

Abstract
An avian ortholog of transforming growth factor β1 (TGFβ1) is the target-derived factor responsible for the developmental expression of large-conductance Ca2+-activated K+(KCa) channels in chick ciliary ganglion (CG) neurons developing in vivo and in vitro. Application of TGFβ1 evokes an acute stimulation of KCathat can be observed immediately after cessation of a 12 hr exposure to this factor, that persists in the presence of protein synthesis inhibitors, and that is therefore mediated by posttranslational events. Here we show that a single 3 hr exposure to TGFβ1 can also induce long-lasting stimulation of macroscopic KCa that persists for at least 3.5 d after the end of the treatment. In contrast to the acute stimulation, this sustained effect is dependent on the transcription and synthesis of new proteins at approximately the time of TGFβ1 treatment. However TGFβ1 does not cause increases in the levels of slowpoke α subunit transcripts in CG neurons, suggesting that induction of some other protein or proteins is required for sustained enhancement of macroscopic KCa. In addition, application of TGFβ1 evoked an almost immediate but transient phosphorylation of the mitogen-activated protein kinase Erk in CG neurons. TGFβ1-evoked Erk activation was blocked by the specific MEK1 inhibitor 2- (2′-amino-3′-methoxyphenyl)-oxanaphthalen-4-one (PD98059). Moreover, application of PD98059 blocked both acute and sustained KCa stimulation evoked by TGFβ1. These results indicate that TGFβ1 elicits a biphasic stimulation of KCa via activation of an MEK1–Erk pathway and raise the possibility that other neuronal effects of TGFβ superfamily members entail Erk activation.
The functional expression of Ca2+-activated K+ channels (KCa) in the large neurons of the developing chick ciliary ganglion (CG) requires interactions with target tissues in the eye (Dourado et al., 1994; Dryer, 1998). This effect is mediated by a soluble trophic factor expressed in the iris, one of the main target tissues of CG neurons (Subramony et al., 1996). We have presented evidence recently indicating that this iris-derived factor is an isoform of transforming growth factor β (TGFβ) (Cameron et al., 1998, 1999; Lhuillier and Dryer, 1999). Transcripts encoding TGFβ4, the principal avian ortholog of TGFβ1 (Burt and Paton, 1992), are expressed in the iris at the appropriate developmental stages (Cameron et al., 1999), and application of recombinant TGFβ1 to CG neurons developing in vitro or in vivo stimulates the functional expression of KCa. Moreover, intraocular injection of a neutralizing pan-TGFβ antiserum inhibits the normal expression of KCa in CG neurons developing in vivo, and the same antiserum blocks the effects of iris extracts on CG neurons developing in vitro (Cameron et al., 1998).
Our previous studies have shown that significant stimulation of KCa in CG neurons is not detectable until at least 5 hr after the onset of a continuous exposure to TGFβ1. In spite of this relatively slow time course, the stimulatory effects of TGFβ1 are not affected by protein synthesis inhibitors and therefore appear to be posttranslational (Subramony et al., 1996; Cameron et al., 1998). These observations raise several questions. For example, it is not known whether the stimulatory effects of TGFβ1 are persistent or, alternatively, whether this factor needs to be present continuously to maintain high densities of functional KCachannels. In addition, the nature of the transduction cascades that mediate the actions of TGFβ1 is unknown.
This last question is of interest because the actions of TGFβs in many systems are caused by SMAD-dependent changes in transcriptional regulation. SMADs are transcription factors that are phosphorylated after stimulation of TGFβ receptors and translocated to the nucleus as heteromeric complexes resulting in changes in gene expression (for review, see Baker and Harland, 1997; Heldin et al., 1997; Massague, 1998). These pathways do not provide an obvious explanation for the posttranslational effects of TGFβ1 on KCa expression in CG neurons. However, a number of reports indicate that TGFβs can also cause activation of mitogen-activated protein kinase (MAP kinase) cascades within minutes after the onset of TGFβ treatment (Hartsough and Mulder, 1995;Hartsough et al., 1996).
We now report that KCa stimulation evoked by a single 3 hr application of TGFβ1 can be observed for almost 4 d after cessation of treatment. The mechanism of this sustained effect differs from the acute stimulation of KCa in that it requires both transcription and translation as well as a longer duration of TGFβ1 treatment. We also show that TGFβ1 evokes a transient activation of the MAP kinase Erk in CG neurons and that Erk activation is essential for both the acute and sustained effects of TGFβ1 on KCa expression.
