

Abstract
Cysteine-string proteins (CSPs) are associated with secretory vesicles and critical for regulated neurotransmitter release and peptide exocytosis. At nerve terminals, CSPs have been implicated in the mediation of neurotransmitter exocytosis by modulating presynaptic calcium channels; however, studies of CSPs in peptidergic secretion suggest a direct role in exocytosis independent of calcium transmembrane fluxes. Here we show that the individual expression of various CSP isoforms in Drosophila similarly rescues the loss of evoked neurotransmitter release at csp null mutant motor nerve terminals, suggesting widely overlapping functions for each isoform. Thus, the structural difference of CSP variants may not explain the opposing putative functions of CSP in neurotransmitter and peptide exocytosis. Consistently, the individual overexpression of each CSP isoform in wild-type Drosophila shows similar effects such as impaired viability and interference with wing and eye development. The dominant effects caused by the overexpression of CSP are suppressed by the simultaneous overexpression of syntaxin-1A but not by the coexpression of SNAP-25. Although overexpression of CSP itself has no apparent effect on the synaptic physiology of larval motor nerve terminals, it fully suppresses the decrease of evoked release induced by the overexpression of syntaxin-1A. A direct protein–protein interaction of CSP with syntaxin is further supported by coimmunoprecipitations of syntaxin with CSP and by protein binding assays using recombinant fusion proteins. Together, the genetic and biochemical interactions of CSP and syntaxin-1A suggest that CSP may chaperone or modulate protein–protein interactions of syntaxin-1A with either calcium channels or other components of the regulatory machinery mediating depolarization-dependent neurotransmitter exocytosis.
- cysteine-string protein
- syntaxin
- synaptic vesicle
- exocytosis
- neurotransmitter release
- synaptic transmission
- secretion
Cysteine-string proteins (CSPs) are highly conserved proteins associated with secretory vesicles and characterized by a unique palmitoylated central string of cysteines (for review, see Buchner and Gundersen, 1997). Extensive homology exists between the N terminus of CSP and the J domain of DnaJ proteins (Caplan et al., 1993), which mediate protein folding and protein complex formation in cooperation with members of the heat-shock protein family (Zylicz, 1993; Kelley, 1998). In a manner analogous to the prokaryotic system, the J domain of CSPs is essential and sufficient to bind the 70 kDa heat-shock cognate protein (Hsc70) and to activate its intrinsic ATPase activity, suggesting that CSP may act as a cofactor of the molecular chaperone Hsc70 (Braun et al., 1996; Chamberlain and Burgoyne, 1997a,b; Zhang et al., 1999).
CSPs were originally described as synapse-associated antigens in the nervous system of Drosophila (Zinsmaier et al., 1990) and, independently, in Torpedo, as positive modulators of N-type calcium channels, because the coexpression of Torpedo RNAs modulated the activity of ectopically expressed N-type channels in frog oocytes (Gundersen and Umbach, 1992). The subsequent localization of CSPs to synaptic vesicles suggested that CSPs may link synaptic vesicles and presynaptic calcium channels to modulate channel activity (Mastrogiacomo et al., 1994). Deletion of the csp gene inDrosophila causes a temperature-sensitive loss of evoked neurotransmitter release at adult first-order interneurons of the visual system (Zinsmaier et al., 1994) and at larval neuromuscular junctions (Umbach et al., 1994). Similarly, intracellular application of CSP antibodies into presynaptic motor neurons inhibits evoked neurotransmitter release at frog neuromuscular junctions (Poage et al., 1999). Further studies of Drosophila mutants showing normal induction of quantal release by calcium ionophores (Umbach and Gundersen, 1997; Ranjan et al., 1998) and reduced relative cytosolic calcium levels after high frequency stimulation (Umbach et al., 1998) are consistent with a defect of depolarization-dependent calcium entry. The idea that CSP may modulate calcium channel activity is further strengthened by the finding that CSP binds to α1A-subunits of P/Q-type calcium channels (Leveque et al., 1998).
Regulation of calcium channel interactions, however, is unlikely to be the only function of CSPs, because the proteins have been localized to secretory vesicles of neuronal and non-neuronal tissues of mammals (for review, see Buchner and Gundersen, 1997) and Drosophila(Eberle et al., 1998). A direct role of CSP in exocytosis independent of calcium transmembrane fluxes has been further substantiated by the overexpression of mammalian CSP in PC12 cells (Chamberlain and Burgoyne, 1998) and insulin-secreting cells (Brown et al., 1998), which both reveal defects of stimulated exocytosis independent of calcium channel activity. Similarly, at peptidergic terminals of cspnull mutant Drosophila, calcium currents are normal, suggesting that CSP may not be a generic modulator of calcium channels (Morales et al., 1999). In this study, we determined whether the differentially expressed CSP isoforms of Drosophila may show significant functional differences to explain the multiple suggested functions of CSP. In addition, we report genetic and biochemical evidence that CSP may mediate depolarization-dependent neurotransmitter release through its interaction with syntaxin-1A.
MATERIALS AND METHODS
Fly stocks. Flies were raised on standard cornmeal–molasses agar medium with dry yeast at the indicated temperatures. The homozygous viable transgene P{elav-GAL4} is described by Lin and Goodman (1994); P{hs-syx} and P{hs-SNAP-25} are described by Wu et al. (1998). The homozygous semi-lethal cspR1,cspU1, andcspX1 deletions are all genetic nulls (Zinsmaier et al., 1994; Eberle et al., 1998) and were kept balanced with a TM6 Tb balancer chromosome. A strain containing a heat-shock hsp70 promoter driving GAL4 (hs-GAL4) has been obtained from the Bloomington Drosophila stock center.
Generation of transgenic fly strains. The three alternatively spliced csp cDNAs, types I–III (Zinsmaier et al., 1990,1994), were EcoRI-restricted and subcloned into the cloning site of the pUAST P-element vector (Brand and Perrimon, 1993). For P-element-mediated germ-line transformation, each pUAST-csp plasmid was injected together with the helper plasmid pUChsπΔ2–3 into 30- to 60-min-old white(w1118) embryos by standard methods (Spradling, 1986). At least two homozygous viable transgenic lines, termed UAS-csp1, UAS-csp2, and UAS-csp3 (w1118; P{UAS-csp1–3}), were established for all three individual csp cDNAs. For CSP overexpression, homozygous UAS-csp lines were crossed with homozygous elav-GAL4 or hs-GAL4 flies to obtain elav-csp (w1118, P{elav-GAL4}/w1118; P{UAS-csp}/+) or hs-csp progeny (w1118; P{UAS-csp}/+; P{hs-GAL4}/+). For csp mutant rescue experiments, UAS-csp, elav-GAL4, or hs-GAL4 transgenes were repeatedly crossed withw1118; Cyo/Sco; cspR1/TM6 Tb to generate UAS-csp, elav-GAL4, or hsp70-GAL4 lines with a heterozygouscsp null background (w1118, P{elav-GAL4}; cspR1/TM6 Tb andw1118; P{hs-GAL4}, cspR1/TM6 Tb andw1118; P{UAS-csp1–3}; cspR1/TM6 Tb). These were then appropriately crossed with each other to obtain elav-csp; csp− and hs-csp; csp− progeny (w1118, P{elav-GAL4}/w1118; P{UAS-csp}/+; cspR1) and (w1118; P{UAS-csp}+; P{hs-GAL4}, cspR1).
