

Abstract
We have explored the plastic ability of neuronal precursors to acquire different identities by manipulating their surrounding environment. Specifically, we sought to identify potential signals involved in the specification of forebrain dopaminergic neurons. Here we describe culture conditions under which tyrosine hydroxylase (TH) expression is induced in neuronal precursors, which were derived directly from the embryonic striatum and adult subependyma (SE) of the lateral ventricle or generated from multipotent forebrain stem cells. TH was successfully induced in all of these cell types by 24 hr exposure to basic fibroblast growth factor (FGF2) and glial cell conditioned media (CM). The greatest magnitude of the inductive action was on embryonic striatal precursors. Although FGF2 alone induced limited TH expression in striatal cells (1.1 ± 0.2% of neurons), these actions were potentiated 17.5-fold (19.6 ± 1.5% of neurons) when FGF2 was coadministered with B49 glial cell line CM. Of these TH-immunoreactive cells, ∼15% incorporated bromodeoxyuridine (BrdU), indicating that they were newly generated, and 95% coexpressed the neurotransmitter GABA. To investigate whether precursors of the adult forebrain subependyma were competent to respond to the instructive actions of FGF2+CM, they were first labeled in vivo with a pulse of BrdU. Although none of the cells expressed TH in control, 0.2% of total cells showed TH immunoreactivity in FGF2+CM-treated cultures. Under these same conditions only, in vitro-generated precursors from epidermal growth factor-responsive stem cells exhibited TH expression in 10% of their total neuronal progeny. Regulation of neurotransmitter phenotype in forebrain neuronal precursors, by the synergistic action of FGF2 and glial-derived diffusible factors, may represent a first step in understanding how these cells are generated in the embryonic and adult brain and opens the prospect for their manipulation in vitro and in vivo for therapeutic use.
- FGFs
- glial-derived diffusible factors
- tyrosine hydroxylase expression
- forebrain precursor cells
- subependymal cells
- catecholaminergic fate
- Parkinson’s disease
Tyrosine hydroxylase (TH) is the first and rate-limiting enzyme in dopamine synthesis. It converts tyrosine to l-dopa. Considerable interest has been generated in regulating the expression of the TH gene, in regions of the CNS that have been depleted of dopamine, as a means of restoring catecholaminergic functions (Mallet, 1996). For example, in models of Parkinson’s disease, in addition to transplanting fetal mesencephalic dopamine cells (for review, see Herman and Abrous, 1994; Olanow et al., 1996), strategies to replace the lost dopaminergic innervation have successfully used cells that have been genetically modified to express TH (Wolff et al., 1989; Horrelou et al., 1990; Fisher et al., 1991;Jiao et al., 1993; for review, see Raymon et al., 1997; Gage, 1998) and directly transferred TH-producing gene cassettes to endogenous striatal cells (During et al., 1994; Horellou et al., 1994; Kaplitt et al., 1994). An alternative approach worthy of consideration is the epigenetic generation of TH-expressing neurons derived from neural multipotential precursor cells (for review, see Anderson, 1992; Baetge, 1993; Brüstle and McKay, 1996; Weiss et al., 1996; Whittemore and Snyder, 1996; Fisher, 1997).
Extrinsic cues control the proliferation of neural precursors and progressively restrict their potential to generate various differentiated progenies (Anderson, 1989). This general concept of neurogenesis emerged from studies of the peripheral nervous system; however, today it is widely accepted as being applicable to the CNS (Edlund and Jessell, 1999). Fibroblast growth factor 2 (FGF2) is a potent mitogen for neuronal precursors of the CNS (Gensburger et al., 1987, Cattaneo and McKay, 1990; Murphy et al., 1990; Ray and Gage, 1994; Daadi et al., 1998). Several in vitro studies have demonstrated that under certain culture conditions, either FGF2 or epidermal growth factor (EGF) can induce extended proliferation of neural precursors derived from embryonic or adult brains [Reynolds and Weiss, 1992; Richards et al., 1992; Kilpatrick and Bartlett, 1993; Ray et al., 1993; Gritti et al., 1996 (for review, see Gage et al., 1995)]. The EGF-responsive stem cells self-renew, produce neurons, astrocytes, and oligodendrocytes, and may participate in repopulating the adult brain (for review, see Weiss et al., 1996; Weiss and van der Kooy, 1998). The principle phenotypes of the neurons generated by these precursors are GABA and substance-P (Ahmed et al., 1995). In preliminary experiments, we found that the coculture of these EGF-generated neuronal precursors with post-natal astrocytes yielded additional neurotransmitter phenotypes, such as neuropeptide Y, somatostatin, methionine-enkephalin, and glutamate (Daadi et al., 1993). Further addition of FGF2 to these precursor/astrocyte cocultures generated TH immunoreactive (TH-IR) neurons (M. Daadi, unpublished results).
Compelling evidence from cell culture studies demonstrates relationships between FGFs and TH-expressing CNS neurons. First, FGF2 regulates the development and differentiation of mesencephalic dopaminergic neurons (Ferrari et al., 1989; Knusel et al., 1990; Mayer et al., 1993; Bouvier and Mytilineou, 1995; Studer et al., 1998). This regulation may be partially caused by the indirect activation of astrocytes by FGF2 (Engele and Bohn, 1991; Gaul and Lubbert, 1992). Second, acidic FGF (FGF1) has been demonstrated to cooperate with catecholamines (Du and Iacovitti, 1995) to induce TH expression in striatal neurons from the embryonic day 13 (E13) mouse. Interestingly, in this study, all of the TH-IR cells were postmitotic (Iacovitti, 1991), and FGF2 was significantly less effective than FGF1 in cooperating with other molecules (Du et al., 1994; Du and Iacovitti, 1995). We have demonstrated previously that the combination of FGF2 and activin or bone morphogenic protein 2 (BMP-2) stimulates TH gene expression in basal forebrain ventricular zone progenitors (Daadi et al., 1998). However, under these conditions, EGF-responsive stem cell progeny did not differentiate into TH-expressing neurons.
The actions of FGF2 and the possible role of astrocytes in secreting factors that induce differentiation (as described above) of neuronal precursors prompted us to examine whether such combinations could induce TH expression in relatively undifferentiated neuronal precursors. We report that FGF2 acts synergistically with glial-derived soluble factor(s) to induce TH expression in neuronal precursors derived directly from the embryonic striatum and adult subependyma (SE) of the lateral ventricles and from the in vitro-propagated multipotent forebrain stem cells.
MATERIALS AND METHODS
Cell cultures
Three separate cell cultures were performed.
Primary neuronal culture of the embryonic brain
Primary neuronal cultures derived from day 14 mouse embryos (E14) were performed as described previously (Daadi et al., 1998). In brief, the dorsal-most aspect of the medial and lateral ganglionic eminences, the cortex, and the mesencephalon were dissected and mechanically dissociated (separately) with a fire-polished Pasteur pipette in serum-free medium composed of a 1:1 mixture of DMEM and F12 nutrient (Life Technologies-BRL). Cells were plated at a density of 106 cells/ml on poly-l-ornithine-coated (15 μg/ml; Sigma, St. Louis, MO) glass coverslips in 24-well Nunclon culture dishes with 0.5 ml/well. The culture medium was a serum-free, chemically defined medium composed of DMEM/F12 (1:1) including glucose (0.6%), glutamine (2 mm), sodium bicarbonate (3 mm), and HEPES buffer (5 mm) [all from Sigma except glutamine (Life Technologies)]. A defined hormone mix and salt mixture (Sigma), including insulin (25 μg/ml), transferrin (100 μg/ml), progesterone (20 nm), putrescine (60 μm), and selenium chloride (30 nm) was used in place of serum. Under these culture conditions ∼98% of cells exhibited neuronal morphology [for detailed description of the bioassay, see Daadi et al. (1998)]. Two hours after plating, growth factors and bromodeoxyuridine (BrdU; Sigma) were added to the culture. In cultures maintained for 1 week, half of the cell culture media was replaced after 3 and 5 d in vitro (DIV). Cells were incubated at 37°C in a 95% air/5% CO2 humidified atmosphere.