MATERIALS AND METHODS
Cell isolation and culture. Ciliary ganglion neurons were dissociated at embryonic day 9 (E9) or E13 as described previously (Subramony et al., 1996; Cameron et al., 1998, 1999; Lhuillier and Dryer, 1999). Experiments on acutely dissociated E13 cells were performed within 3 hr of cell dissociation. Neurons dissociated at E9 were grown for various lengths of time, as indicated in the text and figure legends, on poly-d-lysine-coated glass coverslips in a culture medium described previously (Subramony et al., 1996; Cameron et al., 1998, 1999). Recombinant human TGFβ1 was obtained from R & D Systems (Minneapolis, MN). For experiments designed to examine the role of protein synthesis in the regulation of KCa, the reversible translational inhibitor anisomycin (0.1 mg/ml) and the reversible transcriptional inhibitor 5,6-dichlorobenzimidazole riboside (DRB; 100 μm) were obtained from Sigma (St. Louis, MO) and added to culture media immediately before use. These agents have been shown previously to cause essentially complete inhibition of protein (Subramony et al., 1996) and RNA (Bruses and Pilar, 1995) synthesis in cultured CG neurons. The MEK1 inhibitor 2-(2′-amino-3′-methoxyphenyl)-oxanaphthalen-4-one (PD98059) and the Ca2+/calmodulin-dependent protein kinase II (CaM kinase II) inhibitor KN-62 were obtained from Sigma. Cells were incubated with these inhibitors 30 min before the addition of trophic factors.
Electrophysiology. Whole-cell recordings were made using standard methods as described previously (Dourado and Dryer, 1992;Dourado et al., 1994; Subramony et al., 1996; Cameron et al., 1998,1999). Briefly, 25 msec depolarizing steps to 0 mV were applied from a holding potential of −40 mV in normal and nominally Ca2+-free salines containing 500 nm tetrodotoxin, and the net Ca2+-dependent currents were obtained by digital subtraction using Pclamp software (Axon Instruments, Foster City, CA). Currents were normalized for cell size by computing the soma surface area as described previously (Dourado and Dryer, 1992;Subramony et al., 1996; Cameron et al., 1998, 1999). Similar protocols were used to analyze voltage-activated Ca2+ currents except that KCl in the recording pipettes was replaced with CsCl as described previously (Dourado and Dryer, 1992; Dourado et al., 1994; Cameron et al., 1999). Throughout this paper, error bars represent SEM. Data were analyzed by one-way ANOVA followed by Scheffé's multiple range test using Statistica software (Statsoft, Tulsa, OK), with p < 0.05 regarded as significant.
Immunoblot analysis of mitogen-activated protein kinase (Erk) phosphorylation. For each experimental group, a total of 10 E9 CG were plated onto a single coverslip. TGFβ1 (1 nm) was applied to cultures 3 hr after plating and maintained for varying lengths of time as indicated. Controls did not receive trophic factors. Cells were then washed in ice-cold PBS and lysed in 2× Laemmli sample buffer. Samples were boiled for 5 min and separated by SDS-PAGE on 12% gels. Proteins were transferred to nitrocellulose membranes, which were then blocked in a Tris-buffered saline containing 0.1% Tween 20 and 5% nonfat dried milk before overnight incubation with either a monoclonal antibody specific for diphospho-Erk (Erk-P2; Sigma) or a polyclonal antibody insensitive to the phosphorylation state of Erk (Santa Cruz Biochemicals). Each sample was probed with both antibodies. Blots were analyzed using anti-mouse and anti-rabbit secondary antibodies conjugated to horseradish peroxidase and an ECL detection system (Amersham, Arlington Heights, IL). The ratio of Erk-P2 to total Erk in each culture was determined by densitometry using Scion Image software (Scion Corporation, Frederick, MD). All experiments were repeated three to six times.