Scanning electron microscope. Three-day-old flies were collected and washed in 70% ethanol. Whole adult flies were dried and mounted onto scanning electron microscope stubs and gold-coated with a Denton Vacuum Desk II Sputter Coater (100 Å Au coat). Samples were scanned in a JOEL JSM-T330A scanning microscope and photographed.
Immunoprecipitations and immunoblotting. Fresh or frozen fly heads were homogenized in immunoprecipitation (IP) buffer (50 mm KCl, 0.2% Triton X-100, 1 mm PMSF, 10 mm HEPES, pH 7.0). The homogenate was cleared by centrifugation at 14,000 rpm in an Eppendorf centrifuge for 10 min at 4°C, and DCSP-1 antibody (Zinsmaier et al., 1994) was added in sufficient quantity to bind all CSP within 14 hr at 4°C. The extract was centrifuged four to five times for 10 min at 4°C at 14,000 rpm before sufficient amounts of protein A agarose (Pharmacia) were added to precipitate all IgGs during a 2 hr incubation at 4°C. The proteins immobilized to protein A agarose beads were recovered by low speed centrifugation (5000 rpm). The immunoprecipitate was washed three times with IP buffer before it was resuspended in SDS sample buffer and split. Each half of the precipitate was subjected to SDS-PAGE and immunoblotting as described earlier (Zinsmaier et al., 1990, 1994) and individually stained for CSP and syntaxin. The following antibody dilutions were used: monoclonal antibody DCSP-1 at 1:10 and monoclonal 8C3 against syntaxin at 1:40 (gift of S. Benzer, Caltech, Pasadena, CA).
Glutathione S-transferase binding assay. Drosophila syntaxin-1A and the cytoplasmic part ofDrosophila synaptotagmin were expressed as glutathioneS-transferase (GST) fusion proteins in Escherichia coli and purified by standard methods (Smith and Johnson, 1988). Histidine (His)-tagged Drosophila CSP was expressed in E. coli Bl-21 cells and purified using the pET expression system (Novagen, Madison, WI) essentially as recommended by the supplier. Protein concentrations were estimated by Coomassie blue staining after SDS-PAGE using bovine serum albumin as standard. A protein binding protocol for immobilized GST fusion proteins was adapted from Kee et al. (1995). Briefly, GST-agarose beads were preadsorbed to a protein extract from E. coli cells for 1 hr at 4°C. Beads were collected by centrifugation, and 10–25 pmol immobilized GST fusion proteins and ∼50 pmol soluble His-tagged proteins were added to a total volume of 200 μl binding buffer (150 mm KAc, 1 mmMgCl2, 0.05% Tween-20, 20 mm HEPES, pH 7.4). After rotating for 2 hr at 4° C, the beads were collected by centrifugation and washed twice with 1 ml of binding buffer containing 1 mg/ml gelatin and three times with binding buffer containing 5% glycerol. Proteins bound to the beads were solubilized in SDS sample buffer and subjected to SDS-PAGE and immunoblotting.
Larval body wall muscle preparation. Climbing third instar larvae were dissected to expose their body wall muscles. For dissection, larvae were placed dorsal side up on a small dish with a thin layer of Sylgard resin in calcium-free hemolymph-like (HL-3) medium (Stewart et al., 1994). Larvae were pinned down anteriorly and posteriorly and cut along the dorsal midline, and filleted larvae were pinned out. After removing the viscera, the segmentally repeated larval body wall muscles and the innervating nerve fibers were clearly visible. The muscles were identified as described previously (Johansen et al., 1989).
Electrophysiology. Intracellular whole-cell recordings of miniature excitatory junction potentials (mEJPs) and evoked excitatory junction potentials (EJPs) were made with a single microelectrode (20–40 MΩ) filled with 3 m KCl. All recordings were made from larval muscle 6 of segment 3/4 in the anterior abdomen of dissected third instar Drosophila larvae in HL-3 medium containing 1 mmCa2+. Signals were amplified using an Axopatch-1D amplifier (Axon Instruments) and filtered at 1 kHz. To stimulate evoked excitatory junction potentials, nerve fibers were severed at the base of the ventral ganglion, and EJPs were elicited with a suction electrode (6–10 μm diameter tip) connected to an isolated stimulator (A-M Systems) using a 0.1-msec-long stimulus at three times the threshold response. The temperature of the recording chamber was monitored and controlled using a perfusion system and an in-line heater (SF-27A, Warner Instruments) connected to a TC324-A heater controller (Warner Instruments). The data were digitized at 5 kHz with a Digidata 1200 interface (Axon) and analyzed using pCLAMP 6.0 software (Axon). Mean evoked EJPs were obtained by averaging 15 responses per larva for nlarvae.
RESULTS
Various Drosophila CSP isoforms equally rescue the loss of evoked neurotransmitter release of csp null mutations
The csp gene of Drosophila expresses four different protein isoforms that are derived from at least three alternatively spliced RNAs (Zinsmaier et al., 1990, 1994; Eberle et al., 1998). The CSP variants are differentially expressed in various tissues as revealed by antibodies recognizing only a subset of isoforms and by immunoblotting showing predominant expression of only one isoform in non-neuronal tissues (Zinsmaier et al., 1994; Eberle et al., 1998). All CSP isoforms share three typical and conserved domains: the “J domain” at the N terminus, the centrally located “cysteine-string domain,” and the “linker domain” connecting the J and the string domains (Fig.1A). The CSP variants differ in their C-terminal half, which is poorly conserved betweenDrosophila, Caenorhabditis elegans, and vertebrate CSPs. The C-terminal half of Drosophila CSP1 contains an insertion of 21 amino acids that is absent in CSP2 and CSP3. CSP2 and CSP3 differ in seven amino acids at the C terminus.

A, Protein domain structure of Drosophila CSPs. The different protein isoforms CSP1, CSP2, and CSP3 are derived from alternatively spliced RNA transcripts. All proteins share three conserved domains, the J domain (J, residues 19–82), the linker domain (L, residues 83–113), and the cysteine-string domain (C, residues 114–138). CSPs differ in their C-terminal half; CSP1 contains a 21 amino acid insertion termed variable region 1 (V1), which is not present in CSP2 or CSP3. CSP1 and CSP2 share the same C terminus (V2), which differs by seven residues in CSP3 (V3). B, Analysis of CSP overexpression in wild-type driven by the elav promoter. Immunoblot of fly head protein extracts from wild-type control,cspR1 null mutants and from flies overexpressing CSP1–3 with a elav promoter (elav-csp1,elav-csp2, elav-csp3) or with a csp promoter contained in a genomic csp transgene (csp-promoter). Proteins were resolved by 11% SDS-PAGE, immunoblotted, and stained against CSP with the monoclonal antibody DCSP1 detecting all CSP isoforms. Each lane contains proteins equivalent to one adult head from flies raised at 25°C. elav-csp2 expression shows a prominent band at 32 kDa, elav-csp1 at 33–34 kDa, and elav-csp3 at 36 kDa. Genotypes: elav-csp1 (w1118, {elav-GAL4}/w1118; P{UAS-csp1}/+); elav-csp2(w1118, P{elav-GAL4}/w1118; P{UAS-csp2}/+); elav-csp3 (w1118, P{elav-GAL4}/w1118; P{UAS-csp3}/+); csp-promoter(w1118; P {csp});csp − (w1118; cspR1).