Preparation and differentiation of the EGF-responsive stem cell progeny
Embryonic day 14 medial and lateral ganglionic eminences were obtained as described above. Dissociated cells were plated at a density of 200,000 cell/ml in Corning T75 (Life Technologies/BRL) culture flasks in the defined media together with 20 ng/ml EGF. After 7–8 DIV, floating clusters of cells (neurospheres) were centrifuged (400 rpm), and the EGF-containing media was removed. The pellet was mechanically dissociated and reseeded in fresh EGF-containing media at 50,000 cells/ml for an additional 7–8 DIV until secondary spheres were generated. This entire procedure was performed one additional time. The differentiation of the twice-passaged neurospheres (precursor cell progenies) was performed as follows. Three T75 flasks of 7–8 DIV spheres that had been twice-passaged were spun down for 5 min at 400 rpm. The neurospheres were removed and placed into a 12 ml centrifuge tube and spun down for 5 min at 600 rpm. The EGF-containing supernatant was removed, and the spheres were resuspended in a fresh media (no EGF) plus hormone mix. This step was repeated one more time to ensure the complete removal of EGF from the media. Neurospheres were mechanically dissociated with a fire-narrowed Pasteur pipette and plated under control (media/hormone mix) or TH-inducing conditions (conditioned medium + FGF2) at a density of 106cells/ml on poly-l-ornithine-coated (15 μg/ml; Sigma) glass coverslips in 24-well Nunclon culture dishes with 0.5 ml/well. To induce TH expression in intact neurospheres, dissociated EGF-generated neurospheres were plated at a density of 50,000 cells/ml in Corning T75 culture flasks in the defined media containing 20 ng/ml of EGF, 20 ng/ml of FGF2, and 75% of conditioned medium (CM). After 7 DIV floating neurospheres were rinsed free of growth factors and CM and plated in control media on poly-l-ornithine-coated glass coverslips for a period of 2 hr before fixation and immunocytochemistry.
Dissociated culture of the adult mouse subependyma
Previously described methods (Morshead et al., 1994) were used with modifications. The striata from adult male CD1 albino mice were dissected and cut into 1 mm coronal sections that were transferred into artificial CSF that contained (in mm): 124 NaCl, 5 KCl, 1.3 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 d-glucose, pH 7.35, ∼280 mOsm, and was aerated with 95% O2–5% CO2 at room temperature. The subependyma surrounding the right and left lateral ventricles was microdissected, chopped into smaller pieces, and transferred to a spinner flask (Bellco) with a magnetic stirrer filled with low-Ca2+ artificial CSF that contained (in mm): 124 NaCl, 5 KCl, 3.2 MgCl2, 0.1 CaCl2, 26 NaHCO3, 10 mmd-glucose, pH 7.35, ∼280 mOsm, 1.33 mg/ml of trypsin (9000 U/ml benzoyl-l-arginine ethyl ester), 0.67 mg/ml of hyaluronidase (2000 U/mg), and 0.2 mg/ml of kynurenic acid, and was aerated with 95% O2–5% CO2 at 32–35°C. After 90 min, tissue pieces were transferred to normal artificial CSF for 5 min before trituration. Tissue was then transferred to DMEM/F12 (1:1, Life Technologies) medium containing 0.7 mg/ml ovomucoid (Sigma) and was triturated mechanically with a fire-narrowed Pasteur pipette. Cells were spun down at 400 rpm for 5 min, resuspended, and then plated under control (media/hormone mix) or TH-inducing conditions (CM+FGF2) on poly-l-ornithine-coated (15 μg/ml; Sigma) glass coverslips in 24-well Nunclon culture dishes with 0.5 ml/well.
Growth factors
The growth factors used were human recombinant brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor (GDNF) (PeproTech Inc., Rocky Hill, NJ); human recombinant platelet-derived growth factor ββ (PDGF), transforming growth factors β2 and β3 (TGFβ2, TGFβ3,), activin A (β-subunit), BMP-2, FGF2, and FGF1 (generously provided by Chiron Corporation, Emeryville, CA); human recombinant transforming growth factor α (TGFα; Life Technologies); rat recombinant ciliary neurotrophic factor (CNTF) (generously provided by Drs. R. Dunn and P. Richardson, McGill University); FGF4 and FGF7 (R&D Systems, Minneapolis, MN); Sonic hedgehog (Shh) (kindly provided by Ontogeny, Cambridge, MA); and calcitonin gene-related peptide (CGRP, Sigma).
Preparation of conditioned media
CM was prepared from cultures of postnatal striatal astrocytes and from the B49 rat glial cell line (Schubert et al., 1974) as described by Engele et al. (1991). Astrocyte cultures were prepared from postnatal mice (0–24 hr). Striata were dissected, minced, and transferred into a 15 ml centrifuge tube containing DMEM/F12 (1:1) and 10% fetal bovine serum (FBS). Tissue was dissociated by trituration with a narrow diameter fire-polished Pasteur pipette and plated in 20 ml of DMEM/F12/10% FBS at a density of 150,000 cells/ml in T75 Corning culture flasks. After they reached confluency, the primary astrocyte monolayers were trypsinized and replated at the same density, then once again allowed to reach confluency. B49 cells (kindly provided by Dr. D. Schubert, Salk Institute, San Diego, CA) were cultured in DMEM/10% FBS until confluency. Confluent astrocyte or B49 glial cell cultures were rinsed once with PBS and twice with serum-free DMEM/F12 (1:1) medium containing hormone mix and replaced in the incubator with 20 ml of the same medium. The CM was collected after 24, 48, or 72 hr and centrifuged at 1000 and 2,000 × g to remove cellular debris. The CM was carefully removed, filtered, aliquoted, and stored at −80°C.
In vitro and in vivo BrdU labeling
In vitro labeling. To determine whether neurons were newly generated during the first 24 hr culture period, BrdU (1 μm) was added 2 hr after plating and remained in the media for the duration of the culture, routinely 1 or 3 DIV.
In vivo labeling. In the brains of both embryonic and adult mice, labeling of cells cycling in vivo was performed as follows. Adult male or 13.5 d post-conception pregnant female CD1 Albino mice were injected intraperitoneally with 100 mg/kg BrdU dissolved in sterile saline solution. The injection was repeated five times at 2 hr intervals. Thirty minutes after the last BrdU injection, adult animals or dissected E14 embryos were decapitated and dissected for the subependyma zone or the medial and lateral ganglionic eminences, respectively. Tissues were dissociated and cultured as described above. To localize the subependymal precursors in adult forebrain, BrdU-injected mice were killed by transcardiac perfusion with 4% paraformaldehyde. The brains were cryoprotected in an increasing gradient of sucrose solution, and 10 μm cryostat sections were cut and allowed to adhere to glass slides precoated with gelatin/chrome alum for at least 1 hr before immunostaining with anti-BrdU.