Reverse transcription-PCR analysis of slotranscript expression. The α subunits of large-conductance KCa channels are encoded by theslowpoke (slo) gene. Reverse transcription (RT)-PCR procedures for detection of slo transcripts in chick CG are modified from the methods of Subramony et al. (1996). CG neurons were dissociated at E9, plated at a density of two ganglion equivalents per glass coverslip, and exposed to 1 nm TGFβ1 3 hr after isolation. Control preparations did not receive TGFβ1. Total RNA was isolated from each coverslip using a commercial version of the method of Chomczynski and Sacchi (1987) (Genosys, Woodlands, TX). All of the extracted RNA was random transcribed (1 hr at 37°; Pro-Star RT-PCR kit; Stratagene, La Jolla, CA). All quantitative procedures were performed using the same lot of reverse transcriptase. For PCR analysis of β-actinand slo, cDNAs were amplified in separate tubes using 5 μl of the first-strand product with the following primers: forslo, 5′-GAA-TAC-CTG-AGA-AGG-GAA-TGG-GAG-3′ (forward) and 5′-ATG-CGG-GTC-CAC-ATG-CAA-AG-3′ (reverse) as described previously (Subramony et al., 1996), and for β-actin, 5′-TGC-TGT-GTT-CCC-ATC-TAT-CGT-G-3′ (forward) and 5′-TCT-TTC-TGG-CCC-ATA-CCA-ACC-3′ (reverse). The β-actinprimers yield a 68 bp product whose identity was confirmed by sequencing using methods described previously (Subramony et al., 1996). PCR reactions were performed in 20 μl of a buffer consisting of 2.5 mm MgCl2, 60 mm Tris, 15 mm(NH4)3SO4, 0.25 mm each dNTP, 150 pmol of each primer, and 1 U of Taq polymerase (Promega, Madison, WI). Cycling parameters were 2 min at 92° followed by 25 cycles (slo) or 20 cycles (β-actin) of 1 min at 92°, 3 min at 57°, and 3 min at 72°, followed by a 10 min extension at 72°. These parameters were optimized to remain on the linear phase of the amplification curves for both slo and β-actin. PCR products were separated on 1.5% agarose gels containing ethidium bromide, and band intensity was quantified by densitometry using Scion Image software. To establish that the RT-PCR procedures were quantitative, total RNA was extracted from E9 CG neurons and subjected to serial dilution. For both β-actin and slo, the amount of RT-PCR product was linearly related to the initial total RNA concentration. Quantitation of the effects of TGFβ1 was performed by two different procedures. First, we determined the amount ofslo and β-actin PCR product produced by amplifying from a fixed concentration of total cDNA. Data obtained from this experimental design are expressed as sloβ-actin ratios to normalize for possible differences in the efficiency of RNA extraction, gel loading, etc. Second, total cDNAs obtained from control and TGFβ1-treated cells were subjected to serial dilution to determine the minimum cDNA concentration required to yield a detectable slo PCR product (Tkatch et al., 2000). In these experiments, the cDNA from control and TGFβ1-treated cells had indistinguishable levels of β-actin transcript expression.
RESULTS
Acute and sustained effects of TGFβ1 on KCaexpression in CG neurons
These experiments were performed on CG neurons dissociated at E9. Application of 1 nm recombinant human TGFβ1 for 12 hr caused a robust increase in the functional expression of macroscopic KCa compared with that in controls cultured in the absence of trophic factors (Fig. 1). This increase, which was apparent at the end of the 12 hr treatment (acute stimulation), persisted for at least 3.5 d after the cessation of treatment (sustained stimulation; Fig.1B,C). The acute and sustained effects of TGFβ1 differ in two important ways. First, the acute effect of TGFβ1 could be evoked in the presence of either the reversible transcriptional inhibitor DRB (100 μm) or the reversible translational inhibitor anisomycin (0.1 mg/ml). This indicates that posttranslational mechanisms are sufficient for acute KCa stimulation in CG neurons, as noted in previous reports from our laboratory (Subramony et al., 1996; Cameron et al., 1998). By contrast, the sustained increase in KCa expression was blocked when either anisomycin (Fig. 1C) or DRB (Fig. 1D) were present during and up to 12 hr after cessation of TGFβ1 treatment. These results are statistically significant (p < 0.05). An identical pattern was observed when KCaexpression was stimulated with iris extracts (data not shown). Therefore, the KCa stimulatory action of TGFβ1 in CG neurons is biphasic and has an acute posttranslational component and a sustained component dependent on transcriptional activation and the synthesis of new proteins during or up to12 hr after TGFβ1 treatment. Inhibition of protein synthesis in this manner had no effect on the expression of voltage-activated Ca2+ currents at either time point after TGFβ1 treatment (data not shown). Second, the acute and sustained components of KCa stimulation require different durations of TGFβ1 treatment (Fig. 2). This was established by an experimental design in which 1 nm TGFβ1 was applied to E9 CG neurons for different lengths of time, ranging from 5 min to 12 hr. TGFβ1 was then removed, and CG neurons were maintained in normal medium until either 12 hr or 4 d after the onset of TGFβ1 treatment. Maximal acute stimulation of KCa expression (measured at 12 hr) was achieved with a 1 hr exposure to 1 nmTGFβ1. In contrast, maximum sustained KCaexpression (measured at 4 d) required 3 hr of exposure to 1 nm TGFβ1 and was significantly (p < 0.05) less than maximal with 1 hr of TGFβ1 treatment (Fig. 2).