To determine whether different CSP isoforms may have tissue-specific functions, we individually expressed each isoform inDrosophila using the GAL4/UAS expression system. This system allows targeted gene expression when transgenic flies expressing the yeast GAL4 transcription factor in a tissue-specific pattern are crossed with flies bearing a transgene containing a GAL4-specific upstream activating sequence (UAS) fused to a target gene (Brand and Perrimon, 1993). Full-length cDNAs encoding Drosophila CSP1, CSP2, and CSP3 were subcloned into the fly transformation vector pUAST (Brand and Perrimon, 1993) such that they were expressed under the transcriptional control of a UAS sequence. We established at least two independent homozygous viable transgenic lines for each cDNA (UAS-csp1; UAS-csp2; UAS-csp3). To drive CSP expression we used transgenic flies expressing the transcription factor GAL4 under the control of the heat-inducible heat-shock promoter (hs-GAL4), the neuron-specific elav promoter (elav-GAL4), and the eye-specific glass promoter (gmr-GAL4). We exclusively investigated flies containing one copy of a promoter transgene driving GAL4 and one copy of a UAS-csp transgene that were derived from genetic crosses of strains homozygous for both the promoter and the csp transgene.
The expression of all UAS-csp transgenes was verified by a Western blot analysis showing a significant overexpression of each CSP isoform driven by the elav promoter and the heat-shock promoter. CSPs overexpressed with the elav promoter show molecular weights ranging from 32 to 36 kDa, similar to those of endogenous CSPs indicating normal post-translational palmitoylation of the central string of cysteines (Fig. 1B). The elav-mediated expression of CSP1 shows a dominant band at 33–34 kDa, of CSP2 at 32 kDa, and of CSP3 at 36 kDa. Native CSPs exhibit molecular weights of 32, 33, 34, and 36 kDa (Eberle et al., 1998). Thus, CSP1 and CSP2 may mostly represent the smaller isoforms ranging from 32 to 34 kDa that are presumed to be specifically expressed in the nervous system. In contrast, CSP3 correlates with the native 36 kDa isoform, the only CSP variant expressed in non-neuronal cells at detectable levels (Eberle et al., 1998). To compare levels of CSP overexpression derived from the elav promoter with that from a csp promoter, we used a transgenic fly strain bearing a genomic csp transgene that expresses all CSP isoforms and fully rescues all genetic phenotypes of csp loss of function mutations (Umbach et al., 1994; Zinsmaier et al., 1994). Flies containing one copy of an elav-GAL4 transgene and one copy of a UAS-csp transgene (elav-GAL4/+; UAS-csp1–3/+) show a stronger overexpression for a particular CSP isoform than flies homozygous for the genomic csp transgene (Fig. 1B). A comparable strong overexpression is also observed with the heat-shock promoter 3 hr after inducing expression by a heat-shock at 37°C or by raising flies at a constant temperature of 25°C (data not shown). Interestingly, most of the heat-shock-induced overexpressed proteins show lower molecular weights than endogenous CSPs or elav-expressed CSPs, indicating that the post-translational modification of the cysteine-string domain is incomplete 3 hr after heat-shock induction.
To determine whether the overexpressed CSP isoforms are indeed functional and whether they may have alternative functional requirements, we tested the ability of CSP1, CSP2, and CSP3 to rescue the recessive temperature-sensitive loss of neurotransmitter release at the larval neuromuscular junction of csp null mutant fly strains. The cspR1 mutation deletes the entire csp gene, causing a reduction of evoked neurotransmitter release at room temperature and a complete block above 29°C without affecting spontaneous neurotransmitter release (Umbach et al., 1994). We synthesized flies that expressed one copy of each CSP isoform under the transcriptional control of the elav promoter in a homozygous cspR1 genetic background (elav-GAL4/+; UAS-csp/+; cspR1) and recorded nerve-evoked excatotory junction potentials (EJPs) and spontaneous miniature excitatory junction potentials (MEJPs) from neuromuscular junctions of third instar larvae. Consistent with the normal features of spontaneous release in csp null mutants, we observed no significant effects on spontaneous neurotransmitter release by the individual expression of CSP1, CSP2, and CSP3 at csp mutant neuromuscular junctions (data not shown). However, each individually overexpressed isoform rescues the null mutant phenotype of evoked release. As shown in Figure 2, the mean EJP amplitude of homozygouscspR1 larvae is reduced by ∼50% at 22°C (21.2 ± 3.3 mV, mean ± SEM,n = 5) when compared with wild-type control (44.0 ± 2.0 mV, n = 4). In contrast, homozygouscspR1 larvae expressing CSP1 under the control of the elav promoter (elav-csp1; csp−) exhibit normal amounts of evoked neurotransmitter release (44.3 ± 1.4 mV, n = 4). Similar EJP responses are obtained for the neural expression of CSP2 (49.0 ± 3.4 mV, n = 5) and CSP3 (37.5 ± 1.2 mV, n = 3) in a homozygouscspR1 genetic background, whereas control larvae containing only a UAS-csp transgene show responses similar to the null mutant (UAS-csp1; csp−: 19.8 ± 0.9 mV,n = 6; UAS-csp2; csp−: 23.7 ± 2.0 mV, n = 3; UAS-csp3; csp−: 28.8 ± 1.5 mV,n = 5). All mean EJP amplitudes of CSP1–3-expressing flies are statistically different from amplitudes of homozygouscspR1 flies (for CSP1p = 0.0054; for CSP2 p = 0.0009; for CSP3 p = 0.039; Student's t test) but not significantly different from wild-type control white(p > 0.1). Student's t test also reveals a statistical difference between mean EJP amplitudes obtained from elav-csp1; csp− and UAS-csp1; csp− larvae (p = 0.001), elav-csp2; csp− and UAS-csp2; csp− larvae (p = 0.014), and elav-csp3; csp− and UAS-csp3; csp− larvae (p = 0.026). The individual elav-driven expression of each CSP isoform also rescues the slowly developing temperature-sensitive block of neurotransmitter release ofcspR1 mutants above 29°C (data not shown). Thus, the similar rescue of thecspR1 null neurotransmission phenotype by the neuronal expression of each individual CSP isoform indicates that CSP1, CSP2, and CSP3 may have strongly overlapping functions in evoked neurotransmitter release. In particular, the rescue by CSP3, the presumed non-neuronal CSP isoform inDrosophila, suggests that structural differences of CSP isoforms may not account for the different functions of CSP suggested for evoked neurotransmitter and peptide exocytosis.
