the spontaneous changes in intracellular Ca2+ within individual EP cells showed that actively migrating neurons exhibited only small fluctuations in intracellular Ca2+. In contrast, neurons that had reached the end of migration displayed large, transient Ca2+ spikes. Similar Ca2+ spikes were induced in the EP cells by G protein stimulation, an effect that was reversed by removal of external Ca2+. Stimulation of Go in individual EP cells (by injection of either activated Goαsubunits or mastoparan) also inhibited migration in a Ca2+-dependent manner. These results suggest that the regulation of neuronal migration by G proteins involves a Ca2+-dependent process requiring Ca2+ influx.
Within the developing nervous system, many neurons undergo a transient period of migration, during which their somata actively move to unique and sometimes distant locations. The guidance of motile cells along an appropriate pathway typically involves a combination of repulsive and attractive molecules (Fishman and Hatten, 1993; Muller and Kypta, 1995); however, the intracellular events by which these cues are transduced into an integrated migratory response are unknown. Previously, we demonstrated that Goα, a member of the heterotrimeric class of guanyl-nucleotide binding proteins (G proteins), is expressed by embryonic neurons as they migrate into the enteric nervous system (ENS) of the moth, Manduca sexta. Specifically, we showed that during the formation of the enteric plexus, (a nerve plexus that spans the foregut–midgut boundary), a set of ∼300 neurons (EP cells) (Copenhaver and Taghert, 1989a) first begin to express Goα coincident with their migration along stereotyped pathways on the gut musculature (Horgan et al., 1994). Furthermore, the accessibility of the ENS has permitted direct manipulations of these neurons within the embryo; injections of mastoparan (which stimulates Goα and Giα) into individual EP cells inhibited migration, an effect that was reversed by pertussis toxin (Horgan et al., 1995). These results suggested that Go-mediated events within the EP cells might serve to inhibit or terminate their migratory behavior. However, the mechanism by which G proteins downregulate migration remained undefined.
Work on other systems has indicated that the effects of Goactivation are often linked to the regulation of intracellular Ca2+, either by the modulation of voltage-dependent Ca2+ channels (Schultz et al., 1990; Wickman and Clapham, 1995) or via the regulation of phospholipase C (PLC)- or protein kinase C (PKC)-dependent pathways (Xie et al., 1995; Pan et al., 1997). In turn, the regulation of intracellular Ca2+ is known to play an important role in controlling neuronal motility, although the effects of Ca2+ can be either inhibitory or stimulatory, depending on both cell type and experimental context. For example, neural crest cells migrate precociously when exposed to Ca2+ channel antagonists (Newgreen and Gooday, 1985), whereas cerebellar granule cells are apparently stimulated by Ca2+ influx (Komuro and Rakic, 1996). With respect to Go, the potential role of Ca2+in mediating its inhibitory effect on migration has not been explored.
In this paper, we have examined the interaction of these two intracellular signaling molecules during neuronal migration by examining their respective effects on the EP cells in an in vivo culture preparation. Specifically, we have tested the role that Ca2+ plays in regulating EP cell migration by manipulating extracellular and intracellular levels of Ca2+ during the migratory period. We have also used activated α-subunits to demonstrate the specific effects of Goα on migration, and we have tested whether the inhibition of the EP cells by Goα is Ca2+-dependent. Last, we have examined the effects of G protein activation on spontaneous changes in Ca2+ within individual EP cells as they migrate in the developing embryo.
MATERIALS AND METHODS
Animal preparation and culture. Animal dissection and culture were performed as described previously (Horgan et al., 1994,1995). Briefly, timed egg collections from an M. sextacolony were maintained at 25°C, at which temperature embryonic development lasts ∼100 hr (1 hr = 1% development). Embryos were dissected from the egg shell and underlying membranes and restrained in a Sylgard-coated chamber in either culture medium (50% Schneider’sDrosophila medium, 40% MEM with Hank’s salts, 9.7% heat-inactivated fetal calf serum, 0.2% ecdysone, 0.1% insulin, and 0.01% penicillin-streptomycin) or a defined saline (in mm: 140 NaCl, 5 KCl, 28 glucose, 5 HEPES, 0.2% ecdysone, 0.1% insulin, and 0.01% penicillin-streptomycin, pH 7.4). In experiments in which external Ca2+ concentrations were manipulated, the solutions were osmotically balanced with mannitol. A small incision was made in the dorsal epidermis above the foregut–midgut boundary to expose the premigratory EP cell population. The animals were then allowed to develop in culture for 8–16 hr in a humidified chamber at 28°C.
Bath application of water-soluble compounds, including caffeine (Sigma, St. Louis, MO) and BAPTA (Molecular Probes, Eugene, OR), were diluted into culture medium or defined saline. A23187 (Sigma), BAPTA AM (Molecular Probes), IBMX (Sigma), and ionomycin (Calbiochem, La Jolla, CA) were dissolved into 100% DMSO and then diluted into culture medium or defined saline with a final concentration of 0.5–1% DMSO. AlF4− was made by diluting premixed solutions of 0.6m NaF and 0.5 mm AlCl3 at 1:40 into the culturing solution (Horgan et al., 1994) to give a final concentration of 12.5 μm AlF4−. Equivalent concentrations of NaCl premixed with AlCl3served as a control.
After culturing, animals were dissected completely and fixed with 4% paraformaldehyde in PBS. To visualize the ENS, the preparations were immunohistochemically stained with a monoclonal antibody againstManduca fasciclin II (MFasII, at 1:20,000; a gift of Dr. Paul Taghert, Washington University, St. Louis, MO), followed by the avidin–biotin–HRP protocol from Vector Laboratories (Burlingame, CA). The distribution of postmigratory EP cells was then analyzed using camera lucida techniques. The extent of migration in each experiment was calculated by measuring the distance of the farthest cell from the foregut–midgut boundary on the four middorsal migratory pathways for each preparation. These values were then normalized with respect to the mean value for migration obtained from matched control cultures (to account for any variability in experimental conditions), and the means and SDs for each experimental condition were then calculated from these normalized values. Statistical analyses were performed using a two-tailed Student’s t test to compare the means of matched experimental and control groups.