The acute and sustained effects of TGFβ1 on KCa expression are differentially sensitive to inhibition of protein synthesis. A, Schematic diagram of experimental design. E9 CG neurons were cultured for 12 hr in the presence of 1 nm TGFβ1, in the presence of 1 nm TGFβ1 and protein synthesis inhibitors, or in the absence of any trophic factor. KCa expression was quantified by whole-cell recording immediately or 3.5 d after the cessation of TGFβ1 treatment, as indicated by arrows.B, Representative traces of macroscopic KCa currents in E9 CG neurons treated as indicated.C, Summary of the effects of the reversible translational inhibitor anisomycin in many cells. A single 12 hr application of TGFβ1 evoked a robust and significant (p < 0.05) stimulation of KCa(■) compared with that in control cells (○). A significant (p < 0.05) acute stimulation of KCa persisted in the presence of anisomycin (0.1 mg/ml; ▪). This increase in whole-cell currents is long lasting and can be seen 3.5 d after the end of the TGFβ1 treatment. However, this sustained effect is blocked by treatment with anisomycin during the first 24 hr (p < 0.05). D, Similar results obtained with the reversible transcriptional inhibitor DRB (100 μm; ▪). Therefore transcriptional and translational events during the first 24 hr after the onset of TGFβ1 treatment are necessary to produce a long-lasting stimulation of macroscopic KCa. Data are the mean ± SEM computed from 7 to 15 cells in each group. Data were analyzed by one-way ANOVA followed by Scheffé's post hoc test.

Sustained stimulation of KCa requires a longer duration of TGFβ1 exposure than does acute stimulation. E9 CG neurons were cultured in the presence of 1 nm TGFβ1 for the times indicated on the x-axis. The medium was then changed, and whole-cell KCa was measured 12 hr (○) or 4 d (●) after the start of TGFβ1 treatment. Currents are normalized to the maximum for each group. Note that a single 1 hr exposure to TGFβ1 is sufficient to produce maximal acute KCa stimulation. However a 3 hr treatment with TGFβ1 is needed to produce maximal long-lasting KCa expression. Data are the mean ± SEM computed from 8 to 21 cells for each point.
Although sustained stimulation of KCa expression in CG neurons requires the transcription and synthesis of new proteins at approximately the time of TGFβ1 application, the experiments with DRB and anisomycin do not indicate whether it is the SLO α subunits themselves that are induced. For example, TGFβ1 actions on the functional expression of KCa channels could entail induction of other proteins, e.g., auxiliary subunits or proteins required for plasma membrane insertion. To test this hypothesis, the effects of TGFβ1 on the expression of slotranscripts were analyzed using semiquantitative RT-PCR procedures (Fig. 3). In initial experiments, we established RT-PCR procedures that could detect small differences in the amounts of β-actin and slo transcripts present in a complex mixture of total RNA and that yielded a linear relationship between starting transcript levels and the yield of final products. To test the hypothesis, E9 CG neurons were cultured for 12 hr in the presence or absence of 1 nm TGFβ1, total RNA was extracted, and slo and β-actintranscript expressions were determined. By the use of a fixed cDNA template concentration, treatment with TGFβ1 had no effect on theslo/β-actin transcript ratio (Fig.3A). Moreover, by the use of a range of total cDNA template dilutions, TGFβ1 treatment had no effect on the minimum cDNA concentration required to yield a visible slo PCR product (Fig. 3B). In other words, we find no evidence of the induction of slo α subunit transcripts by TGFβ1. Instead, these data suggest that the sustained effects of TGFβ1 on the functional expression of KCa channels are associated with new synthesis of some other protein or proteins.

TGFβ1 does not stimulate an increase ofslo transcript levels in CG neurons. E9 CG neurons were treated with 1 nm TGFβ1 for 12 hr at which timeslo expression was determined by semiquantitative RT-PCR. A, Top, Examples ofslo and β-actin transcript detection by RT-PCR in control and TGFβ1-treated cells are shown. Note the comparable intensity signals for both transcripts in both groups of cells. Bottom, The mean ± SEM of theslo/β-actin intensity ratios derived from six repetitions of this experiment is shown and indicates that TGFβ1 has no significant effect on slo expression in CG neurons. B, To confirm this result, cDNA from the same cultures used in A was subjected to serial dilution before PCR. Top, Results of a representative serial dilution experiment are shown. Bottom, The mean ± SEM of the log10 of the maximal dilution at whichslo transcripts could be detected is shown. Data are derived from six repetitions of this experiment. This value is not increased in TGFβ1-treated cells, further indicating that TGFβ1 does not increase slo transcript expression.