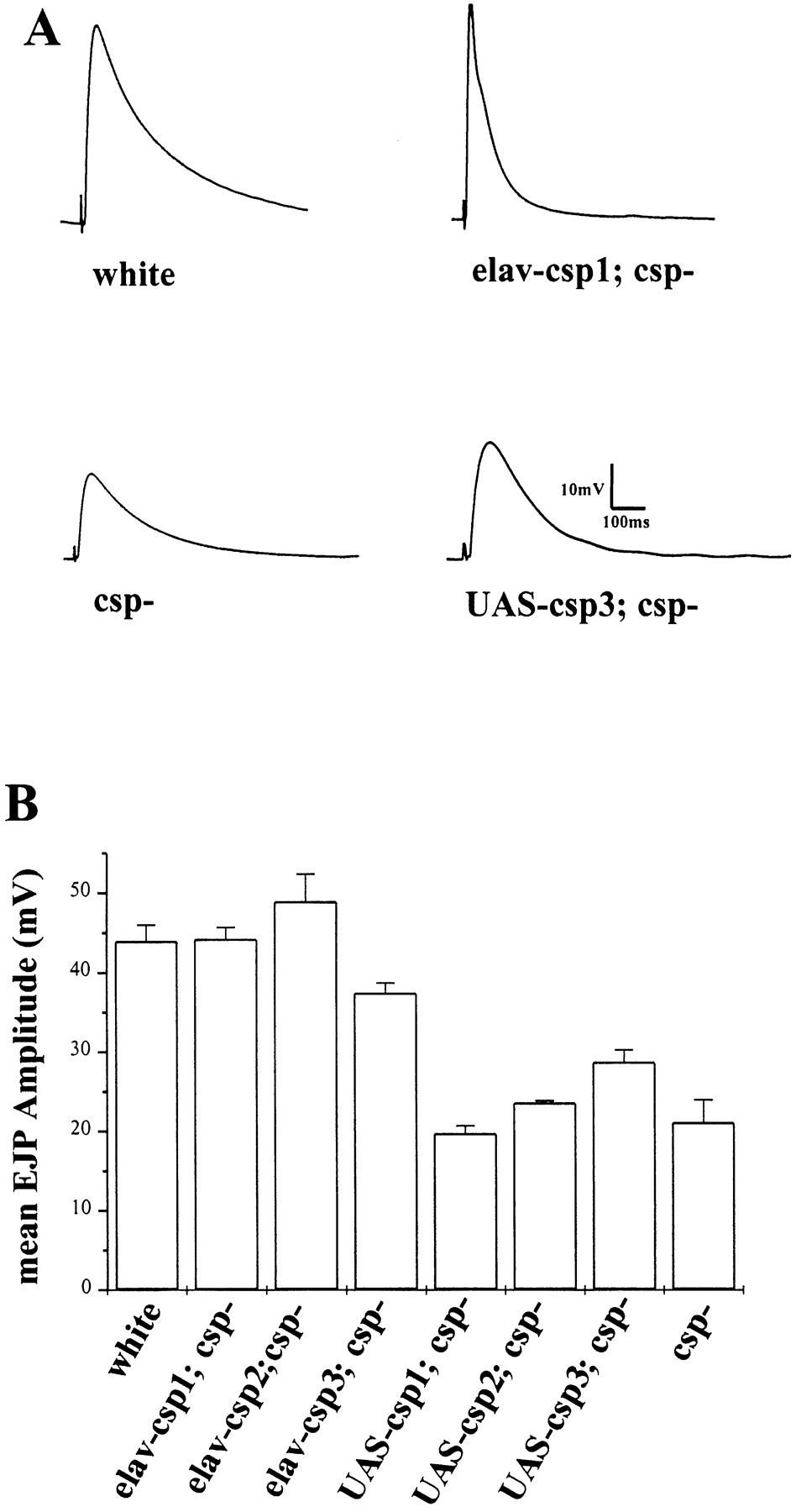
All CSP isoforms individually rescue the loss of evoked release in csp null mutants. A, Representative EJPs are shown for wild-type-like controlwhite, for larvae expressing CSP1 or CSP3 in acsp null mutant genetic background (elav-csp1;csp−and elav-csp3; csp− ), and csp null mutant larvae (csp−). EJPs were recorded from muscle 6 in HL-3 solution at 22°C in the presence of 1 mm Ca2+. Scales for voltage trace and time sweep are as indicated. B, Mean amplitudes of evoked EJP responses at 22°C with 1 mmexternal Ca2+ from wild-type controlwhite, from larvae overexpressing CSP1–3 in a csp minus background (elav-csp1; csp− ,elav-csp2; csp−, elav-csp3; csp−), from controls containing a UAS-csp but not a elav-GAL4 transgene in a csp minus background (UAS-csp1; csp−, UAS-csp2; csp−), and from a csp null mutation (csp−). Error bars represent SEM. Genotypes for A and B: white (w1118), elav-csp1; csp−(w1118, P{elav-GAL4}/w1118; P{UAS-csp1}/+; cspR1), elav-csp2; csp−(w1118, P{elav-GAL4}/w1118; P{UAS-csp2}/+; cspR1), elav-csp3; csp−(w1118, P{elav-GAL4}/w1118; P{UAS-csp3}/+; cspR1), UAS-csp1 (w1118; P{UAS-csp1}/+; cspR1), and the csp−null mutation (w1118; cspR1).
CSP overexpression in Drosophila is semi-lethal and interferes with wing and eye development
After ensuring functional expression of each UAS-csp transgene, we determined the effect of CSP overexpression on the synaptic physiology of wild-type larval motor nerve terminals. The elav-driven overexpression of individual CSPs shows no significant effect on the amplitude of evoked EJP responses at larval neuromuscular junctions (Fig. 3). The mean EJP amplitude recorded from wild-type control is 44.0 ± 2.0 mV (mean ± SEM,n = 4). Larvae overexpressing CSP1–3 with the elav promoter show EJP amplitudes similar to wild type (elav-csp1 39.3 ± 1.9 mV, n = 9; elav-csp2 45.0 ± 4.4 mV,n = 4; elav-csp3 49.9 ± 3.4 mV, n= 4). Larvae containing only a silent UAS-csp transgene also show similar amplitudes (UAS-csp1 39.4 ± 0.2 mV, n = 3; UAS-csp2 40.4 ± 2.1 mV, n = 5; UAS-csp3 44.8 ± 2.3, n = 4). Neither amplitude of CSP1–3-expressing larvae is statistically different from wild-type controls (Student's t test, p > 0.1). Features of spontaneous release are also statistically normal for all examined transgenic flies (data not shown). We tested further whether an overexpression of CSP with the heat-shock promoter after heat-shock induction at 37°C may induce a significant effect on evoked neurotransmitter release. However, neither evoked release nor spontaneous release is significantly affected by the heat-shock-induced overexpression of any CSP isoform (data not shown). Thus, neurotransmitter release at larval neuromuscular junctions appears insensitive to increased levels of CSP.