Intracellular injections. Myristoylated rat recombinant Goα subunits (10 μm; Calbiochem) were activated by a 30 min incubation with 10 μm GTPγS (in mm: 10 MgCl2, 100 NaCl, 1 EDTA, and 20 HEPES, pH 8.0) at 20°C. Control injections were performed with an identical solution lacking Goα. Heat inactivation of activated Goα subunits was performed by boiling for 30 min before injection. Biologically active Gβγ subunits (bovine brain, 1 μm; Calbiochem) was also injected as an additional control. Mastoparan (100 μm; Calbiochem) was initially dissolved in distilled H20 and stored at −80°C. Just before use, injection solutions were diluted 1:10 with an intracellular buffer containing (in mm): 132 K-Asp, 64 mannitol, 5 HEPES, 2 EGTA, 2 MgATP, and 1 CaCl2 plus rhodamine–dextran and biotin–dextran (both at 20 μg/μl; Molecular Probes). Glass electrodes (30–50 MΩ) were used for pressure injection of these solutions into individual EP cells (Horgan et al., 1995). Injected cells were then briefly visualized with a heavily filtered UV lamp source and the intensity and position of the cell noted. In neurons injected with control solutions, this brief UV exposure was not found to disturb normal migration. We estimate that the actual concentration delivered into cells was 1:100 of the pipette concentration. After additional development in culture, preparations were fixed, incubated in avidin–HRP, and reacted in the presence of 1% NiCl to label the injected cells with a black reaction product (Horgan et al., 1995). The preparations were then counterstained with anti-MFasII antibodies (as described above) to label the surrounding EP cells with a brown reaction product. Migratory distances were then calculated for the injected neurons as a ratio of the total distance migrated by the leading EP cell on the same muscle band pathway. Normalized values for each experimental condition were then averaged and compared with the mean values obtained from matched sets of control injections using a two-tailed Student’s t test. For fluorescent labeling of single cells, 10 mg/ml 1,1′-dioleyl-3,3,3′,3′-tetramethylindocarbocyanine methanesulfonate (DiI; Molecular Probes) diluted in methanol was injected as described previously (Copenhaver et al., 1996).
Intracellular calcium imaging. Individual EP cells were injected with the calcium indicator calcium green-1 dextran (Mr 10,000, at 10 mg/ml; Molecular Probes). Embryos were then inverted and restrained in chambers made from Sylgard-coated glass coverslips. Cells were observed through a 60× Plan Apochromat water immersion lens attached to a Nikon Diaphot 200 inverted microscope that was mounted on a vibration isolation table. A heavily filtered UV light source from a 100 W mercury bulb was passed through an FITC (450 nm excitation, 535 nm emission) filter (Omega, Brattleboro, VT). The duration and timing of excitation was controlled with a digital IO board (National Instruments, Austin, TX) connected to a Lambda-10 filter wheel (Sutter Instruments, Novato, CA). A Cohn 12V CCD camera and Hamamatsu Intensifier mounted to the microscope were used to capture and enhance the emitted signal. Data were processed using a Power PC 7100 (Macintosh, Cupertino, CA) interfaced with a PixelPipeline (Perceptics) data acquisition imaging board. The image analysis software used was Cytos III (Applied Scientific Instrumentation Inc., Springfield, OR). Twelve frames were captured every 5–10 sec and averaged to produce individual images. The images were then analyzed for changes in relative fluorescent intensity. Average pixel intensities within a rectangular box surrounding each EP cell (32 × 32 pixels per box), as well as in similarly sized boxes to measure background levels, were monitored simultaneously. Focus and box placement were adjusted approximately every 2–3 min to correct for cell movements. Although this procedure did result in small changes in apparent cell intensity, the fluctuations were not significantly above background noise. The duration of most experiments was 30 min; however, a few neurons were observed for up to 2 hr. During experiments involving AlF4−, baseline images were recorded for 10 min before application. Immediately after AlF4− was added to the bath, cells were refocused and then imaged over the next 20 min.
RESULTS
Neuronal migration of EP cells during embryogenesis
During the formation of the ENS, ∼300 postmitotic EP cells emerge as a group from a neurogenic placode in the foregut epithelium (Copenhaver and Taghert, 1990), and then migrate into the enteric plexus on the surface of the gut musculature (Fig.1) (Copenhaver and Taghert, 1989b). By immunostaining embryos with antibodies to the adhesion molecule fasciclin II (designated in Manduca as MFasII), the sequence of EP cell development could be readily visualized. Before migration commences (at 55% of embryogenesis; Fig. 1A), the EP cells reside in a packet encircling the gut at the foregut and midgut boundary (fg/mg; designated by the small black bar in each panel). As migration begins, individual neurons extend leading processes across the fg/mg boundary and align themselves with one of eight specific longitudinal muscle bands on the midgut (L1–L4 and R1–R4; Figs.1B–D). The EP cell bodies migrate posteriorly for 5–7 hr (Fig. 1, B–D, solid arrows, E), traveling up to 200 μm and distributing themselves along the muscle bands. Anteriorly, a small number of EP neurons also migrate laterally onto the radial muscle bands of the foregut (Fig.1B,C, open arrows). At the end of migration (60–62% of development), the neurons then extend axonal processes posteriorly and laterally to innervate the gut musculature. Of note is the specificity by which the EP cells follow their migratory pathways on the midgut: once aligned with one of the muscle bands, each neuron invariably remains closely apposed to the fibers of that band, never wandering onto the adjacent interband musculature (Fig.1E).

Neuronal migration is essential to the formation of the ENS. A–D, Developmental sequence of EP cell migration (visualized by anti-MFasII immunostaining) to show the stereotyped pattern of migration on preformed muscle band pathways; only the middorsal muscle bands of the midgut (L1, R1) are shown. A, At 55% of embryonic development;B, at 57%; C, at 59%; D, at 61%. One percent of development is equivalent to 1 hr;fg/mg indicates the boundary between the foregut and midgut and is marked by the small black bar in each panel. Solid arrows indicate the leading cells onL1 and R1; open arrows show the lateral pathways on the foregut (out of focus). E, Individual EP cells labeled with DiI during migration onto the midgut; panels show a series of neurons at times and positions indicated by the solid arrows in A–D. Note that the filopodia and subsequent axonal extensions remain primarily restricted to the pathway on which the neuron is migrating. F-G, Normal migration of EP cells when embryos were cultured for 10 hr in serum-free defined saline containing 4 mm Ca2+(F) or 0 mmCa2+ (G) (no Ca2+ added to the saline). The removal of external Ca2+ caused no apparent deleterious effect on neuronal development. Scale bar, 20 μm (A–D, F, G), and 50 μm(E).