Role of mitogen-activated protein kinase cascades in TGFβ1 actions
The fact that acute stimulation of KCachannels by TGFβ1 occurs by posttranslational mechanisms in CG neurons raises an important question as to the nature of the underlying transduction cascade. SMAD activation seems unlikely, because these proteins are transcription factors that have distincttrans-activation and protein interaction domains but that lack motifs that would suggest other enzymatic activities (Kretzschmar and Massague, 1998; Shioda et al., 1998; Johnson et al., 1999). However, there are reports of rapid activation of MAP kinases by TGFβ1 in non-neuronal systems (Hartsough and Mulder, 1995; Hartsough et al., 1996). We have observed that this also occurs in developing CG neurons. These experiments focused on the MAP kinase Erk, which becomes active after phosphorylation of two residues by the dual-specific MAP kinase kinase MEK1 (Matsuda et al., 1992; Seger et al., 1992). Dissociated E9 CG neurons were treated with 1 nmTGFβ1 or normal medium, and the relative abundance of total Erk and Erk-P2 was determined by immunoblot analysis. Note that chicks differ from mammals in that they express only one form of Erk, and therefore immunoblot analysis reveals only a single 42–43 kDa band for this kinase (Perron and Bixby, 1999; Sanada et al., 2000). Treatment with TGFβ1 caused a significant increase in the Erk-P2/total Erk ratio (Fig.4). Interestingly Erk phosphorylation was relatively transient; increases were detectable within 5 min after the onset of TGFβ1 treatment and maintained for at least 1 hr, but returned to baseline after 3 hr, even when TGFβ1 was present continuously. The increase in Erk phosphorylation evoked by TGFβ1 was completely blocked in CG neurons pretreated with the selective MEK1 inhibitor PD98059 (50 μm), an agent widely used in the study of MAP kinase-signaling cascades (Alessi et al., 1995;Dudley et al., 1995). PD98059 also caused a decrease in basal Erk phosphorylation in CG neurons that were not treated with TGFβ1 (Fig.5).
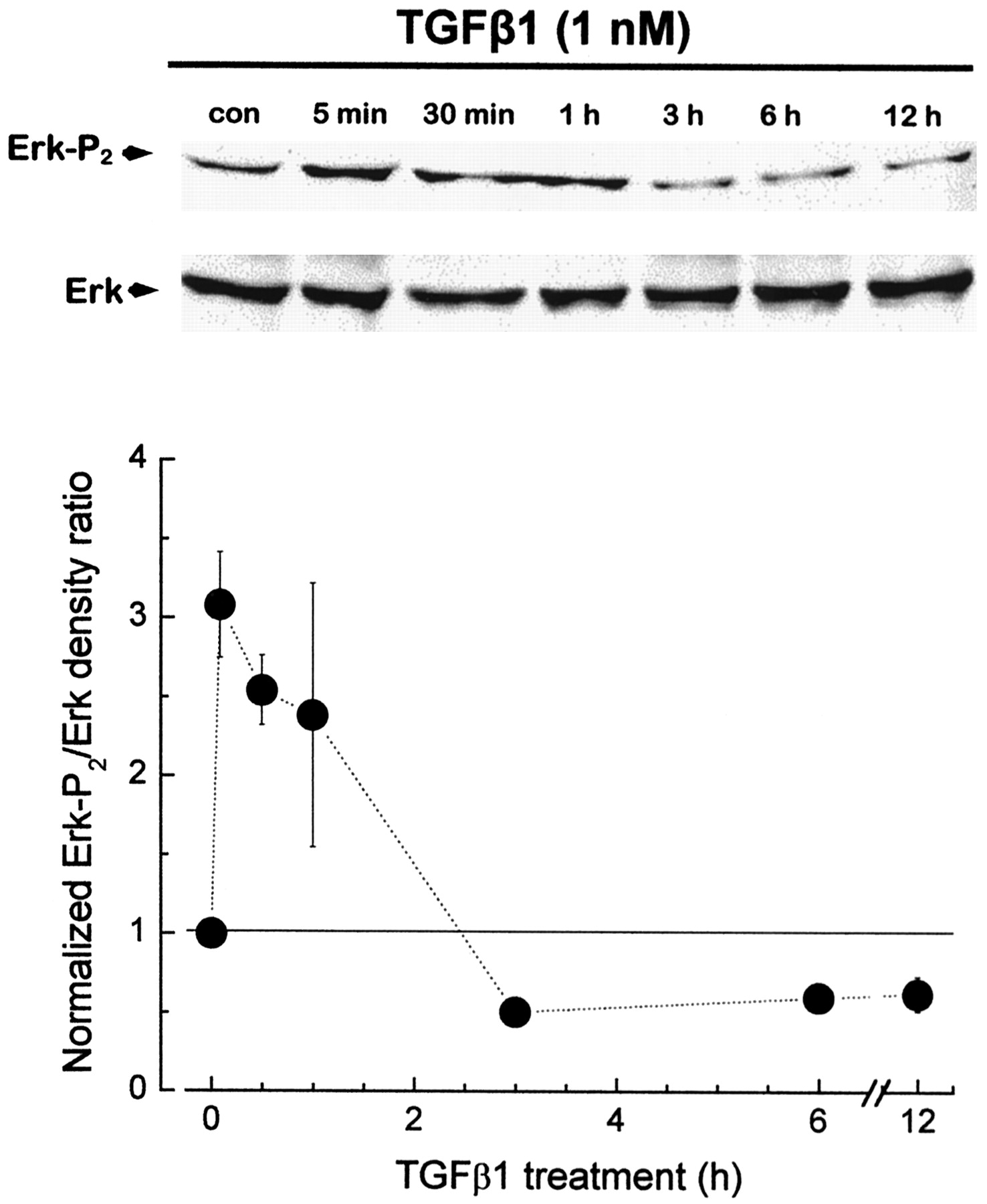
TGFβ1 evokes a transient activation of Erk MAP kinase in CG neurons. E9 CG neurons were cultured in the presence of 1 nm TGFβ1 for various lengths of time as indicated. After treatment, Erk-P2 and total Erk were determined by immunoblot analysis on duplicate gels. Top, Representative blots. Bottom, Mean Erk-P2/Erk ratios ± SEM obtained from three to six independent experiments. TGFβ1 evoked an increase in Erk phosphorylation within 5 min. This increase persisted for >1 hr, but Erk-P2 levels returned to baseline at 3 hr, even in the continuous presence of TGFβ1. con, Control.