Neuronal overexpression of individual CSP isoforms has no effect on neurotransmitter release at larval neuromuscular junctions. A, Representative EJPs recorded from larval neuromuscular junctions of wild-type-like control white, larvae overexpressing CSP2 in the nervous system with the elav promoter (elav-csp2), and control larvae containing only the UAS-csp2 or the elav-GAL4 transgene. EJPs were recorded from muscle 6 in HL-3 solution at 22°C in the presence of 1 mmCa2+. Scales for voltage trace and time sweep are as indicated. B, Mean EJP amplitudes from wild-type-like control white, larvae overexpressing CSP1–3 with the elav promoter (elav-csp1, elav-csp2,elav-csp3), and control larvae containing either an elav-GAL4 or a UAS-csp transgene in trans (UAS-csp1, UAS-csp2,UAS-csp3, elav-GAL4). Fifteen EJPs were averaged for n larvae. Recordings were in the presence of 1 mm Ca2+ at 22°C. Error bars represent SEM. Genotypes for A andB: white (w1118), elav-csp1 (w1118, P{elav-GAL4}/w1118; P{UAS-csp2}/+), elav-csp2 (w 1118 ,P{elav-GAL4}/w1118; P{UAS-csp2}/+), elav-csp3 (w 1118 , P{elav-GAL4}/w1118; P{UAS-csp3}/+), UAS-csp1 (w 1118 ; P{UAS-csp1}/+), UAS-csp2 (w 1118 ; P{UAS-csp2}/+), UAS-csp3 (w 1118 ; P{UAS-csp3}/+), elav-GAL4 (w 1118 , P{elav-GAL4}/w1118).
Although overexpression of CSP has no effect on evoked release at larval neuromuscular junctions, it has significant deleterious effects on normal development of Drosophila. Independent UAS-csp lines for each CSP isoform all show similar phenotypes when placed in trans to any of the examined GAL4 expressing lines. The overexpression of individual CSP variants is semi-lethal in a temperature-dependent manner (Table 1). For flies raised at 29°C, the elav-driven overexpression of each CSP isoform in the nervous system is completely lethal. At 25°C, the overexpression is semi-lethal: 1% of CSP1-, 70% of CSP2-, and 23% of CSP3-expressing flies survive. Raising flies at 18°C significantly increases the survival rate. The continuous overexpression of CSP with the ubiquitously expressing heat-shock promoter is lethal at 29°C and at 25°C but allows a few flies to survive at 18°C. We also noticed that the lethality induced by the elav-driven overexpression of CSP is more severe in male than in female flies. At 18°C, the ratio of male to females overexpressing CSP2 is normal, but for males expressing CSP1 the ratio is only 27%, and for males expressing CSP3 it is 68%. First, we speculated that this may be attributable to the X-chromosomal insertion of the elav-GAL4 transgene. However, the sex-linked lethality is also apparent when CSP is overexpressed with the heat-shock promoter transgene, which is linked to an autosome. Only 25% of the expected number of males survive when either CSP isoform is ubiquitously expressed with the heat-shock promoter.
Summary of lethality induced by overexpression of CSP
Flies escaping from the semi-lethal developmental phenotype caused by the elav-driven overexpression of CSP exhibit motor defects, short adult life spans, and morphological defects. At 25°C, adult wild-type flies live longer than 55 d, but adult escapers expressing CSP1 die within the first 5 d of adulthood (Fig.4). Flies overexpressing CSP2 show life-spans not longer than 51 d, whereas CSP3-overexpressing flies die within 10–21 d. All adult flies escaping the lethal overexpression of either isoform are extremely sluggish, show severely uncoordinated movements, and slip frequently. Morphologically, overexpression of all CSP isoforms reduces body size and impairs wing inflation after pupal eclosion such that the wings appear severely crumpled (Fig.5B–D). Interestingly, overexpression of CSP from the weaker csp promoter using the genomic csp transgene shows no crumpled wing phenotype (Fig.5F), indicating that a high level threshold of overexpressed CSP may be critical for the expression of the wing phenotype. Alternatively, it is possible that the elav promoter may ectopically misexpress CSP. We tested these possibilities and reduced the levels of overexpressed CSP by removing the endogenouscsp gene. Removal of one copy of endogenous csp in flies heterozygous for the cspR1deletion (elav/+; UAS-csp2/+; cspR1/+) has little effect on the crumpled wing phenotype (Fig. 5G). However, homozygous cspRI null mutant flies overexpressing CSP2 (elav/+; UAS-csp2/+; cspR1/cspR1)exhibit normal wings (Fig. 5H), suggesting that a critical threshold level of overexpressed CSP is necessary for the expression of the wing phenotype.

Overexpression of CSP reduces adult life span. Survival curves of adult flies overexpressing CSP1–3 and wild-type controls. Adult elav-csp1 flies die prematurely after 4–5 d, elav-csp3 within 10–25 d, and elav-csp2 within 51 d. Controls exhibit life spans longer than 55 d. Flies were raised at 25°C. Newly emerged flies were collected within 12 hr, kept at 25°C, and dead flies were counted every 12 hr.

Wing defects caused by the elav-driven overexpression of individual CSP isoforms. A–L, Light microscopic images of wings from 2- to 5-d-old flies raised at 25°C unless otherwise indicated. Flies overexpressing CSP1–3 in the nervous system by the elav promoter fail to inflate their wings after eclosion such that wings appear “crumpled” (B–D). This phenotype is not observed in flies overexpressing CSP from a csp promoter contained in a genomic csp transgene, csp-csp (F). Reduction of CSP levels by removing one copy of endogenous csp (elav-csp2; csp−/+) partially suppresses the crumpled wing overexpression phenotype (G), and abolishment of endogenous csp (elav-csp2; csp−) fully suppresses the overexpression phenotype (H). Simultaneous overexpression of syntaxin-1A driven by a heat-shock promoter (elav-csp; hs-syx) partially suppresses the elav-CSP wing phenotype in flies raised at 25°C (K). Higher levels of syntaxin induced by the application of a daily heat-shock during late larval and pupal development fully suppresses the wing phenotype for flies raised otherwise at 25°C (L). Genotypes: (A) wild type; (B) elav-csp2 (w1118,P{elav-GAL4}/w1118;P{UAS-csp2}/+); (C) elav-csp1 (w1118, P{elav-GAL4}/w1118;P{UAS-csp1}/+); (D) elav-csp3 (w1118,P{elav-GAL4}/w1118;P{UAS-csp3}/+); (E) elav-GAL4 (w1118,P{elav-Gal4}); (F) csp-csp (w1118; P{csp}—a genomic csp transgene expressing all isoforms); (G) elav-csp3; csp−/+ (w1118, P{elav-GAL4}/w1118; P{UAS-csp2}/+; cspR1/TM3 Sb); (H) elav-csp3; csp−(w1118, P{elav-GAL4}/w1118; P{UAS-csp2}/+; cspR1); (I) elav-csp2 raised at 18°C -compare to (B); (J) hs-syx control (w1118, P{hs-syx}/+; P{UAS-csp2}/+); (K) elav-csp2; hs-syx at 25°C (w 1118 , P{elav-GAL4}/w 1118 ; P{hs-syx}/+; P{UAS-csp2}/+); (L) elav-csp2; hs-syx raised at 25°C and heat-shocked for 1 hr at 37°C per day.