Immunocytochemistry
Rabbit polyclonal antisera and mouse monoclonal antibodies directed against neurotransmitter phenotypes and neural antigens were used as primary antibodies for indirect immunofluorescence. Polyclonal anti-tyrosine hydroxylase (1:1000) and dopamine-β-hydroxylase (1:200) were purchased from Eugene Tech, Inc. Identical results were obtained with another polyclonal and monoclonal anti-tyrosine hydroxylase obtained from Pel-Freez Biologicals (Rogers, AR) and Incstar, respectively. Monoclonal anti-β-tubulin (type III, 1:1000) and polyclonal anti-GABA (1:5000) were purchased from Sigma. Monoclonal antibody against GFAP (1:100) was purchased from Boehringer Mannheim (Mannheim, Germany). Anti-BrdU (1:5) used in proliferation assays was obtained from Amersham (Arlington Heights, IL). Secondary antibodies raised in goat against mouse and rabbit immunoglobulins, conjugated to the fluorophore rhodamine isothiocyanate (1:200) or fluorescein isothiocyanate (1:100), were purchased from Jackson ImmunoResearch (West Grove, PA). Indirect immunocytochemistry was performed on cells that had been cultured for 24 hr or longer on glass coverslips. Coverslips were fixed with 4% paraformaldehyde (with 0.1% glutaraldehyde for anti-GABA) for 20 min followed by three washes (10 min each) in PBS. After the PBS rinse, coverslips were processed for dual labeling and incubated with the primary antibodies generated from different species, which were added together in PBS/10% normal goat serum/0.3 Triton X-100 for 2 hr at 37°C. After three rinses in PBS, secondary antibodies were applied in PBS for 30 min at room temperature. Coverslips were then washed three times (10 min each) in PBS, rinsed with water, placed on glass slides, and coverslipped using Fluorsave (Calbiochem, La Jolla, CA) as the mounting medium. Fluorescence was detected and photographed with a Nikon Optiphot photomicroscope. For each experimental condition, the number of TH-IR, BrdU-IR, β-tubulin-IR, or GABA-IR cells and the total number of live cell nuclei stained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) were determined by examining the entire surface area of each coverslip at 400× magnification or counting the number of cells in 15 randomly chosen microscopic observation fields per coverslip. The total counts were then expressed as a percentage of the total DAPI-stained nuclei or of the total number of cells expressing the neuronal marker class III β-tubulin (Lee et al., 1990; Ahmed et al., 1995; Qian et al., 1997). Data represent the mean ± SEM of experiments performed three or four times on independent culture preparations, each performed in duplicate. Statistical analysis of the data was performed using a one-way ANOVA, and significance of intergroup differences was determined by applying Student’s t test. Differences were considered significant at p < 0.05 andp < 0.01.
Northern analysis and RT-PCR for neurotransmitter-synthesizing enzyme expression
Total RNA extraction. After removal of the culture medium, plated cells were lysed in situ using 1 ml of TRIzol (Life Technologies-BRL) per well. Lysates from like samples were pooled and placed on ice for 15 min. Two-tenths volume of chloroform was added, shaken vigorously, and allowed to stand at room temperature for 5 min. The phases were separated in a clinical centrifuge spinning at 2500 rpm for 20 min. The aqueous phase was transferred into a fresh tube, an equal volume of isopropanol was added, the contents were mixed by inversion, and the RNA was allowed to precipitate overnight at −20°C. The following day, the tubes were centrifuged at 3500 rpm for 20 min, the supernatant was decanted off, and the pellet was dissolved in 4 m guanidinium isothiocyanate solution (Life Technologies-BRL). The RNA was precipitated using 2 vol of absolute ethanol and incubation at −20°C overnight. When the RNA was required, the tubes were spun at 3500 rpm for 20 min. The pellets were washed in 70% ethanol, and the tubes were spun again. The air-dried pellets were dissolved in water, and the concentration of each sample was determined spectrophotometrically.
Northern hybridization. Aliquots (20 μg) of total RNA were fractionated on agarose formaldehyde gels. The RNA was transferred by capillary action from the gel matrix to Hybond-N+ (Amersham) using 10 × SSC (1.5 m NaCl, 0.15 m sodium citrate, pH 7.0), and the RNA was fixed onto the membrane by baking. To make a probe to tyrosine hydroxylase, a 48-mer complementary to the mRNA at nucleotides 1435–1482 (GenBank accession M69200) was synthesized and then labeled by 3′-end tailing with α-33P-dATP (DuPont, Billerica, MA) using terminal transferase (Boehringer Mannheim). The dATP tails, on average, were from three to five nucleotides long per oligonucleotide molecule. The membranes were prehybridized for 30 min in QuikHyb hybridization mix (Stratagene, La Jolla, CA) at 71°C. Herring sperm DNA (1 μg per membrane) and labeled probe (10 ng/ml hybridization) were added to the prehybridization mix, and the membranes were allowed to incubate for 60 min at 71°C. The posthybridization washes consisted of one quick wash in 2× SSC, 0.1% SDS at room temperature, two 15 min washes in 2× SSC, 0.1% SDS at room temperature, and finally one 30 min wash in 0.1× SSC, 0.1% SDS at 60°C. BioMaxMR autoradiographic film (Kodak) was placed against the air-dried filters and exposed over a 5 d period at −80°C. The sizes of the bands on the developed autoradiographs were verified by calculating the size of RNA corresponding to the apparent migration of the band down the gel relative to standard molecular weight markers.
RT-PCR. Aliquots (1 μg) of total RNA from the cells were reverse-transcribed in the presence of 50 mm Tris-HCl, pH 8.3, 75 mm KCl, 3 mm MgCl2, 10 mm DTT, 0.5 mm dNTPs, and 0.5 μg oligo-dT(12–18) (Pharmacia, Dorval, Québec, Canada), with 200 U Superscript RNase H-Reverse Transcriptase (Life Technologies-BRL). PCR primers specific to glutamic acid decarboxylase (GAD67; GenBank accession number S61897), to TH (GenBank accession number M69200), and to β-actin (GenBank accession number M12481) were designed using the Primer Designer software, Version 2.0 (Scientific and Educational Software), and synthesized in the Oligo 1000 DNA Synthesizer (Beckman Instruments). The GAD67 upstream primer corresponded to nucleotides 764–785, whereas the downstream primer corresponded to nucleotides 1047–1026 on the complementary strand. The TH upstream primer corresponded to nucleotides 1055–1076, whereas the downstream primer corresponded to nucleotides 1308–1289 on the complementary strand. The β-actin sense strand corresponded to nucleotides 512–536 and to nucleotides 950–930 for antisense strand. Aliquots of cDNA equivalent to 40 ng of total RNA were amplified in 25 μl reactions containing 10 mm Tris-HCl, pH 8.3, 50 mm KCl, 1.5 mm MgCl2, 50 pmol of each primer, 400 μm dNTPs, and 0.5 U AmpliTaq DNA polymerase (Perkin-Elmer, Emeryville, CA). PCR was performed using the following thermal profile: 4 min at 94°C; 1 min at 94°C, 1 min at 60°C, 2 min at 72°C, for 30 cycles; 7 min at 72°C, and finally a soak at 4°C overnight. The following day, 15 μl aliquots of the amplified products were run on a 2% agarose Tris–acetate gel containing 0.5 μg/ml ethidium bromide. The products were visualized through a UV transilluminator, captured in a digital format using the DC40 camera, and analyzed with the BioMax 1D Image Analysis software (Kodak digital Science EDAS, Eastman Kodak Company, Rochester, NY) on a Macintosh LC 575 computer.
RESULTS
FGF2 induces the expression of TH in neuronal precursors derived from the E14 striatum and cortex
We examined the actions of the mitogenic growth factors EGF and FGF2 on the number of cells expressing TH in dissociated cultures derived from the E14 mouse cortex, striatum, and ventral mesencephalon (VM) after 24 hr (1 DIV). Of the three regions examined, as expected under control conditions (no mitogen, no serum), the ventral mesencephalic cultures demonstrated an abundance of TH-IR (∼2000/cm2, 2% of the total cells, 7% of total β-tubulin-IR neurons) (Fig. 1). Cultures of the cortex or striatum contained few TH-IR cells. Neither serum nor EGF showed any appreciable effects, whereas exposure of striatal or cortical cells to 20 ng/ml FGF2 resulted in a significant increase in the number of TH-IR cells (Fig. 1). The greatest increase seen in TH-IR cells in the presence of FGF2 was in the striatal cultures that contained 380 ± 77 TH-IR cells/cm2, equivalent to 0.31 ± 0.06% of the total cells and 1.1 ± 0.2% of total β-tubulin-IR neurons. Under these conditions, however, there was no change in the number of TH-IR cells in cultures derived from the VM. Examination of BrdU incorporation, as an index of newly generated cells (Gratzner, 1982), demonstrated that none of the VM-derived TH-IR cells incorporated BrdU; however, 25–30% of the TH-IR striatal cells were born during the 24 hr culture period (Fig. 1). The addition of 0.1% FBS potentiated the TH-inducing effects of FGF2 on striatal and cortical cells 2.5-fold, without increasing the proportion of BrdU incorporation (Fig. 1).