TGFβ1-evoked Erk activation in CG neurons is blocked by the MEK1 inhibitor PD98059. Erk phosphorylation in E9 CG neurons was determined by immunoblot analysis of cells cultured in the presence or absence of PD98059 (50 μm) and/or TGFβ1 (1 nm). Top, A representative immunoblot.Bottom, Summary of the mean ± SEM from three independent experiments. Note that PD98059 completely blocked Erk phosphorylation induced by TGFβ1 and also caused a marked fall in basal Erk phosphorylation.
These results raise the possibility that TGFβ1 acts via MAP kinase cascades to regulate the functional expression of KCa channels. To test this hypothesis, the effects of 1 nm TGFβ1 were examined in E9 CG neurons cultured in the presence or absence of 50 μm PD98059. Cells were incubated with this inhibitor beginning 30 min before the addition of trophic factors. In initial experiments, KCa expression was monitored by whole-cell recording 12 hr after the start of TGFβ1 treatment (Fig.6A). As expected, treatment with TGFβ1 evoked a robust increase in macroscopic KCa compared with that in controls cultured in the absence of trophic factor. These increases were completely blocked in CG neurons pretreated with PD98059, and KCaexpression in those cells was indistinguishable from that in controls. PD98059 also blocked the KCa stimulatory effects of iris extracts (data not shown). Treatment with PD98059 by itself had no effect on KCa expression. These results are statistically significant (p < 0.05). We have shown previously that KCa density is maximal in CG neurons by E13 (Dourado and Dryer, 1992; Cameron et al., 1998,1999), and it is worth noting that 50 μmPD98059 had no effect on whole-cell KCa in CG neurons isolated at that stage (data not shown). Therefore, the effects of PD98059 cannot be attributed to direct blockade of KCa channels, a point that can also be ascertained in Figure 7B. Moreover PD98059 had no effect on the expression or gating of voltage-activated Ca2+ currents in E9 CG neurons growing either in the presence or absence of trophic factors (data not shown). PD98059 has been reported to inhibit increases in CaM kinase II associated with the induction of long-term potentiation in CA1 hippocampal neurons (Liu et al., 1999). Therefore, to exclude the possibility that PD98059 actions were caused by inhibition of CaM kinase II, E9 CG neurons were treated with the CaM kinase II inhibitor KN-62 (10 μm). This drug had no effect on the stimulation of KCa evoked by TGFβ1 in CG neurons (Fig. 6B). In summary, these data strongly suggest that signaling via Erk is required for the acute stimulation of the KCa expression evoked by TGFβ1.

Inhibition of Erk signaling blocks the stimulatory effects of TGFβ1 on KCa expression in CG neurons. E9 CG neurons were cultured for 12 hr in medium containing 1 nmTGFβ1 in the presence or absence of protein kinase inhibitors. Cells were incubated with these inhibitors beginning 30 min before the addition of trophic factors. KCa was measured by whole-cell recording at the end of the 12 hr trophic factor treatment.Numbers in parentheses indicate the number of cells recorded for each condition. A, The MEK1 inhibitor PD98059 (50 μm) completely blocked stimulation of KCa expression evoked by a 12 hr treatment with 1 nm TGFβ1. B, The CaM kinase II inhibitor KN-62 (10 μm) had no effect on TGFβ1-stimulated KCa expression. Asterisks denotep < 0.05 compared with control cells, andn.s. denotes no significant difference. Data were analyzed by one-way ANOVA followed by Scheffé's post hoc test.