Overexpression of CSP also interferes with eye development, causing a significantly misshaped eye. Externally, the surface of the fly compound eye forms a smooth and regular surface of ∼800 facets, or ommatidia. Small mechanosensory bristles project from alternate facet vertices over most of the eye's surface (Fig.6A). The overexpression of all CSP isoforms disrupts this regular ommatidial pattern such that the surface of the eye appears rough (Fig.6B–D). A closer examination of the rough eye reveals a slightly elliptical shape of the compound eye and the loss of some bristles; a fusion of several ommatidial units is occasionally observed (data not shown). As for the wing and the lethal phenotypes, overexpression of CSP from the csp promoter causes no eye phenotype. However, a reduction of CSP levels caused by removing endogenous CSP reduces the severity of the rough eye phenotype caused by the elav-driven CSP overexpression (Fig. 6F), indicating that the phenotype requires a critical threshold of overexpressed CSP. In addition, we examined CSP overexpression mediated by the glass promoter driving expression in photoreceptors and pigment cells (Ellis et al., 1993). Glass-mediated expression causes a more severe eye phenotype than the overexpression with the elav promoter, showing a very elliptical shape, lacking most bristle cells, and appearing glassy (data not shown).

Eye defects caused by the elav-driven overexpression of individual CSP isoforms. A–I, Scanning electron microscopic images of adult eyes from 2- to 5-d-old flies raised at 25°C unless otherwise indicated. The elav-GAL4 driven overexpression of all three CSP isoforms in the nervous system disrupts eye development, causing a rough surface of the eye (B–D). Flies overexpressing CSP from a genomic csp gene fragment with the csp promoter (csp-csp) show normal eyes (E). The rough eye phenotype of elav-CSP2 is partially suppressed by the removal of endogenous CSP (F). Simultaneous overexpression of syntaxin-1A (hs-syx; elav-csp2) driven by the heat-shock promoter at 25°C partially suppresses the rough eye phenotype of CSP overexpressing flies (H). Higher levels of syntaxin-1A coexpression induced by the application of a daily heat-shock during late larval development completely suppress the rough eye phenotype (I). Genotypes for (A) wild type; (B) elav-csp1 (w1118, P{elav-GAL4}/w1118; P{UAS-csp1}/+); (C) elav-csp2 (w 1118 , P{elav-GAL4}/w1118; P{UAS-csp2}/+); (D) elav-csp3 (w 1118 , P{elav-GAL4}/w1118; P{UAS-csp3}/+); (E) csp-csp (w 1118 ; P{csp} - a genomic csp transgene expressing all isoforms); (F) elav-csp2; csp− (w1118, P{elav-GAL4}/w1118; P{UAS-csp2}/+; cspR1); (G) hs-syx control (w1118, P{hs-syx}/+; P{UAS-csp2}/+); (H) hs-syx; elav-csp2 raised at 25°C (w 1118 , P{elav-GAL4/w1118; P{hs-syx}/+; P{UAS-csp2}/+); (I) hs-syx; elav-csp2 raised at 25°C and heat-shocked for 1 hr at 37°C per day. Scale bar, 100 μm.
Simultaneous overexpression of syntaxin-1A and CSP suppresses the effects of their individual overexpression
Because the overexpression of CSP may cause a “titration” effect of CSP interacting proteins, one may consequently expect that the simultaneous expression of an interacting protein suppresses the effects of CSP overexpression. Thus, we tested whether the coexpression of syntaxin suppresses the CSP overexpression phenotypes because syntaxin has been suggested as a possible target of CSP chaperone action to indirectly modulate presynaptic calcium channel activity (Seagar et al., 1999). To overexpress syntaxin, we used a transgenic line expressing syntaxin-1A (hs-syx) under the direct transcriptional control of a heat-shock promoter (Wu et al., 1998). To exclude nonspecific effects by the overexpression of a synaptic protein, we used a transgenic line expressing SNAP-25 with the heat-shock promoter as a control (Wu et al., 1998). Both fly lines are homozygous viable and show normal wings and eyes when raised at 25°C. Flies were synthesized containing a hs-syx (or hs-SNAP-25), an elav-GAL4, and a UAS-csp transgene in trans to simultaneously overexpress CSP from an elav promoter and syntaxin or SNAP-25 from a heat-shock promoter. At 25°C, coexpression of syntaxin partially suppresses the crumpled wing (Fig. 5K) and the rough eye phenotype (Fig. 6H) induced by the overexpression of CSP. Both the wing and the eye phenotypes are completely rescued by the application of daily heat-shocks during late larval and pupal development to elevate syntaxin expression (Figs. 5L,6I). In contrast, flies coexpressing SNAP-25 show no effect on the mutant wing or eye phenotype induced by the overexpression of CSP (data not shown). This suggests that CSP and syntaxin may specifically interact in vivo.
As shown, CSP overexpression itself does not significantly interfere with evoked neurotransmitter release at motor nerve terminals. However, heat-shock-induced overexpression of syntaxin-1A reduces evoked neurotransmitter release at Drosophila neuromuscular junctions (Wu et al., 1998), raising the possibility that CSP overexpression may suppress the syntaxin overexpression phenotype. Thus, we analyzed evoked release at neuromuscular junctions of larvae overexpressing CSP and syntaxin-1A (Fig.7). Although wild-type larvae exhibit mean EJP amplitudes of 44.0 ± 2.0 mV (mean ± SEM,n = 4), larvae continuously expressing syntaxin from one copy of the hs-syx transgene at 25°C show reduced EJP amplitudes of 29.2 ± 2.2 mV (n = 6). This 34% decrease of evoked release is statistically significant (p = 0.0086, Student's t test) and comparable with the originally described 67% decrease of evoked release where syntaxin-1A overexpression has been induced from two copies of the transgene by a heat-shock at 37°C (Wu et al., 1998). The effect of syntaxin overexpression is not modulated by the presence of either an elav-GAL4 (data not shown) or a UAS-csp2 transgene (30.4 ± 3.2 mV,n = 5). However, larvae containing all transgenes and simultaneously overexpressing syntaxin and CSP (hs-syx; elav-csp2) show normal EJP amplitudes (42.7 ± 3.1 mV, n = 6) at 25°C. Statistical comparison of hs-syx; elav-csp2 with hs-syx or hs-syx; UAS-csp2 responses shows that they are significantly different (p = 0.029 and 0.0031, respectively), whereas hs-syx; elav-csp2 responses are not significantly different from wild-type control (p > 0.05, Student'st test). A similar suppression of syntaxin overexpression phenotypes has also been observed for the coexpression of CSP3 (data not shown). Thus, CSP overexpression fully suppresses the 34% decrease of evoked release induced by the overexpression of syntaxin-1A, indicating that both proteins interact through a common protein complex at synaptic terminals.

Overexpression of CSP suppresses the decrease of evoked release induced by the overexpression of syntaxin-1A. Continuous overexpression of syntaxin-1A from the heat-shock promoter at 25°C reduces neurotransmitter release in a similar manner as the induction of syntaxin expression by a heat-shock. This loss of evoked release is suppressed by the simultaneous overexpression of CSP2.A, Representative EJP recordings from larval neuromuscular junctions of wild-type-like control white, larvae overexpressing CSP2 (elav-csp2), larvae overexpressing syntaxin-1A and containing a csp transgene but not an elav transgene (hs-syx; UAS-csp2), and for larvae overexpressing syntaxin-1A and CSP2 (hs-syx; elav-csp2). All larvae were raised at 25°C and recordings are from muscle 6 at 1 mm Ca2+ at 25°C. B, Mean amplitudes of evoked EJP responses at 25°C with 1 mmexternal Ca2+ from control white, larvae overexpressing CSP2 (elav-csp2), larvae overexpressing syntaxin-1A (hs-syx and hs-syx; UAS-csp2), and larvae overexpressing CSP and syntaxin-1A (hs-syx; elav-csp2). Recordings were as inA. Bars indicate SEM. Genotypes: white (w1118); elav-csp2 (w1118, P{elav-GAL4}/w1118; P{UAS-csp2}/+); hs-syx (w1118; P{ hs-syx}/+); hs-syx; UAS-csp2 (w 1118 ; P{ hs-syx}/+; P{UAS-csp2}/+); and hs-syx; elav-csp2 (w 1118 , P{elav-GAL4/w1118; P{hs-syx}/+; P{UAS-csp2}/+).