Effects of calcium manipulations on migration
To determine the effect of altering extracellular levels of Ca2+ on EP cell migration, an in vivoculture preparation was used that permitted access to the migrating neurons and their surrounding environment. Staged embryos were isolated before the onset of migration and cultured for 12–24 hr in a serum-free, defined saline containing 4 mmCaCl2 (a concentration similar to that estimated in complete culture medium containing serum). The extent of migration in these conditions, as measured by the distance traveled by the leading neuron, was slightly reduced (by ∼10%) but otherwise appeared normal when compared to dissected and undissected controls (compare Fig.1C,F). This effect was likely caused by the slight overall reduction in the rate of embryogenesis seen in the absence of serum (data not shown). When the external concentration of Ca2+ was elevated in the defined saline, the development of the EP cells proceeded normally; however, there was a statistically significant decrease in the extent of migration when compared with 4 mm CaCl2 saline controls (Fig.2). In 12 mmCaCl2, the extent of migration was 85% of that measured in 4 mm CaCl2(p < 0.05), whereas in 20 mmCaCl2, migration was only 53% of controls (p < 0.001). The inhibitory effect of elevated Ca2+ was reversible: when embryos were exposed to 20 mm CaCl2 saline for 30 min and then incubated in 4 mm CaCl2 saline for the duration of the culture period, we found that the normal extent of migration had been restored (Fig. 2). Because manipulations of external Ca2+ concentrations have been shown to cause proportional changes in intracellular Ca2+ in a variety of preparations (Rehder et al., 1991; Komuro and Rakic, 1992;Gomez et al., 1995), these results suggested that elevating intracellular Ca2+ caused a corresponding reduction in EP cell motility.

Increased extracellular Ca2+causes a concentration-dependent inhibition of migration. Effects of increasing external levels of Ca2+. Embryos were cultured in defined saline containing 0, 4, 12, or 20 mmCa2+ during the normal period of EP cell migration. An additional set was initially incubated in 20 mmCa2+ saline for 30 min and then cultured in 4 mm Ca2+ saline (histogram labeled20/4 mm ). Extent of migration was subsequently calculated by measuring the distance traveled by the leading neuron on each pathway from the fg/mg boundary as a proportion of control measurements taken from cultures incubated in complete medium. Averaged data are presented as ± SEM. Histograms were normalized to values in 4 mm Ca2+.n ≥ 16 for each condition. *p< 0.05; **p < 0.001.
To test the effects of reduced intracellular Ca2+ on EP cell development, we cultured embryos in saline containing no added CaCl2 (“0 mm”). In contrast to the effects of elevated Ca2+, the extent of EP cell migration was not significantly different from that seen in control cultures incubated with 4 mm CaCl2 (Figs. 1G,2). During these experiments, defined saline was applied to the ENS via a temporary incision in the body wall, which typically resealed during subsequent development in culture. Consequently, we did not attempt to quantify the exact Ca2+ concentration in the 0 mm saline, because we assume that the level of Ca2+ surrounding the EP cells was gradually augmented by local release from cellular stores. We therefore consider the 0 mm Ca2+ saline to be nominally Ca2+-free. Complete removal of trace levels of external Ca2+ with EGTA caused a general dissociation of the migratory neurons and embryonic death (data not shown). Nevertheless, Ca2+ levels were sufficiently depleted in the 0 mm Ca2+ saline to prevent the Ca2+-dependent effects of several ionophores and toxins (described below).
Besides measuring the distance of the leading neuron on each of the migratory pathways (indicated in Fig. 2), we also quantified the extent of migration in these experiments by (1) measuring the average distance traveled by each neuron on a given pathway and (2) measuring the total number of neurons that had migrated on each pathway (data not shown). However, because these alternative methods yielded histograms that were essentially identical to those shown in Figure 2, we used this first form of quantitation for all subsequent manipulations. These results demonstrated that the defined saline used in these studies could support normal EP cell migration in the absence of serum, thereby allowing us to vary external levels of Ca2+ in a controlled manner. In addition, increasing Ca2+levels above 4 mm caused a dose-dependent decrease in migration, suggesting that proportional elevations in intracellular Ca2+ within the EP cells may inhibit their motility.
As a more direct means of manipulating intracellular Ca2+, we used the ionophores A23187 (Reed and Lardy, 1972) and ionomycin (Liu and Hermann, 1978) to permeabilize the neurons to Ca2+. The ionophores were applied externally to the EP cells by their addition to the culture medium at the beginning of the migratory period. When applied in normal culture medium, both of these compounds caused a dose-dependent decrease in EP cell migration (Fig. 3A). In low ionophore concentrations (1 μm), the distance traveled by the EP cells was reduced by 20% (p < 0.01) but the neurons proceeded along their normal pathways. At higher concentrations, even a brief exposure to either of the ionophores completely inhibited the migration of most neurons, with only a few EP cells traveling for short distances from the original foregut packet onto the midgut. As an additional means of increasing intracellular levels of Ca2+ in the EP cells, we also applied caffeine (Palade et al., 1989; Mironov and Usachev, 1991) to cultured embryos at the onset of migration. Similar to the ionophores, caffeine caused a statistically significant and concentration-dependent inhibition of migration (Fig. 3A). As with the effects of elevated extracellular Ca2+ (Fig. 2), the inhibitory action of caffeine was reversed when preparations were rinsed after 30 min with normal culture medium (Fig. 3A). Besides inducing the release of internal Ca2+ stores, caffeine has also been reported to inhibit phosphodiesterase activity (Nehlig et al., 1992). However, when we applied IBMX (100 μm) to control for this effect, we observed no change in the extent of EP cell migration (data not shown). Because IBMX and caffeine have generally been found to have similar membrane permeabilities (Kehoe, 1990;Usachev et al., 1995; Gommerat and Gola, 1996), this observation supports our conclusion that the effect of caffeine on EP cell migration resulted from the release of intracellular Ca2+.