Acute and sustained effects of TGFβ1 on KCa expression in CG neurons require early Erk activation. E9 CG neurons were cultured for 12 hr in a medium containing no trophic factor (○), 1 nm TGFβ1 (■), or a combination of 1 nm TGFβ1 and 50 μm PD98059 (▪).A, Application of PD98059 (filledhorizontalbar) during and after exposure to TGFβ1 (hatchedhorizontalbar) blocked both the acute and sustained stimulatory effects of TGFβ1 on KCa expression. B, In contrast, application of PD98059 starting after cessation of the 12 hr TGFβ1 treatment had no effect on the sustained stimulation of KCa. Therefore, both the acute and long-lasting stimulatory effects of TGFβ1 require transient activation of Erk, and PD98059 does not cause direct blockade of KCa.
Is Erk activation also required for the sustained stimulation of KCa evoked by TGFβ1? In these experiments, the effects of 1 nm TGFβ1 were examined in neurons cultured in the presence or absence of 50 μm PD98059. An additional group of control neurons was cultured in the absence of trophic factor (Fig. 7). Both the acute and sustained effects of TGFβ1 on KCa expression were blocked by PD98059 when it was present during the 12 hr TGFβ1 treatment (and for the next 3.5 d thereafter). These results are statistically significant (p < 0.05) and suggest that MAP kinase signaling is required for both components of TGFβ1 action (Fig. 7A). However PD98059 had no effect on either acute or sustained stimulation of KCa when it was applied immediately after cessation of the 12 hr TGFβ1 treatment (Fig.7B). This result is consistent with the immunoblot analyses indicating that TGFβ1-induced Erk phosphorylation returns to baseline after 3 hr of continuous treatment (Fig. 5). Therefore, activation of Erk MAP kinase during the first 12 hr of treatment is essential for the induction of KCa stimulation, but ongoing activation of this pathway is not required for the sustained effects of TGFβ1.
DISCUSSION
Previous studies from our laboratory have shown that an avian ortholog of TGFβ1, known as TGFβ4, is essential for the developmental expression of functional plasma membrane KCa channels in chick ciliary neurons (Subramony et al., 1996; Cameron et al., 1998, 1999; Lhuillier and Dryer, 1999). In the present study we have shown that TGFβ1 has a biphasic action comprised of an acute stimulation of KCaexpression mediated by posttranslational processes and a sustained effect that requires the transcription and synthesis of new proteins. Both effects require activation of the MAP kinase Erk. The sustained effect of TGFβ1 suggests that continued exposure to this trophic factor may not be required for the maintenance of functional KCa channels in vivo. In this regard, TGFβ4 transcripts are detectable in the chick eye by E9, remain robust until E12, but subsequently decline to substantially lower levels by the time of hatching (Jakowlew et al., 1992).
The precise mechanism of the TGFβ1-evoked posttranslational stimulation of KCa could entail covalent modification of channels that are already in the plasma membrane, modification of other proteins that interact with KCa channels, recruitment of KCa channels into the plasma membrane, or some combination of these processes. Maximal stimulation of macroscopic KCa does not occur until 5–7 hr after the onset of TGFβ1 treatment, and TGFβ1 needs to be present for 1 hr to observe maximal acute stimulation of KCa. This time course is faster than that of neuromodulation events that entail direct phosphorylation or dephosphorylation of channel molecules. This raises the possibility that translocation of some membrane protein, possibly the SLO α subunits themselves, is required. We have obtained preliminary evidence that supports this theory; specifically we have found that application of microtubule-disrupting agents such as colchicine can block the acute KCa stimulatory effects of TGFβ1 (L. Lhuillier and S. E. Dryer, unpublished data). In addition, Xia et al. (1998) and Schopperle et al. (1998) have identified two proteins, known as Slob and dSLIP-1, that interact with the SLO channels of Drosophila. Coexpression of either of these proteins with SLO channels in heterologous systems prevents translocation of SLO channels to the plasma membrane and thereby results in SLO accumulation in intracellular pools. It is possible that TGFβ1 regulates the plasma membrane targeting of chick ciliary neuron SLO channels by modulating their interaction with molecules that have a similar mode of action. Because of this, it also is worth noting that TGFβ1 does not induce a detectable increase in the expression ofslo transcripts in E9 CG neurons, even though transcription and translation at approximately the time of TGFβ1 treatment are required for the sustained stimulation of KCa. This suggests that the synthesis of other proteins is required for the sustained TGFβ1-evoked stimulation of functional KCa channels, possibly proteins required for normal processing or trafficking of these channels.