Growth factor treatment increases the number of tyrosine hydroxylase-immunoreactive cells in primary cultures derived from different brain regions. Dispersed cells derived from the ventral mesencephalon, striatum, and cortex were grown on poly-l-ornithine-coated glass coverslips in the presence of the indicated factors (FBS, 0.1%; EGF, 20 ng/ml; FGF2, 20 ng/ml). Cells were fixed after 1 DIV and processed for TH and BrdU immunocytochemistry, as described in Materials and Methods. Neurons were identified by their immunoreactivity to anti-β-tubulin. The TH-IR neurons were expressed as a percentage of the number of TH-IR neurons present in FGF2-treated cultures. In FGF2-treated cultures from the ventral mesencephalon, 1.86 ± 0.12% of the total DAPI-stained nuclei and 6.6 ± 0.4% of the total neurons were TH-IR, respectively. In FGF2-treated cultures from the striatum, 0.31 ± 0.06% of the total cells and 1.1 ± 0.2 of the total neurons were TH-IR. Cultures derived from the cortex and treated with FGF2 showed the fewest TH-IR cells: ∼0.06% of the total number of live cells and 0.2% of the total number of neurons. Results are mean ± SEM of experiments performed three times on independent culture preparations, each performed in duplicate. Because of the represented scale of the x-axes, the error bars do not appear in some of the histograms. The asterisk indicates the level of significance with respect to FGF2-treated cultures;p < 0.05.
These data suggest that FGF2 had two actions on striatal and cortical neuronal precursors. First, FGF2, as has been demonstrated previously, was mitogenic for neuronal precursors that could be induced to express TH. Quantitation of total BrdU-labeled cells confirmed this conclusion. After 24 hr, control cultures contained 29 ± 4% BrdU-labeled cells, whereas those exposed to FGF2 contained 72 ± 11% BrdU-labeled cells. Second, FGF2 (but not EGF, which was equally capable of inducing cell proliferation; 69 ± 8% BrdU-labeled cells) could induce the expression of TH in both newly generated and recently born (see below) striatal and cortical cells, an action that could be potentiated by factors (such as those in serum) that were not inductive alone. Interestingly, this study also revealed that the VM-derived TH-IR cells did not incorporate BrdU. This finding corroborates previous studies (Engele et al., 1991) and demonstrates that the E14 midbrain dopaminergic neurons are all postmitotic. It also supports our hypothesis that for FGF2 to induce TH expression, the precursor cells need to be at the newly generated or recently postmitotic stage.
TH induction by FGF2 is dramatically enhanced by media conditioned by astrocytes, and in particular the B49 glial cell line
Given the well known actions of astrocytes in enhancing the differentiation and survival of mesencephalic dopaminergic neurons (Denis-Donini et al., 1984; Engele et al., 1991; O’Malley et al., 1992; Takeshima et al., 1994) and forebrain neuronal precursors (Daadi et al., 1993), we examined the putative cooperative actions of media conditioned (CM) by astrocytes or the B49 glial cell line on FGF2 induction of TH in cultured striatal cells. The CM was collected into serum-free media, as described in Materials and Methods, over a 24 or 72 hr period. The addition of CM (either one as listed above) had no effect on the number of TH-IR cells in 24 hr striatal cell culture (Fig. 2). When CM from striatal astrocytes collected after 24 or 72 hr was combined with FGF2, the number of TH-IR cells increased from 0.31 ± 0.06% of the total cells (1.1 ± 0.2% of total neurons) in cultures treated with FGF2 alone to 1.13 ± 0.16% (4.0 ± 0.5% of total neurons) and 1.63 ± 0.34% (5.8 ± 1.2% of total neurons), respectively (Fig. 2). CM derived from mesencephalic glia did not further increase the number of TH-IR cells (data not shown). However, a greater cooperative action was observed when FGF2 was combined with CM derived from a confluent culture of B49 glial cells. Coapplication of FGF2 and CM from B49 cells resulted in a 17.5-fold increase (to 5.50 ± 0.43% of total cells; 19.6 ± 1.5% of total neurons) in the number of TH-IR striatal cells (Figs. 2,3A,D).

Conditioned media from glial cells potentiates the actions of FGF2 on the number of tyrosine hydroxylase-immunoreactive cells in cultures of striatal neuronal precursors. Dissociated striatal cells (5 × 105) were grown on poly-l-ornithine-coated glass coverslips under the indicated conditions (CM, 75%; FGF2, 20 ng/ml). Cells were fixed after a 24 hr culture period and processed for indirect immunocytochemistry for TH, as described in Materials and Methods. Because control culture contained between 0 and 8 TH-IR cells and to allow for multiple comparison, the number of TH-IR neurons present in FGF2-treated cultures was taken as 100% (1.1 ± 0.2 TH-IR of the total number of neurons). Results are the mean ± SEM of three independent experiments, each performed in duplicate. Because of the represented scale of the x-axes, the error bars do not appear in some of the histograms. The asterisk indicates the level of significance with respect to CM-treated cultures;p < 0.01.

The combination of FGF2 and CM induces TH immunoreactivity in striatal neuronal precursors. Dissociated cells (5 × 105) of the E14 striatum were cultured on poly-l-ornithine-coated glass coverslips in serum-free medium without (A, B) or with (C–G) 75% of B49 glial cell line-conditioned media and FGF2 (20 ng/ml) for 24 hr (A–D, F, G) and 3 d (E). Two hours after plating, all culture wells received 1 μm BrdU (a marker for DNA synthesis) (Gratzner, 1982). Fixed cells were processed for dual immunocytochemistry (as described in Materials and Methods) for TH and BrdU (A–D), TH and GABA (F, G), or single TH immunocytochemistry (E). In control cultures (A, B) the newly generated cells that had incorporated BrdU do not express TH. In FGF2+CM-treated cultures (C, D), the arrow shows one example of a cell that had incorporated BrdU and expressed TH. After 3 DIV, TH-IR cells developed typical neuronal morphology (E).F, G, Example of a 24-hr-old TH-IR cell that also coexpressed the neurotransmitter GABA. Scale bar (shown inE): A–D, 40 μm: E, 25 μm: F, G, 10 μm.
Despite the difference in collection time observed for CM from striatal astrocytes, there was no increase in TH-inducing activity (as assessed by the numbers of TH-IR cells) when B49 CM was collected for 72 hr (data not shown). As was the case for the addition of serum, CM did not increase the proportion of TH-expressing cells that had incorporated BrdU. In fact, the proportion of TH-IR cells that were newly generated during the 24 hr culture period decreased from 25–30% in the presence of FGF2 to 12–15% with FGF2+CM. The significance of these proportions is discussed further below.