CSP directly binds syntaxin-1A
To gather independent biochemical evidence for a protein interaction of CSP with syntaxin, we analyzed whether syntaxin coimmunopurifies with CSP from Drosophila protein extracts and compared CSP immunoprecipitations from wild-type extracts with those from cspX1 null mutations where CSP is absent because of a partial gene deletion (Zinsmaier et al., 1994; Eberle et al., 1998). Proteins of wild-type and homozygouscspX1 extracts were solubilized, and CSP was immunoprecipitated with a monoclonal antibody detecting all CSP isoforms (Zinsmaier et al., 1994; Eberle et al., 1998). Each immunoprecipitate was split and analyzed by immunoblotting for the presence of CSP and syntaxin-1A. CSP antibodies immunoprecipitated CSP and coimmunoprecipitated syntaxin from wild-type protein extracts but not from controls where the antibody, protein A agarose, or the fly extract were omitted (Fig.8A). Consistently, immunoprecipitations from protein extracts ofcspX1 deletion mutants showed no coimmunopurification of syntaxin (Fig. 8B), suggesting that the coimmunopurification of syntaxin specifically depends on the presence of CSP. Controls showed no coimmunopurification of synaptotagmin or synapsin with CSP (data not shown).

In vitro binding of CSP and syntaxin -1A. A, Coimmunoprecipitation of syntaxin-1A from Drosophila wild-type extracts.Drosophila head protein extracts were solubilized with Triton X-100 and immunoprecipitated with DCSP1 antibodies detecting all CSP isoforms (IP CSP). Control experiments omitted the monoclonal antibody (no Ab), the fly extract (no extract), or protein A-coupled beads (no protein A). Bound proteins were recovered by centrifugation, washed, and resuspended in SDS buffer. Three percent of the soluble fraction (S) of the immunoprecipitation and one-half of the immunoprecipitated pellet fraction (P) were analyzed on separate immunoblots for the presence of CSP and syntaxin-1A (Syx). CSP antibodies immunoprecipitate CSPs and coimmunoprecipitate syntaxin. Controls show neither a CSP- nor a syntaxin-specific signal. B, Similar coimmunoprecipitation of syntaxin-1A from protein extracts of wild-typeDrosophila and cspX1deletion mutants where CSP is absent shows copurification of syntaxin from wild-type extracts but not from mutant extracts. C,Recombinant protein binding of syntaxin-1A with CSP. Soluble His-CSP was incubated in a 2:1 molar ratio with immobilized GST-syntaxin-1A (GST-Syx), with immobilized GST-synaptotagmin (GST-Syt), or with immobilized GST protein (GST control) for 2 hr at 4°C. The affinity precipitate was recovered by centrifugation, washed, and resuspended in SDS-PAGE buffer. Recombinant His-CSP (1/25) used for the binding assay and all of the affinity precipitate was analyzed by immunoblotting for the presence of recombinant CSP. Note that <4% of total His-CSP binds by GST-syntaxin (GST-Syx + His-CSP) but not to GST-synaptotagmin (GST-Syt + His-CSP) or the GST control.
Because coimmunopurifications do not indicate a direct protein–protein interaction, we performed protein binding assays with recombinant CSP and syntaxin-1A fusion proteins. For these experiments, we used His-CSP and GST-tagged syntaxin-1A. GST-syntaxin fusion protein was immobilized to glutathione agarose beads and incubated with defined amounts of purified His-tagged CSP fusion protein. Controls contained equal amounts of immobilized GST-synaptotagmin fusion protein or immobilized GST protein. As shown in Figure 8C, His-CSP fusion protein binds to GST-syntaxin but not to GST-synaptotagmin or to GST protein alone, indicating that CSP directly binds to syntaxin. Comparing the amount of His-CSP retained by GST-syntaxin with the total amount of His-CSP used reveals that <4% of the incubated His-CSP is bound to syntaxin. Together, the genetic interactions and the in vitro binding of CSP and syntaxin support the speculation that syntaxin may be a target of CSP chaperone function.
DISCUSSION
It has been suggested that CSP modulates depolarization-dependent calcium fluxes in neurotransmitter exocytosis (Gundersen and Umbach, 1992; Mastrogiacomo et al., 1994) and mediates a direct step in peptidergic exocytosis independent of calcium transmembrane fluxes (Brown et al., 1998; Chamberlain and Burgoyne, 1998; Zhang et al., 1998; Morales et al., 1999). Because several slightly different CSP variants are differentially expressed in invertebrates and mammals (Zinsmaier et al., 1994; Chamberlain and Burgoyne, 1996; Brown et al., 1998; Eberle et al., 1998), it appears possible that these different protein isoforms may mediate the contrasting functions of CSP for the secretion of neurotransmitters and peptides. To test whether the differentially expressed Drosophila CSP isoforms may equally mediate neurotransmitter release, we individually expressed each isoform in the nervous system of csp null mutantDrosophila. As shown here, the individual expression of each CSP variant is able to restore the loss of evoked neurotransmitter release at larval motor nerve terminals of csp null mutants. The full rescue of evoked release by CSP3, which we identified to encode a 36 kDa isoform abundantly expressed in non-neuronal tissues but not at neuromuscular junctions (Eberle et al., 1998), especially indicates that the three examined CSP variants may have largely overlapping functions in neuronal and non-neuronal cells despite their differential expression patterns. Because CSP has been shown to mediate a direct step of exocytosis independent of transmembrane calcium fluxes in PC12 and insulin-secreting cells, it now appears possible that CSP may mediate a similar step in neurotransmitter exocytosis in addition to the suggested modulation of calcium channels.
We further investigated the effects of CSP overexpression for regulated neurotransmitter release in Drosophila. Although the overexpression of CSP potentiates dopamine release in permeabilized neuroendocrine PC12 cells (Chamberlain and Burgoyne, 1998), it depresses stimulated insulin release in insulin-secreting cell lines (Brown et al., 1998; Zhang et al., 1999). In contrast to the opposing effects of CSP overexpression in endocrine and neuroendocrine secretion, the overexpression of CSP at Drosophila motor nerve terminals has no significant effect on evoked neurotransmitter release as shown by normal EJP amplitudes in larvae overexpressing CSP1–3. This difference between PC12, β-cells, and fly motor nerve terminals may be attributable to the different approaches used, or it may be influenced by different levels of endogenous CSP. Alternatively, the different observations may reflect different functional requirements of CSP for stimulated exocytosis in endocrine, neuroendocrine, and nerve terminals.