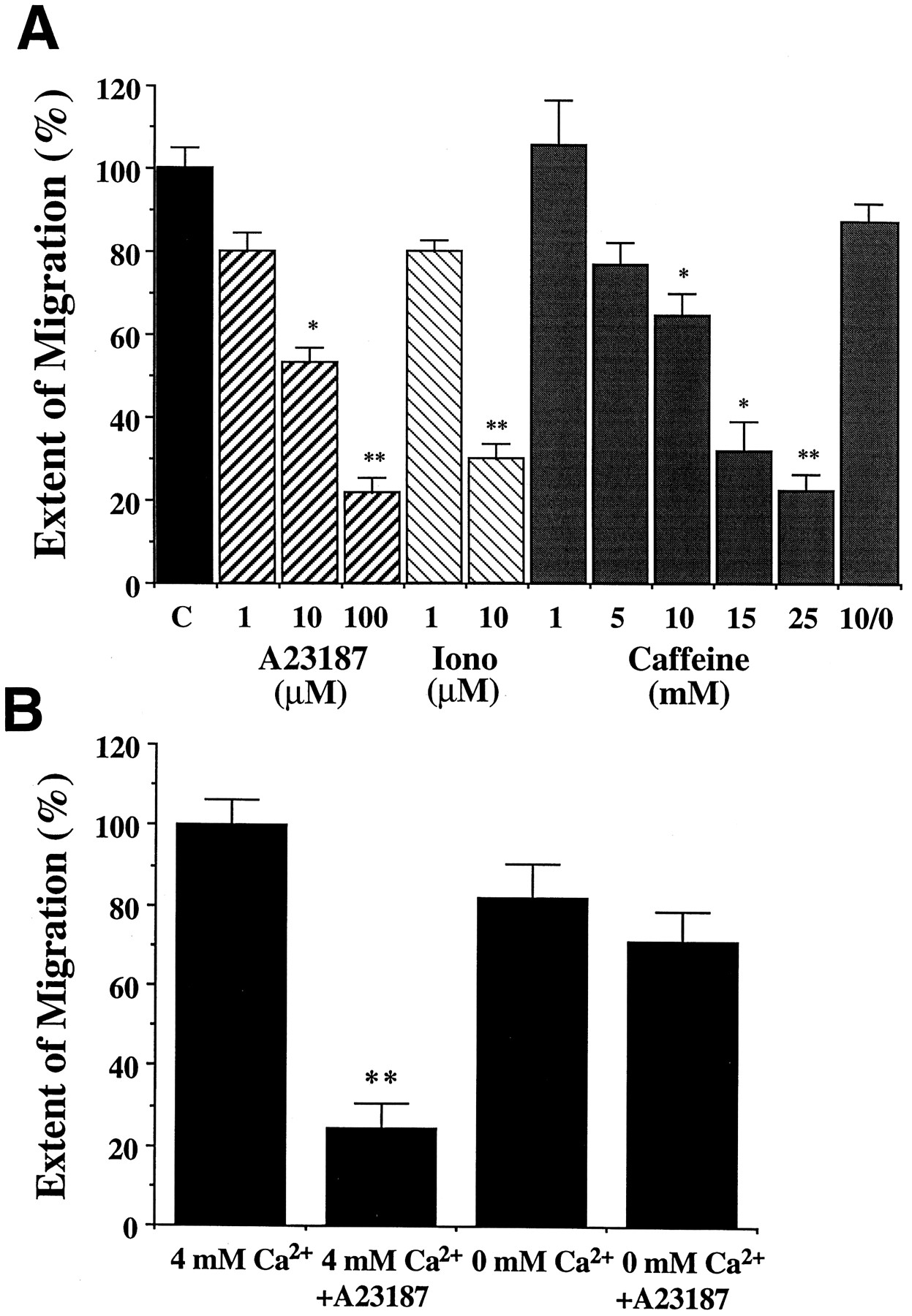
Increased intracellular Ca2+causes a concentration-dependent inhibition of migration.A, Effects of increasing internal levels of Ca2+. Embryos were cultured in complete culture medium containing A23187, ionomycin, or caffeine at indicated concentrations during the migratory period. Preparations cultured in 100 μm A23187 were rinsed after 2 hr and incubated in normal culture medium until the completion of the experiment. An additional set was treated with 10 mm caffeine for 30 min and then cultured in caffeine-free medium (histogram labeled10/0 mm ). The distance of migration was normalized to values taken from matched cultures. Averaged data are presented as ±SEM; n = 8–20. *p < 0.05; **p < 0.001.B, The inhibitory effect of A23187 is markedly attenuated in 0 mm Ca2+ saline. Preparations were cultured in serum-free defined saline containing 4 or 0 mm Ca2+ ± 3 μm A23187. The distance of migration was normalized to that of control embryos cultured in saline containing 4 mm Ca2+. Averaged data are presented as ±SEM; n ≥ 12. **p < 0.001.
Because all of our experiments were conducted within the developing embryo, we were unable to use UV-activated compounds such as caged Ca2+ to induce selective changes in intracellular Ca2+ within individual neurons. It is therefore possible that some of the manipulations described above might have affected migration indirectly, either by causing a general disruption of the developing ENS or by perturbing the surrounding musculature on which the EP cells migrate. However, none of the treatments that we used to manipulate intracellular Ca2+ altered the morphology of the EP cells or their expression of MFasII, indicating that these manipulations did not overtly damage the neurons. Rather, the elevation of intracellular Ca2+ levels (either by increasing the cell permeability to Ca2+ or by releasing Ca2+ from intracellular stores) consistently caused a dose-dependent decrease in cell migration. These results are consistent with the effects of exposing the EP cells to high Ca2+ saline (Fig. 2), suggesting that elevation of intracellular Ca2+ inhibits or downregulates the migratory process selectively.
To show that the effect of the ionophores was dependent on external Ca2+, we performed an additional experiment with A23187 applied in defined saline with and without added Ca2+ (Fig. 3B). When an intermediate concentration of A23187 (3 μm) was applied to cultured embryos in defined saline containing 4 mmCaCl2, there was a robust inhibition of migration. The more dramatic effect of A23187 in defined saline compared with A23187 in normal culture medium (Fig. 3A) was caused by the absence of serum, and might reflect either nonspecific binding of the ionophore by serum proteins (not present in defined saline) or stabilization of the neuronal membranes by components included in the more complex culture medium. In this experiment, control preparations cultured in saline without Ca2+ showed a slight reduction in migration but were not significantly different from controls. In contrast, when A23187 was applied in 0 mmCa2+, its inhibitory effect was greatly attenuated (Fig. 3B). The reduction in migration caused by the ionophore changed from an 86% inhibition in 4 mmCa2+ to only a 26% inhibition in 0 mmCa2+, which was not significantly different from the 0 mm Ca2+ controls. These results indicated that the inhibitory effect of the ionophore was dependent on extracellular Ca2+, supporting our conclusion that manipulations that increase cytoplasmic Ca2+ in the EP cells cause a concentration-dependent inhibition of neuronal migration.