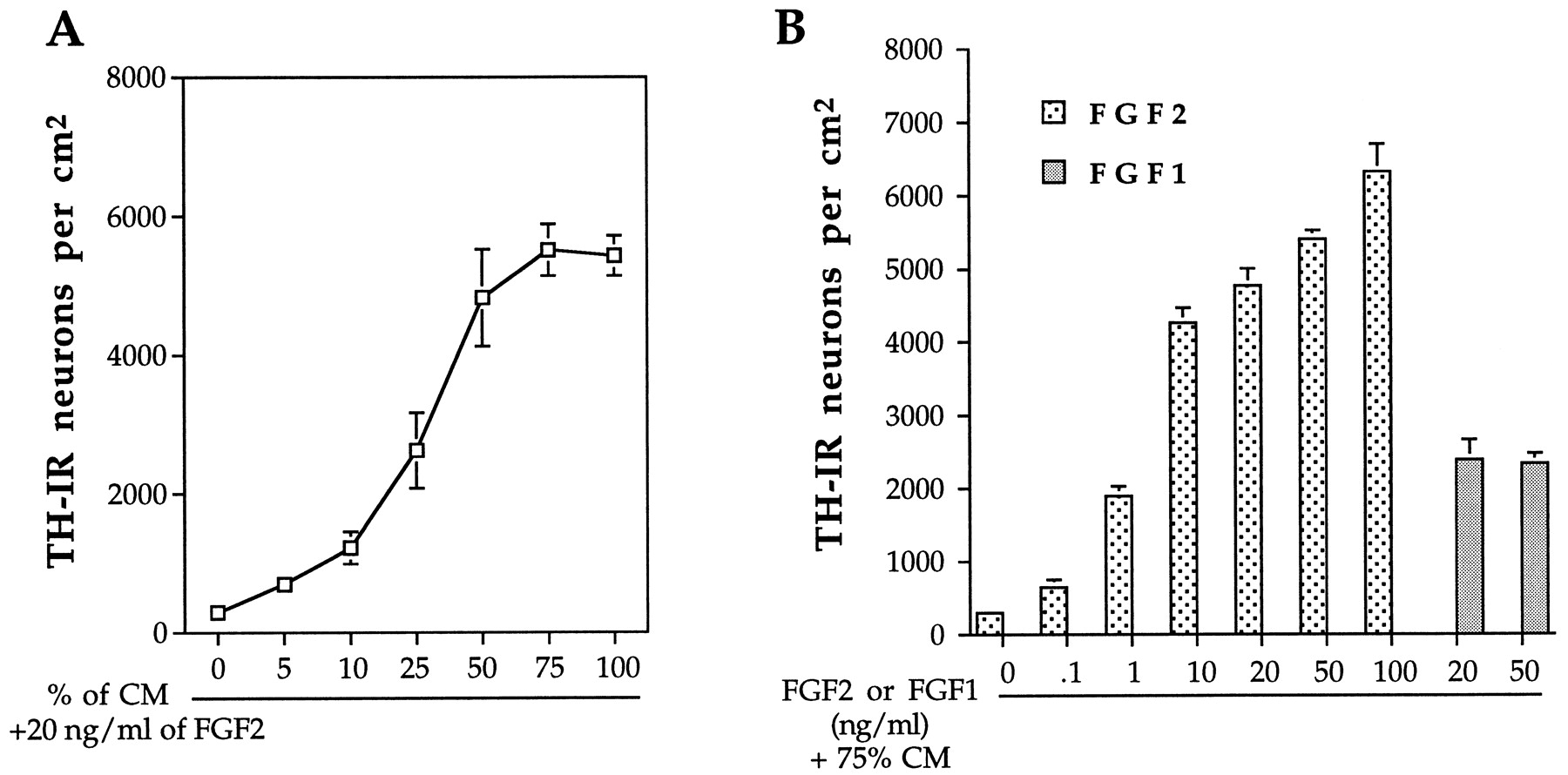
The individual actions of FGF2 and CM (henceforth synonymous with B49 CM) were dose dependent (Fig. 4). While using a fixed concentration of FGF2 (20 ng/ml), increasing proportions of CM gradually potentiated the number of TH-IR cells observed after 24 hr. Maximal effects of CM were achieved when used at 50–75% of the total media (Fig. 4A). Similarly, at a fixed concentration of 75% CM, FGF2 actions were first detected at 1 ng/ml, and the maximal effect was achieved with 50–100 ng/ml (Fig.4B). Fixed concentrations of FGF1 (examined at 20 and 50 ng/ml) yielded ∼50% of the efficacy of FGF2 (Fig.4B).

CM and FGF2 cooperate synergistically in a dose-dependent manner to induce TH expression in striatal neuronal precursors. A, Striatal cells were cultured in the presence of 20 ng/ml FGF2 and increasing concentrations of CM for a period of 24 hr and processed for TH immunocytochemistry, as described in Materials and Methods. A fluorescent-field microscope with 40× objective was used to count the total number of TH-IR cells in the entire area of each coverslip. B, Dissociated cells were cultured in the presence of 75% CM and increasing concentrations of either FGF2 or FGF1, and then processed for TH immunocytochemistry and quantified as described in Materials and Methods. Data represent the mean ± SEM of experiments performed three times on independent culture preparations, with two replicates for each condition within the independent experiments.
Because the B49 glial cell line fueled the discovery of GDNF, a potent survival factor for midbrain dopaminergic neurons (Engele et al., 1991;Lin et al., 1993), we asked whether GDNF is the glial-derived factor that cooperates with FGF2 to induce TH expression. The results shown in Figure 5 demonstrate that GDNF is not involved in the rapid induction of TH expression in striatal neuronal precursors.

GDNF does not mimic the actions of CM on the appearance of tyrosine hydroxylase-immunoreactive neurons in cultures of striatal cells. Striatal cells were plated at a density of 5 × 105 cells per well on poly-l-ornithine-coated glass coverslips and cultured under the indicated conditions for 24 hr before being processed for TH immunocytochemistry. Similar to Figures 1 and 2, the number of TH-IR neurons was represented as a percentage of the FGF2-treated cultures in which 1.1 ± 0.2 of the total number of neurons are TH-IR (see Results). Results are mean ± SEM of four independent experiments, each performed in duplicate.
Relationship between proliferation, differentiation, and TH induction in striatal neuronal precursors
The TH-inducing actions of FGF2+CM, particularly in cells that also incorporated BrdU, suggest that newly generated or recently postmitotic neurons would be most susceptible to manipulation. To directly demonstrate that actively mitotic neuronal precursors are induced to express TH, a 2 hr pulse of BrdU was applied at 8 and 22 hr in culture, and cells were examined at 24 hr. These experiments revealed that 4.3 ± 1.4% and 3.9 ± 0.8% of the total TH-IR cells have progressed through the S phase of the cell cycle during the period of BrdU availability (2 hr) after 8 and 22 hr in culture, respectively. To further understand the mechanism of the TH induction, we examined the relationship between time dependence and proliferative actions of FGF2+CM on the induction of TH expression. Although FGF2+CM could induce 5.11 ± 0.73% of the total cells to express TH when applied at plating, if delayed by only 24 hr this was reduced to 0.10 ± 0.02% cells. If delayed by 4 d, FGF2+CM was entirely without effect. Examination of endogenous proliferation (i.e., of control cultures) showed a correlative, precipitous decline in the numbers of proliferating cells. Although 25.50 ± 0.68% BrdU-labeled cells were detected over the first 24 hr, if detection was delayed by 2 d only 4.34 ± 0.06% of the total cells incorporated BrdU in the last 24 hr culture period. This dropped to 0.39 ± 0.08% if BrdU was added after 4 DIV, and the cultures were processed for BrdU immunocytochemistry after 5 DIV. Thus, the ability to induce TH declines with the proliferative capacity of the striatal precursors. Although control cultures showed between 25 and 29% of cells incorporating BrdU, this increased almost threefold in response to FGF2, yet it actually decreased threefold (8.73 ± 1.7%) in the presence of CM alone. Further addition of FGF2 to CM-containing medium did not further increase the number of BrdU-labeled cells (9.08 ± 1.91%). Thus, it appears that the mechanism that underlies the differentiation actions of CM in inducing TH may be associated with attenuating the endogenous proliferative capacity of the striatal precursors.
To ascertain whether the most recently generated neuronal precursors are the most susceptible to express TH, we prelabeled this populationin vivo. This was performed by a series of BrdU injections into E13.5 pregnant female mice. Dissociated cultures of the embryonic striata were grown for 24 hr in the absence or presence of FGF2+CM, and an examination of TH and BrdU coexpression followed. Of the 4.0 ± 0.13% cells that expressed TH, 44% were also BrdU-IR. This result confirms that both mitotically active and most recently generated neuronal precursors are particularly sensitive to TH induction.
FGF2+CM stimulates TH mRNA expression
The induction of TH expression in response to FGF2+CM could occur either by an activation of the gene encoding the enzyme or by posttranslational actions and enhancement of protein levels. To examine these possibilities, we isolated total RNA from control cultures of striatal cells and those treated with FGF2+CM and then measured the levels of TH mRNA with Northern blot analysis (Fig.6A). A single band corresponding to the 1.8 kb TH transcript (Ichikawa et al., 1991) was detected only in the cultures treated with FGF2+CM. Thus, the induction of TH expression occurs at the level of gene transcription.