A number of previous studies have shown that the duration of Erk activation has profound consequences on subsequent cellular responses (for review, see Widmann et al., 1999). For example, in most pheochromocytoma 12 (PC12) cell lines, transient Erk activation [evoked by epidermal growth factor (EGF)] results in cell proliferation, whereas sustained Erk activation (evoked by NGF) causes these cells to leave the cell cycle and differentiate toward a neuronal phenotype (Traverse et al., 1992; Yaka et al., 1998; York et al., 1998). Moreover EGF does cause neuronal differentiation in certain PC12 cell lines in which EGF evokes abnormally sustained Erk activation (Traverse et al., 1994; Yamada et al., 1996). Here we have observed that TGFβ1 evokes an almost immediate but transient stimulation of Erk phosphorylation in developing CG neurons. Maximal sustained KCa stimulation requires 3 hr of continuous exposure to TGFβ1, even though Erk activation has returned to baseline levels by that time. This suggests that Erk activation is a relatively early event in the cascades that lead to either acute or sustained stimulation of KCa expression. Activated Erk can phosphorylate a host of cytosolic and nuclear target molecules and thereby initiate a variety of intracellular events, including transcriptional activation and modulation of other signal transduction cascades (Widmann et al., 1999). In some cells this can take the form of a negative feedback loop, because activated Erk can directly inhibit molecules, such as ras and MEK1, that are required for its own activation (Whitmarsh and Davis, 1996). Our data on the timing of TGFβ1 receptor stimulation and Erk phosphorylation are consistent with a model in which TGFβ1 activates multiple signaling cascades, including pathways that feed back to limit the duration of Erk activation, along with separate sustained cascades involved in transcriptional control.
Many of the actions of TGFβs and related factors are mediated by a family of intracellular transduction proteins known as SMADs (Baker and Harland, 1997; Heldin et al., 1997; Massague, 1998). SMADs are transcription factors that are translocated to the nucleus as heteromeric complexes after the activation of TGFβ receptors. These molecules have distinct trans-activation and/or protein interaction domains but do not have any other (known) enzymatic activities or sequence motifs that would suggest additional activities (Kawabata and Miyazono, 1999). Therefore, a mechanism by which SMADs could mediate the acute posttranslational effects of TGFβ1 is not obvious from the current literature. However SMADs could well be involved in the sustained effects of TGFβ1, even though Erk activation is required for this effect. For example, it is possible that sustained stimulation of KCa requires the binding of SMADs and Erk-sensitive transcription factors to thecis-acting regulatory elements of a single gene (but probably not the slo gene). In this regard, the formation of complexes between Smad2/4 and an Erk-sensitive basic leucine zipper transcription factor is required for transcriptional activation by TGFβ in vascular endothelial cells (Topper et al., 1998). Alternatively, sustained stimulation of KCa may require transcriptional activation of multiple genes that are regulated independently by SMADs and Erk-sensitive transcription factors. Finally, it is possible that Erk causes direct phosphorylation of SMAD-containing complexes. This occurs in intestinal epithelial cells, in which Erk-dependent phosphorylation of Smad1 appears to be essential for TGFβ1-induced transcriptional activation (Yue et al., 1999).
It is worth noting that other members of the TGFβ superfamily of growth factors produce effects on developing CG neurons. For example, activin induces expression of somatostatin immunoreactivity in CG neurons. During normal in vivo development, secretion of follistatin from the iris ensures that only those CG neurons that innervate the choroid layer are normally exposed to this factor (Darland et al., 1995; Darland and Nishi, 1998), but all CG neurons are competent to respond to activin (Coulombe et al., 1993). In agreement with these observations, type IIA activin receptors are expressed in CG neurons (Kos and Coulombe, 1997). In addition, we have observed that target-derived TGFβ3 causes inhibition of KCastimulation evoked by either TGFβ1 or β-neuregulin-1 and that this effect is physiologically significant for the normal in vivoexpression of these channels in ciliary neurons (Cameron et al., 1999). The inhibitory effect of TGFβ3 is not caused by competitive inhibition with TGFβ1 for a common pool of receptors. These observations suggest that several potentially SMAD-dependent pathways control the differentiation of CG neurons. It is possible that activation of additional pathways, such as the Erk MAP kinase pathway, by one or more of these factors provides an additional measure of specificity in signaling. In other words, the sum total of transduction pathways activated by a given trophic factor may be the key parameter that determines the final response, and therefore different factors that cause activation of a common signaling molecule may evoke very different responses.
In summary, we have observed that TGFβ1 evokes an acute posttranslational stimulation of functional KCachannels in chick ciliary neurons, along with a more sustained effect that requires the transcription and synthesis of new proteins. Both of these effects are associated with transient activation of Erk MAP kinase. Activation of Erk may be a common feature of the actions of TGFβs and related growth factors on neuronal cells.
Footnotes
This work was supported by National Institutes of Health Grant NS-32748. We are grateful to Patrick Callaerts and Claudio Punzo for assistance with immunoblot analysis, Laurence Dryer for reading previous drafts of this manuscript and assistance with RT-PCR, and Jill Cameron for helpful discussions.
Correspondence should be addressed to Dr. Stuart E. Dryer at the above address. E-mail: SDryer{at}UH.EDU.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}