To examine the effects of reducing intracellular levels of Ca2+ within the EP cells, we applied BAPTA AM, a membrane-permeable form of the Ca2+ chelator BAPTA (Tsien, 1980; Dickens et al., 1990), which is activated intracellularly after hydrolysis by cytosolic esterases. BAPTA AM was applied in 0 mm Ca2+ saline to the EP cells at the onset of migration, and the embryos were subsequently cultured for 8 hr. Both embryonic development and EP cell migration proceeded in these experimental conditions; however, in many of the embryos, the normal guidance of the EP cells was severely disrupted. Whereas neurons cultured in 0 mm Ca2+ saline exhibited a normal pattern of migration (Fig.4A), EP cells exposed to BAPTA AM under these conditions displayed extensive misrouting onto inappropriate musculature. Figure 4B shows an example of a plexus treated with BAPTA AM, in which a large number of neurons wandered off their longitudinal muscle band pathways (arrows) and migrated onto the interband musculature. The severity of this effect was decreased if BAPTA AM was applied in saline containing 4 mm CaCl2 (Fig. 4C), presumably because of the ability of EP cells to compensate for buffered intracellular Ca2+ concentrations by a mechanism involving Ca2+ influx. However, some signs of aberrant pathfinding were still seen on the foregut under these conditions (arrowheads). In contrast, when a cell-impermeable form of BAPTA (100 μm) was applied in 0 mm Ca2+ saline as a control, no defects in migration were observed (Fig. 4D). Although BAPTA AM might affect migration indirectly by inhibiting the formation of the muscle band pathways, previous data have shown that removal of the pathways by surgical means completely prevents EP cell migration rather than promoting their dispersal onto the interband musculature (Copenhaver et al., 1996). Although it is conceivable that BAPTA AM might selectively perturb the expression of particular guidance cues on the visceral musculature, thereby creating an abnormally permissive environment for undirected migration, we think it more likely that BAPTA AM acts by disrupting the normal intracellular mechanism by which the neurons respond to their normal guidance cues, a conclusion that is supported by the experiments described below.

Buffering intracellular Ca2+disrupts normal pathfinding in migration. Camera lucida drawings of individual preparations representative of experiments in which embryos were cultured during the migratory period in the following conditions:A, 0.5% DMSO control in defined saline with 0 mm Ca2+ (n = 24);B, 100 μm BAPTA AM in 0 mmCa2+ saline (n = 15);C, 100 μm BAPTA AM in 4 mmCa2+ saline (n = 8);D, 100 μm BAPTA (noncell permeable) in 4 mm Ca2+ saline (n = 8). Solid arrows indicate disrupted migration of the EP cells; arrowheads indicate misdirected process outgrowth. Scale bar, 75 μm.
The effect of BAPTA AM could be attributable to a reduction in the overall cytoplasmic level of Ca2+ (Dickens et al., 1990) or to the ability of this compound to suppress fluctuations of Ca2+ within the cell (Kuijpers et al., 1992; Gu and Spitzer, 1995). Our results support the latter, because simply lowering Ca2+ levels by incubating the EP cells in 0 mm Ca2+ saline (Fig. 2) or in external BAPTA (Fig. 4D) did not by itself alter their migration. However, the fact that neither of these treatments disrupted migration could be attributable to either the incomplete removal of Ca2+ or the release of Ca2+ into the medium from the embryo itself. Residual levels of Ca2+ in our nominally free Ca2+saline might therefore be sufficient to support migration, although not the inhibitory action of the Ca2+ ionophore A23187 (Fig. 3B). Alternatively, the compensatory release of Ca2+ from intracellular stores might be sufficient to sustain neuronal motility, even when external Ca2+ had been substantially reduced. Nevertheless, these results suggest that buffering intracellular Ca2+ in the EP cells as they migrate may alter a Ca2+-dependent signaling mechanism involved in restricting the neurons to their normal pathways.
G protein-dependent inhibition of migration requires calcium
Previously, we presented evidence that G protein activity within the EP cells can regulate their motility. Both GTPγS (which activates all G proteins; Gilman, 1987) and AlF4− (which stimulates heterotrimeric G proteins but not the Ras-related family of small G proteins; Sternweiss and Gilman, 1982; Anderson et al., 1991;Kahn, 1991) caused a dose-dependent inhibition of neuronal migration (Horgan et al., 1994). More specifically, we showed that intracellular injections of the wasp toxin mastoparan (which specifically activates Go and Gi family subtypes; Higashijima et al., 1990) inhibited migration in a pertussis toxin-sensitive manner (Horgan et al., 1995). Because Goα is the only G protein in insects known to be sensitive to pertussis toxin (Quan et al., 1989;Thambi et al., 1989) and is the only G protein that we have detected in the developing EP cells (Horgan et al., 1994), these results suggested that Goα serves an inhibitory function with respect to neuronal motility. However, to verify the specificity of this effect, we injected constitutively activated Goα subunits (Goα*) into individual EP cells at the onset of their migration. As shown in Figure5A, injections of Goα* caused a 42% decrease in the extent of EP cell migration when compared with injected control neurons, an effect that was highly significant (p < 0.001). In contrast, injections of heat-inactivated Goα* had no significant effect on migration. Injections of biologically active Gβγ subunits similarly did not perturb EP cell migration or differentiation (data not shown). These results definitively show that activation of Goα within the EP cells regulates their migratory behavior in an inhibitory manner.

Goα stimulation inhibits migration and requires the presence of external Ca2+.A, Individual EP cells were injected at the onset of migration with a solution of dextran-conjugated dyes plus or minus GTPγS-activated Goα subunits (1 μm). Control injections were performed using the activation solution without Goα. Heat-inactivated solutions of Goα were boiled for 30 min before injection. After culturing the embryos for 12–16 hr, the percent of maximal migration was calculated by measuring the distance that the injected neuron had migrated as a proportion of the distance traveled by the leading neuron on the same pathway. n ≥ 30 for each histogram. Averaged data are presented ± SEM; **p < 0.001. B, Embryos were cultured in defined saline containing 4 or 0 mmCa2+, and individual EP cells were injected at the onset of migration with a solution of dextran-conjugated dyes ± 100 μm mastoparan. After 12–16 hr in culture, the percent of maximal migration was calculated as described in Figure4A. n ≥ 15 for each histogram. Averaged data are presented ± SEM; **p < 0.001.
Because Go has been shown to act via the modulation of intracellular Ca2+ concentrations in other systems, we examined whether its effect on neuronal migration might also be mediated by Ca2+ in the developing ENS. When mastoparan was injected into migrating EP cells cultured in 4 mm Ca2+ saline (Fig. 5B), we observed a significant decrease (52% of controls) in their migration when compared with injected control neurons (p< 0.001); these results were similar to the effects of mastoparan in the presence of culture medium containing serum (Horgan et al., 1995). However, when embryos were cultured in 0 mmCa2+ saline, mastoparan no longer inhibited the migratory behavior of injected neurons (Fig. 5B). Therefore, the inhibitory effects of both Ca2+ ionophores (Fig.3B) and mastoparan are dependent on the presence of extracellular Ca2+, indicating that Ca2+ influx is required for the G protein-mediated regulation of EP cell migration.