Regulation of TH and GABA expression in the striatal neuronal precursors. A, Total RNA was isolated from the harvested 1-d-old cells and assayed (20 μg of RNA per lane) for the expression levels of TH transcripts (1.8 kb) by Northern blot analysis (see Materials and Methods for description of probe used and the conditions of hybridization). B, Total RNA samples were extracted from 1- and 3-d-old cultures that had been treated as indicated. The relative abundance of TH, GAD67, and β-actin transcripts was assessed by RT-PCR (see Materials and Methods for details about the primer sequences and the conditions of the RT-PCR).
FGF2+CM regulates both GABA and TH expression in the striatal neuronal precursors
Given that the majority of the striatal cells express GABAin vitro (Mizuno et al., 1994; Max et al., 1996; Daadi et al., 1998) and in vivo (Mugnaini and Ortel, 1985), we investigated whether GABA expression might also be regulated by FGF2+CM treatment. Application of FGF2+CM for 24 hr simultaneously stimulated the TH and GABA expression at both the protein and mRNA levels (Fig.6B; Table 1). By contrast, after 3 DIV, control cultures showed an increase in the number of GABA-IR neurons, whereas those exposed to FGF2+CM demonstrated a decrease in the numbers of both TH- and GABA-IR cells (Table 1). This finding was confirmed at the mRNA level using semiquantitative RT-PCR analysis (Fig. 6B). We then asked whether TH and GABA coexist in the same cells. Double-labeling immunocytochemistry with antibodies to TH and GABA revealed that 95% of the total TH-IR cells coexpressed GABA (Fig. 3F,G; Table1). These data confirmed our previous finding (Daadi et al., 1998) and demonstrated at both the cellular and molecular level that a subset of the striatal neuronal precursors express TH transiently after 24 hr exposure to FGF2+CM. The percentage of neuronal precursors that expressed TH after 3 DIV remained the same after 7 DIV (data not shown), suggesting a stable induction of TH gene expression and a heterogeneity within the striatal precursors (see Discussion).
Coexpression of GABA and TH in cultures of striatal precursors after treatment with FGF2 and conditioned media
FGF2+CM induces precursors of the adult forebrain subependyma to express TH in vitro
The SE is the adult equivalent of the embryonic subventricular zone (SVZ) (Boulder Committee, 1970), and it contains a population (0.2–0.4%) of neural stem cells that under normal conditions are relatively quiescent and generate the constitutively proliferating progeny (10% of the SE cells) that have a cell-cycle time of 12.7 hr (Morshead and van der Kooy, 1992; Morshead et al., 1998). Therefore, in the next series of experiments, we asked whether FGF2+CM could induce TH in neuronal precursors derived from the adult SE (presumably produced by stem cells within the SE). To accomplish this, we first labeled the SE precursors of the adult mouse forebrainin vivo by six injections of BrdU, given at 2 hr intervals (Morshead et al., 1994). Figure7B shows the location of the SE precursors (BrdU-IR) surrounding the lateral ventricle of the adult forebrain. Thirty minutes after the last BrdU injection, the SE was dissected, enzymatically dispersed, and cultured in the absence or presence of FGF2+CM. The cells were fixed and examined for TH and BrdU immunoreactivity 1 and 3 d after plating. In control conditions, many cells were labeled with BrdU, but none were TH-IR. However, cultures treated with FGF2+CM showed newly generated TH-IR cells. Figure 7 illustrates examples of cells that expressed both TH and BrdU immunoreactivity after 1 (Fig. 7C,D) and 3 d (Fig.7E,F) in culture. Younger TH-IR cells (1 d old) exhibited features of undifferentiated cells, with small, round cell bodies and no or short processes. After 3 DIV, the TH-IR cells gained features typical of differentiated neurons, with small bipolar or multipolar cell bodies, diameters varying between 8 and 14 μm, and long, thin, and aspiny neuritic processes (Fig. 7F). Quantitative data from three independent experiments indicated that of the total number of cells plated, 0.23 ± 0.03% exhibited TH-IR. Of those cells exhibiting TH immunoreactivity, 63% were BrdU-IR, which suggests that they were derived from the proliferating cells of the adult SE and were born during the 12 hr that preceded the primary culture. In contrast to the embryonic striatal precursors, the number of SE-derived TH-IR cells did not decrease over time (data not shown).

Induction of TH expression in precursors derived from the adult subependyma. Constitutively proliferating cells in the subependyma (SE) of adult mice were first labeledin vivo by five intraperitoneal injections of BrdU (see Materials and Methods). To confirm the in vivo location of the constitutively proliferating cells, a group of mice were perfused, and the brains were cryoprotected and cryostat-sectioned. The 10 μm sections were processed for BrdU immunocytochemistry. A, B, Drawing and photomicrograph of coronal section through the striatum of adult mouse. B, Photomicrograph of BrdU-IR cells within the SE surrounding the lateral ventricle. The location of this photomicrograph is outlined by the dotted lines inA. C–F, The SE cells were dissected, enzymatically dispersed, suspended in complete medium with or without FGF2+CM, and plated in the absence of BrdU. After culture periods of 1 DIV (C, D) and 3 DIV (E, F), the cells were fixed and processed for dual-label indirect immunocytochemistry for TH and BrdU (as outlined in Materials and Methods). Photomicrographs are of newly generated cells that had incorporated BrdU in vivo (C, E) and were TH-IR (D, F) in culture treated with FGF2+CM.aca, Anterior commissure, anterior; cc, corpus callosum; CTX, cortex; LV, lateral ventricle; SE, subependyma; STR, striatum. Scale bar (shown in F):B, 70 μm; C–F, 15 μm.
Generation of TH-expressing neurons from embryonic forebrain multipotent stem cells
In the final series of experiments, we asked whether neuronal precursors derived from EGF-responsive stem cells could be induced to express TH. Clones (or neurospheres) of undifferentiated cells derived from the EGF-responsive stem cells (see Materials and Methods) were propagated (passaged) numerous times, dissociated, and plated in serum-free medium containing 1 μm BrdU, in the absence or presence of FGF2+CM. After 24 hr the cultures were processed for TH and BrdU immunocytochemistry. As was the case for isolated cells of the adult SE, TH-IR cells were observed only in cultures treated with FGF2+CM (Fig. 8A). TH immunoreactivity was not observed in control cultures nor when 20 ng/ml FGF2 was coincubated with 50 ng/ml BDNF, CNTF, PDGF, TGFα, TGFβ2, TGFβ3, GDNF, activin A, BMP-2, GDNF, Shh, or CGRP (data not shown). In agreement with previous studies (Ahmed et al., 1995; Arsenijevic and Weiss, 1998), we have found that most of cells (>80%) in these cultures were astrocytes. Thus, only a minor proportion of dissociated cells (1–2% in both control and treated cultures) expressed the neuronal marker β-tubulin. Quantitative data from three independent culture experiments indicated that 10 ± 2% of the total number of β-tubulin-IR neurons were TH-IR. Moreover, in a proportion similar to that seen with newly generated TH-IR cells of the adult SE, ∼65% of the TH-IR cells were also BrdU-IR. In older cultures of dissociated neurospheres, and again analogous to the SE-derived precursors, the number of TH-IR cells did not change over time (data not shown). In these cultures, after 3 DIV, TH-IR cells acquired typical neuronal morphology, with small bipolar or multipolar cell bodies and extended long neuritic processes (Fig. 8B). After 7 DIV, a small percentage of these neurons (<2%) exhibited a “projection neuron-like” morphology with complex neuritic arborization and long process outgrowth exceeding 0.5 mm (Fig. 8C). Neurospheres were also grown in suspension (intact) in media containing EGF (20 ng/ml), FGF2 (20 ng/ml), and CM (75%) (see Materials and Methods). After 7 DIV, individual clones were isolated and plated in control medium for 30 min to adhere to the glass coverslip. After fixation and immunocytochemistry for TH, the clones remained in spherical shapes, which rendered the assessment of the total number of TH-IR cells per clone difficult. We counted the total number of clones that had exhibited at least one visible TH-IR soma on the surface. These data revealed that 15 ± 2% of the plated multipotential stem cells had the potential to generate secondary clones containing TH-IR neurons (Fig. 8D).