Although CSP overexpression does not modulate neurotransmission at fly motor nerve terminals, it has otherwise severe effects that reduce viability during development and adult life. Adult escapers exhibit motor defects such as uncoordinated walking and impaired balance control, which may indicate that synaptic transmission at synapses of the CNS is impaired, whereas the robust larval neuromuscular junctions are less sensitive to increased levels of CSP. Escapers also exhibit morphological defects in wing and eye development that may be a result of improper secretion of protein factors that influence cell–cell communication in the developing eye or wing disk. Alternatively, the defects may arise from anomalies in membrane biogenesis. As for the overexpression of mammalian CSPs in insulin-secreting cell lines (Zhang et al., 1999), the overexpression of individual Drosophila CSP isoforms causes similar effects, although some slight quantitative differences are observed. These could be caused by position effects at different insertion sites of individual transgenes (Spradling and Rubin, 1983), but this is unlikely because we examined at least two independent lines for each transgene. Thus, it may indicate that the three CSP isoforms may indeed somewhat differ in their function, although this difference is apparently too subtle or not relevant for evoked neurotransmitter release at motor nerve terminals.
We tested further whether the effects of CSP overexpression may be suppressed by the simultaneous overexpression of syntaxin, which has earlier been speculated to interact with CSP (Seagar et al., 1999). We provide genetic and biochemical evidence that supports an interaction of CSP with syntaxin. Coimmunoprecipitation of syntaxin with CSP fromDrosophila protein extracts and further protein binding assays using recombinant proteins demonstrate a direct protein–protein interaction between CSP and syntaxin-1A. The weak nature of this interaction is not unexpected because transient protein–protein interactions are often a critical feature among molecular chaperones and their substrates (Gething and Sambrook, 1992; Silver and Way, 1993). This biochemical evidence is further strengthened by genetic interactions because the coexpression of syntaxin-1A, but not that of the SNAP-25 control, suppresses the wing and eye phenotypes caused by the overexpression of CSP, indicating that syntaxin specifically interacts with CSP. The suppression of the eye phenotype by syntaxin is consistent with our speculation that overexpression of CSP may titrate its binding partners, because loss-of-function mutations of syntaxin also show a rough eye phenotype (Schulze and Bellen, 1996). Although the overexpression of CSP itself does not modulate evoked neurotransmitter release, it fully suppresses the decrease of neurotransmitter release at Drosophila neuromuscular junctions induced by the overexpression of syntaxin-1A alone. Together, the genetic interactions and the in vitro binding of CSP and syntaxin-1A suggest that CSP and syntaxin-1A are associated with a common protein complex, possibly to mediate evoked neurotransmitter release and other steps of membrane traffic.
Two alternative models of CSP function have been suggested. For peptidergic secretion, CSP has been shown to mediate a direct step of stimulated exocytosis independent of calcium transmembrane fluxes (Brown et al., 1998; Chamberlain and Burgoyne, 1998; Zhang et al., 1998; Morales et al., 1999). An increase or decrease of CSP levels in PC12 and insulin-secreting cells severely reduced exocytosis without affecting transmembrane calcium fluxes. Because these effects persisted in permeabilized cells, any regulation via soluble second messengers can be excluded, suggesting that CSP may function directly in exocytosis (Chamberlain and Burgoyne, 1998; Zhang et al., 1998, 1999). In contrast, for fast neurotransmitter release, CSP has been suggested to link synaptic vesicles with calcium channels and to modulate channel activity because CSP is associated with synaptic vesicle membranes (Mastrogiacomo et al., 1994) and modulated N-type calcium channel currents when ectopically coexpressed in frog oocytes (Gundersen and Umbach, 1992). This idea has been further supported by the in vitro binding of CSP to P/Q-type calcium channels (Leveque et al., 1998) and by the consistent defects observed in csp mutantDrosophila like the reduction of evoked but not spontaneous neurotransmitter release (Umbach et al., 1994; Zinsmaier et al., 1994) and reduced presynaptic cytosolic calcium levels after repetitive stimulation (Umbach et al., 1998).
It has been further speculated that CSP may coordinate sequential protein–protein interactions between calcium channels and their associated synaptic proteins (Seagar et al., 1999) because CSP is presumably a vesicular, membrane-bound cofactor of the molecular chaperone Hsc70 (Braun et al., 1996; Chamberlain and Burgoyne, 1997a,b) and because CSP binds to the same cytoplasmic loopII–III of calcium channels that contains the synprint site mediating channel interactions with SNAP-25, synaptotagmin, and syntaxin (Leveque et al., 1998). Interestingly, disruption of these interactions by the presynaptic injection of the synprint binding peptide in rat neurons (Mochida et al., 1996) causes strikingly similar defects of evoked neurotransmitter release as the deletion of the Drosophila csp gene (Umbach et al., 1994;Heckmann et al., 1997): both reduce synchronous release but increase asynchronous release and paired pulse facilitation. Because the syntaxin/channel interaction reduces channel activity by prolonging an inactivated state (Bezprozvanny et al., 1995; Wiser et al., 1996), it has been speculated that CSP could promote the dissociation of a syntaxin/channel complex (Seagar et al., 1999), if CSP promotes calcium channel activity as originally suggested (Gundersen and Umbach, 1992).
Here we provide experimental evidence for this speculation showing that CSP interacts with syntaxin in vitro and in vivo. This interaction may modulate the dissociation of syntaxin from calcium channels as previously speculated (Seagar et al., 1999). Alternatively, CSP may modulate protein interactions with other syntaxin-interacting proteins to mediate a calcium-dependent step of exocytosis as implied by the direct role of CSP in peptidergic exocytosis (Chamberlain and Burgoyne, 1998; Zhang et al., 1998, 1999). An obvious candidate for this interaction is synaptobrevin, which has been shown to bind CSPin vitro (Leveque et al., 1998). Both speculations are consistent with our results and with the recent characterization of a mutation in Drosophila syntaxin-1A deleting a multiple protein binding domain that reduces CSP, synaptotagmin, and calcium channel binding (Wu et al., 1999). A third possible role for the CSP/syntaxin interaction is that CSP may simply chaperone protein folding or protein transport of syntaxin. Such a function should cause reduced levels of syntaxin in csp null mutants and proportionally reduce evoked and spontaneous release as observed inDrosophila syntaxin mutants (Schulze et al., 1995; Wu et al., 1998). However, this possibility is unlikely because only evoked release but not spontaneous release is abolished in csp null mutants at restrictive temperatures (Umbach et al., 1994). To finally determine whether the CSP/syntaxin interaction is required for a modulation of presynaptic calcium channel activity or for a step of evoked exocytosis independent of calcium transmembrane fluxes, or both, will require the analysis of mutations in CSP that specifically interrupt the interaction with syntaxin.
Footnotes
This work was supported by grants to K.E.Z. from the National Science Foundation, National Institutes of Health, the March of Dimes Birth Defects Foundation, and the Whitehall Foundation. We thank Drs. T. L. Schwarz, M. Wu, and H. Bellen for sharing cDNA constructs and fly stocks, and S. Benzer for the gifts of antibodies used in this investigation.
Z.N. and R.R. contributed equally to this work.
Correspondence should be addressed to Konrad E. Zinsmaier, Department of Neuroscience, 234d Stemmler Hall, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6974. E-mail:zinsmaie{at}mail.med.upenn.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}