As shown in Figure 5, injection of either Goα* or mastoparan caused only a partial reduction in the average distance of EP cell migration. Possibly, this result was attributable to the limited volume that could be injected into the embryonic neurons without damaging the cells. In addition, however, the partial inhibition of the EP cells by Go might reflect the presence of other, complementary mechanisms that regulate the extent of neuronal migration in vivo. For example, previous attempts to inhibit Goα activity in the EP cells with pertussis toxin did not perturb the normal termination of their migration, although this treatment did cause exuberant axon outgrowth (Horgan et al., 1995). Within the developing embryo, therefore, more than one signaling mechanism may be sufficient to terminate EP cell migration during normal development; alternatively, Goα may serve a more modulatory function, possibly acting as part of an intracellular response to inhibitory cues located on inappropriate pathways.
Effects of G protein activity on intracellular Ca2+ levels in migrating neurons
Having shown that the inhibitory effects of Goα on EP cell migration require Ca2+ influx, we next investigated whether G protein activity induces specific changes in intracellular Ca2+ within the developing neurons. To monitor spontaneous changes in intracellular Ca2+levels, individual neurons were injected with the Ca2+ indicator dye calcium green-1 dextran at selected times during their normal migration. The embryos were then transferred to a microscope equipped with a CCD camera, and the cells were imaged in vivo every 5 sec for 30–120 min periods to monitor changes in fluorescent intensity. During the normal migratory period, we observed small fluctuations in intracellular Ca2+ levels within most of the dye-injected EP cells (Figs. 6A,7A), but no large transients were observed in any of the neurons examined (Fig.7D).

Intracellular Ca2+ activity imaged in migratory neurons before and after AlF4−. Embryo cultured in defined saline containing 4 mmCa2+ in which three EP cells were injected with calcium green-1 dextran. The cells were then imaged for 20 min before (A) and after (B) bath application of 12.5 μm AlF4−. Images were generated by averaging 12 individual frames collected over ∼2 sec. Pseudocolor representation shows relative changes in fluorescent intensity: blue, low; red, high. Each row of time points represents consecutive 30 sec intervals during the imaging period. Arrows highlight increased Ca2+ concentrations in the cell bodies;arrowheads, increased Ca2+concentrations in their leading processes. Scale bar, 10 μm.

AlF4-induced Ca2+spike activity is greatly diminished in the absence of external Ca2+. A–C, Each graph shows the time course of changes in fluorescent intensity measured in three separate neurons (numbered traces). Trace labeledB in each panel represents simultaneous measurements of the background intensity. A, Imaging during the period of migration in 4 mm Ca2+.B, Imaging after the addition of 12.5 μmAlF4− in saline containing 4 mmCa2+. C, Imaging after the addition of AlF4− in saline containing 0 mmCa2+. A, B, Time course of Ca2+ activity in the same three cells shown in Figure 6 (double-headed arrows indicate times when images were captured for generating Fig.6A,B); traces in Crepresent cells from a separate preparation. D–F, Histograms of the number of spikes displayed per injected cell during 20 min of continuous imaging: D, before AlF4− application; E, after AlF4 in 4 mm Ca2+ saline;F, after AlF4− in 0 mmCa2+ saline. A spike was defined as a transient increase in fluorescence >50% of baseline intensity in the cell. The total number of cells in each condition was 56, 27, and 29, respectively.
Because all of the imaging studies were performed in semi-intact embryos on an inverted microscope, it was not possible to monitor changes in intracellular Ca2+ simultaneously with the injection of individual neurons. As an alternative, we applied AlF4− (an activator of all heterotrimeric G proteins) at the same concentration that previously was shown to cause a ∼50% inhibition of EP cell migration (Horgan et al., 1994). When AlF4− was added to the cultured embryos in the presence of 4 mm Ca2+, large, transient increases in Ca2+ occurred in the majority of cells imaged (Fig. 6B). Figure 7, A andB, shows the changes in the average pixel intensities of the same three cells injected in Figure 6 before and after AlF4− administration. After a short delay, all three cells began to spike intermittently. In 81% of the EP cells examined (Fig. 7E), AlF4− induced at least one Ca2+ spike that transiently elevated the fluorescent intensity of the cells by an average of 75% above baseline. The spikes were characterized by a relatively steep average rise time of 12.8 sec, followed by a slower recovery of 54.5 sec before returning to baseline. The number of spikes displayed by an individual cell within the recording period was variable; 11 of 22 cells displayed only a single transient, whereas the rest exhibited multiple spikes (Fig.7E). To control for nonspecific effects of aluminum on the cells, we substituted AlF4− with AlCl3, a compound that does not stimulate G protein activity (Northrup et al., 1983) and has no effect on EP cell migration (Horgan et al., 1994). This treatment did not induce changes in intracellular Ca2+ within any of the migrating neurons examined (0 of 16 cells; data not shown).
To determine whether the AlF4−-induced spikes were dependent on extracellular Ca2+, we also imaged neurons in embryos that were cultured in saline containing 0 mm added Ca2+. Under these conditions, AlF4− no longer induced spiking activity in most of cells observed (Fig. 7C,F). Both the total number and frequency of Ca2+ transients observed in 0 mm Ca2+ were decreased: only 17% of cells responded to AlF4− with any Ca2+ transient activity, and most of those displayed only a single spike over the duration of the experiment. In addition, the overall size of the residual spikes was somewhat reduced (only a 47% increase in fluorescent intensity in 0 mmCa2+ vs a 75% increase in 4 mmCa2+). These observations indicate that Ca2+ influx from extracellular sources is required for initiating the periodic spiking activity stimulated by AlF4− and contributes to these transient increases in intracellular Ca2+. They also suggest that the initial influx of Ca2+ may normally be amplified by the subsequent release of intracellular stores, resulting in a Ca2+ spike. A similar amplification of Ca2+ influx by the release of intracellular stores has recently been demonstrated in cultured hippocampal neurons, in which transient Ca2+ spikes continued to be elicited in extracellular Ca2+ levels as low as 50 μm (Jacobs and Meyer, 1997).
In a variety of mature cell types, Go has been shown to modulate intracellular Ca2+ via the regulation of voltage-sensitive Ca2+ currents (Hille, 1994). In the EP cells, however, we found that (1) a number of specific Ca2+ channel blockers applied in embryonic culture had no effect on migration; (2) the application of high-K+ saline solutions to the embryos (which should cause the depolarization of neurons and lead to the opening of voltage-sensitive Ca2+ channels) also had no effect; (3) an analysis of the electrophysiological characteristics of the EP cells indicated that the neurons do not acquire voltage-gated inward currents until the end of migration; and (4) a large variety of neurotransmitters and peptides that might depolarize the neurons via the activation of ligand-gated currents neither stimulated nor inhibited EP cell motility (data not shown). As an alternative, we are currently investigating whether Goα may regulate intracellular Ca2+ levels in the EP cells via the activation of one of a variety of voltage-independent Ca2+ channels (discussed in the following section).