CM+FGF2 induces TH expression in neuronal precursors derived from multipotential neural precursor cells. Seven-day-old neurosphere clones, grown as described in Materials and Methods, were collected and mechanically dissociated in defined medium. Dispersed cells were either plated on poly-l-ornithine-coated glass coverslips (adherent culture conditions, A–C) or replated into a 75 cm2 tissue culture flask (suspension cultures,D) in complete medium supplemented with growth factors and CM. Culture periods are indicated for each photomicrograph. Fixed cells or clones were processed for TH immunocytochemistry, as described in Materials and Methods. TH-IR cells were observed only in cultures treated with FGF2+CM (A–D). A–C, Time course evolution of morphological characteristics acquired by the TH-IR neurons in FGF2+CM-treated cultures (see Results).D, Photomicrograph of 7-d-old TH-IR clones grown in suspension in the presence of EGF (20 ng/ml), FGF2 (20 ng/ml), and CM (75%) (see Results). Scale bar (shown in D): A, B, 20 μm; C, 50 μm; D, 30 μm.
DISCUSSION
The present study demonstrates that FGFs and glial-derived diffusible factors act synergistically to direct the forebrain neuronal precursors to a cathecholaminergic fate. This finding represents a further step in understanding how embryonic and adult precursors could be manipulated in regard to their neurotransmitter phenotype choices. Our data also support the notion that combinatorial signaling involving glia could differentially regulate neuronal lineage decisions within the mammalian CNS.
In addition to having mitogenic actions, FGF2 (and not EGF) can also stimulate TH gene expression in subpopulations of striatal and cortical neuronal precursors and their postmitotic progeny, cultured in a chemically defined media. These forebrain-derived precursors do not express TH under normal conditions. The instructive TH-inducing action of FGF2 increased 2.5-fold in the presence of 0.1% serum and was dramatically potentiated by 17.5-fold in the presence of glial CM. Rather than a direct action, it is possible that FGF2+CM may act to increase the proliferation of a specific neuronal population susceptible to express TH. However, when the striatal precursors were first labeled in vivo with a pulse of intraperitoneal BrdU injection, the number of double-labeled TH/BrdU cells in FGF2+CM-treated culture increased by fourfold (in comparison with cultures treated with BrdU at plating). These data indicate that the TH-inductive action of FGF2+CM is independent of a direct mitotic action and that it is rather closely related to the inherent proliferative nature of the striatal precursors. In fact, delaying the application of FGF2+CM to the cultures resulted in the loss of striatal neuronal precursor competence to respond to the instructive actions. In aged cultures, the loss of responsiveness to the TH-inducing factors directly correlated with a precipitous drop in the number of proliferating cells. This phenotypic plasticity of striatal precursors is similar to that of the cerebral cortex (McConnell, 1992; Levitt et al., 1993; Ghosh and Greenberg, 1995; Götz et al., 1995) whereby newly born precursors are endowed, during an early critical period, with an intrinsic plasticity that may be related to the cell cycle/postmitotic nature of the cell. Although the exact mechanism by which FGF2+CM regulates TH expression is not clear, our observations suggest that CM promoted differentiation, at least in part, by driving the striatal precursors out of the cell cycle. We show that CM repressed FGF2-induced proliferation. One possibility is that FGF2 simultaneously activates signaling molecules required for proliferation and TH expression in striatal neuronal precursors, whereas CM modulates these two FGF2-induced intracellular signaling pathways by inhibiting the mitogenic and potentiating the TH-activated signaling pathways. It remains possible, however, that FGF2+CM supports the survival of a population of TH-expressing cells that would otherwise die in the defined medium. Similar to our previous study (Daadi et al., 1998), to examine the selective (survival) versus the instructive (differentiation) actions of FGF2+CM, we used the life cell marker DAPI. We found that there is no difference in the number of DAPI-stained fragmented nuclei (characteristic of apoptotic cells) (Raff, 1992) between control and FGF2+CM-treated cultures (data not shown). Nevertheless, we cannot totally exclude a minimal survival action of FGF2+CM on certain neuronal precursors, including those that are TH-IR. Even if this occurred, the minimal survival effect would not account for the dramatic increase in TH-IR cell production (Fig. 2). The number of TH-IR neurons declined after 3 DIV, which suggests either a cell death mechanism occurring within the TH-IR subpopulation or a plasticity in neurotransmitter phenotype expression. This decline was not caused by cell death, because in our culture condition none of the TH-IR cells were undergoing apoptosis after 3 DIV, as assessed by the analysis of the structure of DAPI-stained nuclei (data not shown). This finding suggests that a downregulation of TH gene expression occurs within a subpopulation of the TH-IR cells. It could be that under physiological circumstances the selection of a certain phenotype requires the sustained presence of the instructive molecules and/or other factors that intervene sequentially. Indeed, specification of the dopaminergic lineage in the midbrain appears to require the continual presence (at least for a certain period of time) of FGF8 (Ye et al., 1998). Moreover, throughout adult life in the forebrain, the maintenance of TH expression in the olfactory bulb requires the continual presence of the synaptic activity of the olfactory afferents (Nadi et al., 1981; Baker and Farbman, 1993).
The induction of TH expression has been reported previously in noncathecholaminergic neurons. Iacovetti (1991) first demonstrated that striatal neurons could express TH when incubated in the presence of muscle differentiation factor (MDF). Subsequently, these investigators found that FGF1 cooperates with MDF (Du et al., 1994) or catecholamines (Du and Iacovitti, 1995) to induce TH expression. Noteworthy in these studies was that (1) newly generated cells never expressed TH, (2) striatal cells had to withdraw from the cell cycle to respond to the TH-inducing actions of MDF (Iacovetti, 1991), and (3) FGF2 was significantly less effective than FGF1 in cooperating with other molecules (Du et al., 1994). We have shown that 25 and 15% of the TH-IR striatal cells in cultures treated with FGF2 and FGF2+CM, respectively, were proliferating. Furthermore, BrdU pulse-labeling studies have provided direct evidence that TH is induced in the mitotically active neuronal precursors (see Results). Together, these data indicate that our conditions targeted a neuronal precursor cell population. However, we cannot exclude the possibility of an overlapping between subsets of postmitotic striatal TH-IR cells in both conditions.
The vast majority (95%) of TH-IR cells coexpressed GABA. Most of these TH-IR cells exhibited small bipolar or multipolar somas (8–14 μm) with aspiny neuritic processes. Similar morphological characteristics define the striatal- and SVZ-derived GABAergic and dopaminergic interneurons that migrate tangentially to the neocortex and the olfactory bulb, respectively (Betarbet et al., 1996; De Carlos et al., 1996; Anderson et al., 1997; Luskin et al., 1997; Tamamaki et al., 1997). Interestingly, subpopulations of striatal and olfactory bulb neurons coexpress dopamine and GABA (Gall et al., 1987; Kosaka et al., 1995; Betarbet et al., 1996, 1997; Max et al., 1996). Furthermore, the neocortical neurons have the potential to express dopamine when treatedin vitro with BDNF and dopamine (Zhou et al., 1994). Together with our present and previous findings (Daadi et al., 1998), these observations suggest the existence of a ganglionic eminence-derived bipotential neuronal precursor that can express either dopamine or GABA under specific epigenetic conditions. It is particularly noteworthy that in the adult primate, the TH/GABA-IR neurons described by Greenmayre and colleagues (Betarbet et al., 1997) appeared intrinsically in the striatum, and their number increased after 1-methyl-4-phenyl-1,2,3,6-tetrahydro pyridine-induced dopaminergic cell loss. It would be of great interest to determine whether these cells are derived from the SE (see below for further discussion). Current experiments, whereby EGF and subsequently FGF2+CM were infused into the forebrain of adult Parkinsonian rats, favor this hypothesis (Daadi et al., 1997).
The work of Morshead and colleagues (1994, 1998) demonstrated that a relatively quiescent population of multipotential stem cells exists in the mammalian adult forebrain SE. This stem cell population generates the constitutively proliferating cells of the SE in vivo. When these mitotically active precursors (presumptive stem cell progeny) were labeled in vivo, dissected, and plated in the presence of FGF2+CM, they expressed TH after 24 hr in culture. Over 3 d, these TH-IR cells developed neuronal morphology. A subpopulation of the in vivo constitutively proliferating cells, localized in the rostral part of the SVZ, migrates postnatally and throughout the adult life to the olfactory bulb (Luskin, 1993; Lois and Alvarez-Buylla, 1994; for review, see O’Rourke, 1996). Some of these neuronal precursors migrate to the glomerular cell layer and differentiate into dopaminergic interneurons (Betarbet et al., 1996). It could be that the SE cells we induced to express TH in vitro correspond, at least in part, to the olfactory bulb dopaminergic interneurons. This hypothesis is supported by two observations. First, the low percentage in TH-expressing cells is similar to the in vivo conditions, whereby only 25% of the SE-derived neuronal precursors migrate to the olfactory bulb (Morshead et al., 1998) and only a minimal number of this subpopulation (1:30, ratio that may be affected by cell survival; A. Alvarez-Buylla, personal communication) reaches the glomerular layer (and hence have the potential to express TH) (Lois and Alvarez-Buylla, 1994). Second, there is a similarity between the morphological characteristics of the SE-derived TH-IR cells we observed and those of the in vivo migrating olfactory bulb interneuron precursors (Luskin, 1993; Menezes et al., 1995; Rousselot et al., 1995). However, numerous studies have reported that the expression of dopamine in the olfactory bulb is regulated by the olfactory sensory afferents and/or CGRP (Nadi et al., 1981; Baker et al., 1983; Denis-Donini, 1989). When tested alone or in combination with FGF2 in our bioassay, CGRP was not able to induce the TH expression in the forebrain precursor cells. Yet, previous studies suggested that neither the olfactory afferent innervation nor CGRP is sufficient to induce dopamine phenotype (Baker, 1990; Biffo et al., 1990; Finger and Böttger, 1992; Baker and Farbman, 1993). This mechanism is rather complex and may also involve indirect actions of the olfactory receptor afferents (McLean and Shipley, 1988; Finger and Böttger, 1992). The work of Baker and Farbman (1993) suggests that these olfactory afferents may regulate TH gene expression indirectly through the stimulation of other cell types within the glomerular cell layer and local release of growth factors. Interestingly, the olfactory bulb periglomerular and dopaminergic interneurons develop around the time of birth and postnatally (Halazs et al., 1981; Specht et al., 1981; Luskin, 1993), which suggests a possible role of glia in the differentiation process. In support of this hypothesis is the finding that the newly generated neurons of the adult forebrain SE migrate in vivo along a restricted pathway, called the rostral migratory stream (Altman, 1969), that is particularly enriched in astrocytes (Lois et al., 1996; Peretto et al., 1997). Within both the SE of the lateral wall of the lateral ventricle and the migratory stream, the newly generated neuronal precursors destined to migrate to the olfactory bulb are ensheathed by astrocytes (Lois et al., 1996; Doetsch et al., 1997; Peretto et al., 1997). Moreover, phenotypic specification of migrating neuronal precursors could occur through instructive actions of the migratory pathway (for review, see Hatten, 1993). Together these data demonstrate that astrocytes form an early local microenvironment for the SE neuronal precursors and suggest that glia-derived molecules may restrict the fate of these SE progeny in vivo.
In the presence of EGF, a single germinal zone-derived precursor cell will proliferate and give rise to a cluster of undifferentiated cells with the properties of neuroepithelial stem cells. We have demonstrated previously that under specific culture conditions, stem cell progenies are able to express various neurotransmitter phenotypes, including GABA, substance P, NPY, somatostatin, met-enkephalin, and glutamate (Daadi et al., 1993). The present data extend our knowledge of the developmental potential of the forebrain stem cells and demonstrate that their progeny differentiate into TH-producing cells when grown in the presence of EGF, FGF2, and CM. In dissociated cultures, 10% of the total number of neurons were induced to express TH. Using the same treatment, a similar percentage of TH-IR neurons was obtained from neurospheres derived from fetal human tissues. After numerous (>45) passages, and similar to the murine neurospheres, human neurospheres were always responsive to the TH-inducing conditions (M. Daadi and A. Vescovi, unpublished results). Although 10% of the neurons express TH, this translates to a relatively small number of neurons per coverslip. This observation is not surprising because most (∼80%) of the EGF-generated progeny of stem cells are astrocytes (see Results). However, it is important to stress that in contrast to the embryonic striatal cells, we have never seen a single TH-IR neuron in control cultures derived from both EGF-generated precursors and those derived directly from the adult SE. This observation renders the instructive actions on the later two sources of cells (although only a small percentage are TH-IR cells) to be rather significant. To increase the number of TH-expressing neurons, it may be useful to first promote the neuronal lineage of precursor cells (Ahmed et al., 1995; Johe et al., 1996; Mayer-Proschel et al., 1997; Williams et al., 1997; Arsenijevic and Weiss, 1998). Nevertheless, it remains unresolved why, in general, only a small proportion of neuronal precursors are susceptible to the induction of TH. This could be attributable to the presence of other instructive molecules in CM that are more potent in promoting other lineage species or to the low concentration of the TH-inducing factors present in the CM. Alternatively, it could be that the majority of neuronal precursors are endowed with a restricted range of fate. The weak potency of CM derived from embryonic astroglia (relative to B49 CM) favors the second hypothesis. Moreover, after partial purification of glial CM, and when combined with FGF2, the isolated active fraction was more potent and consistent (than CM) in inducing TH expression in stem cell progeny (M. Daadi, unpublished results).
When FGF2 was combined with BDNF, CNTF, PDGF, TGFα, TGFβ2–3, activin-A, BMP-2, GDNF, Shh, or CGRP, we were not able to see any induction of TH expression in the in vitro-generated forebrain multipotential stem cell progeny. These observations suggest that a novel glial differentiation factor(s) is responsible for this action. However, given the small number of TH-IR neurons derived from stem cells, it is difficult to make a firm assessment regarding the involvement of the other known molecules in TH gene regulation. It could be that these molecules, in cooperation with FGF2, had a minimal inductive effect that was undetectable by our immunocytochemical techniques. Because Shh is required for the midbrain dopaminergic neuron specification (Hynes et al., 1995a,b, 1997; Wang et al., 1995), the fact that it did not induce TH expression in forebrain-derived neuronal precursors is intriguing. There are many possible explanations. The difference in the embryonic age, the spatial location, and in vitro culture conditions may explain the different responsiveness of neuronal precursors to the same set of molecules. Second, it could be that during development Shh acts earlier than the glial-derived differentiation factors (described in this report) in concert with FGFs. Third, the midbrain and forebrain dopaminergic neurons may require different factors and likely separate mechanisms for their specification. In favor of this last hypothesis is the finding that in contrast to the olfactory bulb dopaminergic cells, the midbrain dopaminergic neurons failed to develop in the orphan nuclear receptor Nurr1 null mice (Zetterström et al., 1997; T. Perlmann, personal communication).
Significance of the finding
It has been well established that astroglial cells function as a support for migrating neuronal precursors in the developing brain and provide physiological assistance to neuronal functions in the mature brain. The present study suggests that glial diffusible factors may also instruct neuronal precursors to commit to a particular neuronal lineage. This represents a further step in understanding the inherent ability of the mammalian brain to generate diverse cell types and could help in developing successful therapeutic strategies for the treatment of Parkinson’s disease.
Footnotes
This work was supported by Novartis Canada Limited and the Medical Research Council of Canada. We thank Dr. D. Schubert for providing the B49 glial cell line, M. Y. A. Panlilio for Northern blot technical assistance, and S. E. Daadi for proofreading this manuscript. We also thank the following individuals for their generous supply of the reagents used in this study: CNTF, Drs. R. Dunn and P. Richardson; Sonic hedgehog, Ontogeny Inc.; PDGF, TGFβ2, TGFβ3, activin A, BMP-2, FGF2, and FGF1, Chiron Corporation.
Correspondence should be addressed to Dr. Marcel M. Daadi, Center for Neuronal Survival, Montreal Neurological Institute, McGill University, 3801 Rue University, Montreal, Canada H3A 2B4.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}