The demonstration that AlF4− leads to large, global increases in Ca2+ within the EP cells explains the inhibitory effect of AlF4− on migratory behavior (Horgan et al., 1994), because increases in intracellular Ca2+ levels also reduced migration (Figs. 2-4). In contrast, we only observed relatively small fluctuations in intracellular Ca2+ during the active phase of EP cell migration, as already noted (Fig. 7A). However, when individual neurons were imaged near the end of migration, ∼20% of the EP cells exhibited large, spontaneous Ca2+spikes similar in size to those induced by our treatments with AlF4− (Fig. 8). Because there is considerable variability in the extent of migration normally exhibited by the EP cells (Copenhaver and Taghert, 1989a), we could not determine whether Ca2+ spikes of this type always accompany the termination of migratory behavior. Nevertheless, these results suggest that transient increases in intracellular Ca2+ may normally regulate the extent of EP cell migration within the developing embryo. Together with the inhibitory effect that we have demonstrated for Goα in the EP cells, these results support a model for the Ca2+-dependent regulation of neuronal migration by Go, as summarized below.

Spontaneous Ca2+ spike observed toward the end of EP cell migration. Graph shows the time course of changes in fluorescent intensity measured in an EP cell at the end of its migration. Bottom trace (B) represents changes in background fluorescence. Embryo at ∼65% of embryogenesis was imaged in 4 mm Ca2+saline over 1 hr. Spikes of this magnitude have been observed in 3 of 14 cells imaged.
DISCUSSION
The experiments described in this paper demonstrate a functional relationship between intracellular levels of Ca2+and the activity of G proteins in the control of neuronal migration. By using an embryonic preparation of Manduca, we have been able to investigate the role of these two signaling molecules within individual migratory neurons in vivo. Manipulations designed to increase intracellular Ca2+ concentrations consistently caused dose-dependent decreases in migration. In contrast, lowering Ca2+ levels within the neurons promoted nonselective or misdirected migration and outgrowth onto inappropriate musculature. As shown previously, the stimulation of G protein activity in the EP cells negatively regulates their migratory behavior (Horgan et al., 1994, 1995). In this paper we have expanded these findings by showing that Goα, the one G protein that we have detected in the undifferentiated EP cells (Horgan et al., 1994), specifically inhibited migration. Moreover, G protein stimulation caused large, transient increases in Ca2+ within the EP cells, and the inhibitory effect of G protein activity on neuronal migration required the presence of external Ca2+. Taken together, our results support the following model by which Go may regulate the motile behavior of the EP cells. During the course of active EP cell migration, the neurons come in contact with an inhibitory signal in the local environment. This molecule (or molecules), once recognized by its G protein-coupled receptor, would cause the activation of Go, either locally within the leading filopodia or globally throughout the migratory neuron. Go activation subsequently leads to an increase in intracellular Ca2+, a process that requires Ca2+ influx. The results of this Ca2+ increase would include the downregulation of the ongoing cytoskeletal dynamics that underlie migratory behavior.
Several aspects of this model remain to be clarified, including the identity of the putative ligand (or ligands) that may normally lead to the activation of Go in the migratory EP cells and its distribution in the developing ENS. For example, inhibitory molecules of this type might be expressed only at the termination sites for migration along the muscle band pathways, serving to end the migratory phase of EP cell development and possibly induce the transition from migration to axon outgrowth (Copenhaver and Taghert, 1989b). The spontaneous appearance of large Ca2+ spikes in EP cells during the normal termination period for migration (Fig. 8), similar to those induced by G protein stimulation with AlF4− (Fig. 7), would support this possible role for Go. Alternatively, a growth-inhibitory ligand might be expressed on the nonsupportive interband musculature of the midgut, where it would induce the collapse of exploring filopodia that had extended off their migratory pathway. In this manner, localized activation of Go within the EP cell would act to steer it away from an inappropriate substrate. This more subtle inhibition could contribute to the small Ca2+ fluctuations we observed during normal migration (Fig. 7A). Moreover, this hypothesis is supported by the effects of BAPTA AM, which should dampen any transient changes in intracellular Ca2+ and that induced the misdirected migration of EP cells onto abnormal regions of the gut (Fig. 4). It is therefore possible that the regulation of intracellular Ca2+ levels may affect multiple aspects of neuronal migration. Last, we have not yet investigated the specific mechanism by which elevated Ca2+ may regulate EP cell motility, although we have found that their migration requires actin polymerization but not microtubule assembly (data not shown). Likely target proteins may therefore include one of a variety of actin-binding proteins the activity of which can be modulated by Ca2+ (McLaughlin et al., 1993; Sun et al., 1995), leading to changes in cell shape and behavior (Lankford and Letourneau, 1989; Letourneau et al., 1994).
Calcium as a regulator of neuronal motility
As with other instances of cellular motility, the actions of Ca2+ during neuronal migration may be both context- and cell type-specific (Caterina and Devreotes, 1991). During axon outgrowth, for example, normal growth cone motility requires the maintenance of intracellular Ca2+ levels within a specific range, above or below which motility is inhibited (Kater and Mills, 1991; Man-Son-Hing and Haydon, 1992). In contrast, the effects of intracellular Ca2+ with respect to neuronal migration are less well understood. In avian neural crest cells, for example, Ca2+ seems to act in an inhibitory manner, because blocking voltage-sensitive Ca2+ channels stimulated their migration (Newgreen and Gooday, 1985). In addition, measurements of intracellular Ca2+ during migration showed that migrating crest cells had lower levels of Ca2+ than did their nonmotile counterparts (Dickens et al., 1990). However, in cerebellar granule cells of the CNS, elevations in Ca2+ have been suggested to play a stimulatory role, possibly in response to the neurotransmitter glutamate (Rakic and Komuro, 1994). Using a brain slice preparation,Komuro and Rakic (1992, 1993) found that a variety of treatments intended to lower Ca2+ levels caused a decrease in granule cell migration. In support of this model, they subsequently used a fluorescent Ca2+ indicator to reveal oscillatory increases in Ca2+ (between 5 and 25% above baseline) within granule cell somata that lasted ∼1–2 min and that correlated with the saltatory movements of these neurons in vitro (Komuro and Rakic, 1996).
In contrast, we did not observe any regular, periodic oscillations in the relative levels of Ca2+ within the EP cells during their migration in vivo, although brief, spontaneous changes on the order of 5–10% were seen at irregular intervals throughout the recording period (Figs. 6A,7A). It is possible that these relatively small Ca2+ fluctuations in the EP cells are similar to those observed by Komuro and Rakic, or they may represent Ca2+ oscillations within the filopodia of the EP cells that were beyond the resolution of our detection system. Conversely, the much larger Ca2+ spikes that we observed after G protein stimulation appear to play an inhibitory role, curtailing migration. Thus, Ca2+ oscillations may serve both stimulatory and inhibitory functions during neuronal migration, depending on the size and possibly the location of the event within the cell. However, it remains to be determined in our system whether the oscillation of intracellular Ca2+per se (Gu and Spitzer, 1995) or a concomitant, sustained elevation in basal levels of Ca2+ (Kater and Mills, 1991) is ultimately responsible for regulating neuronal migratory behavior.
Calcium-mediated effects of G proteins
Although a variety of interactions between G proteins and Ca2+-dependent processes have been described in other systems, the mechanism by which Go might regulate Ca2+ in the EP cells is still unclear. One obvious explanation would involve a Go-mediated enhancement of voltage-sensitive Ca2+ currents (Hille, 1994). Such a scenario seems unlikely, however, based on our preliminary investigations of EP cell physiology (described above). Moreover, Go activity is normally associated with theinhibition of voltage-activated Ca2+currents in other preparations (Hille, 1994). Alternatively, it is possible that Go might exert its effects via the modulation of non-voltage-gated ion channels. For example, in mast cells, a ∼50 pS cation conductance has been identified the activation and inhibition of which could be mimicked by nonhydrolyzable analogs of GTP and GDP (Penner et al., 1988; Fasolato et al., 1994). A similar inward Ca2+ current is activated by purinergic-receptor agonists in PC-12 cells (Reber et al., 1992), and it may also underlie the influx of Ca2+ after muscarinic receptor activation in fibroblasts (Felder et al., 1992, 1993). More recently, a long-lasting, nonselective cation current has been identified in hippocampal pyramidal neurons that is stimulated by a G protein-mediated process (Crepel et al., 1994; Congar et al., 1997). G protein-mediated inhibition of migration might also involve one of the store-operated Ca2+ channels (Berridge, 1995), which open in response to the depletion of Ca2+ from intracellular stores and may be regulated by a variety of intracellular signaling molecules (Berridge, 1993). Channels of this type, and their possible regulation by Go, warrant further investigation with respect to the control of neuronal migration.
Cytosolic oscillations in Ca2+ are widespread phenomena resulting from the temporal coordination of Ca2+ influx and Ca2+ release from intracellular stores (Berridge, 1992). These events require a complex interaction between multiple Ca2+ channels that display both calcium-induced calcium release and calcium-induced current inactivation (Ehrlich, 1995; Murphy et al., 1995). It is possible that multiple sources of Ca2+ are responsible for the Ca spikes that were induced in the EP cells by G protein stimulation. For example, the few AlF4-induced spikes that we observed in 0 mm Ca2+saline (Fig. 7C) displayed a similar time course and shape as the spikes recorded in the presence of Ca2+, although the peak magnitude was decreased by 38%. This result suggests that intracellular pools of Ca2+ might act to amplify an initial Ca2+ influx (Berridge, 1997), which occasionally may be sufficient to initiate a spike even when external Ca2+ levels have been substantially reduced.
Alternatively, internal Ca2+ stores require external Ca2+ for their replenishment (Hoth and Penner, 1993;Putney and Bird, 1993). Therefore, in a cell undergoing spontaneous Ca2+ fluctuations, low levels of external Ca2+ might be necessary to maintain such activity for any significant length of time (Berridge, 1992). Thus, it is possible that during EP cell migration, the function of Goinvolves the cyclic regulation of intracellular Ca2+release, but that transmembrane Ca2+ influx is indirectly required in the refilling of these same Ca2+ stores. Evidence from other systems has shown that both the Goα subunit (Moriarty et al., 1990) and Gβγ (Blank et al., 1992; Katz et al., 1992) can induce phosphoinositol hydrolysis and the subsequent release of Ca2+ from IP3-sensitive stores (Strittmatter et al., 1994; Evans et al., 1995). Go has also been shown to act via regulation of PKC (Strittmatter et al., 1994; Xie et al., 1995; van Biesen et al., 1996; Pan et al., 1997) and therefore may participate in the control of Ca2+modulation by way of a PLC- or PKC-mediated effect. By using selective agonists and antagonists of the PLC and PKC pathway, we are currently investigating whether such a mechanism might be involved in controlling EP cell migration.
G protein regulation of migration
Unlike the well characterized mechanisms of action for Go in the mature nervous system, the roles that Go may serve during neuronal development are only partially understood. However, information from a number of studies has implicated G proteins related to Goα in the control of cellular motility. For example, process outgrowth in N1E-115 neuroblastoma and PC-12 cells can be stimulated by Goactivation (Strittmatter et al., 1994; Xie et al., 1995), whereas process outgrowth is inhibited by pertussis toxin (Schuch et al., 1989;Doherty et al., 1991; Williams et al., 1992). In cultured neurons, by contrast, pertussis-sensitive G proteins have been linked to the inhibition of growth cone motility after treatment with brain membrane extracts (Igarashi et al., 1993), the active component of which has been identified as collapsin (Luo et al., 1993). Goα is a highly abundant protein in the nervous system (Brabet et al., 1988;Strittmatter et al., 1990); its onset of expression coincides with the initial differentiation of embryonic neurons (Pituello et al., 1991;Otte et al., 1992) and is maintained throughout the periods of their migration and outgrowth (Garibay et al., 1991; Wolfgang et al., 1991;Schmidt et al., 1994), making Goα a likely participant in the control of these processes. However, genetic deletions of Goα in the nematode have resulted in relatively subtle abnormalities of the mature nervous system (Mendel et al., 1995;Segalat et al., 1995), and the specific functions of Goαduring neuronal differentiation have remained elusive. The appearance of Goα in the EP cells coincident with their migration suggests that it may participate in the regulation of their motile behavior (Horgan et al., 1994, 1995). In the present work, we have confirmed the inhibitory role of G proteins with respect to EP cell migration and demonstrated that Goα can itself downregulate the migratory process in a Ca2+-dependent manner. Elucidation of the mechanisms by which Goα and Ca2+ regulate the migratory process should further our understanding of the signal transduction pathways that control neuronal motility in vivoand the role of particular G proteins in the developing nervous system.
Footnotes
This work was supported by National Institutes of Health Grant NS-35369. We are indebted to Drs. Steven Matsumoto and Marc Carey for their assistance and generosity during the calcium imaging studies. We thank Drs. Michael Forte, Edwin McClesky, and Steven Matsumoto for critical reviews of this manuscript and other members of our laboratory for their support and helpful discussions.
Correspondence should be addressed to Philip F. Copenhaver, Department of Cell and Developmental Biology, L215, 3181 Southwest Sam Jackson Park Road, Portland, OR 97201.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}