

Abstract
Several studies have shown that activation of α2-adrenergic receptors (α2ARs) leads to mild analgesic effects. Tricyclic antidepressants (TCAs), such as desipramine (DMI), which block norepinephrine transporters (NETs), also produce mild antinociception. The coadministration of either α2AR agonists or TCAs with opiates produces synergistically potentiated antinociception. It has been postulated that the analgesic effects of TCAs are determined by their ability to inhibit norepinephrine reuptake via interactions with the NET. To test this idea, we studied mice lacking a functional NET in spontaneous and morphine-induced antinociceptive paradigms. Morphine (10 mg/kg, s.c.) treatment produced greater analgesia, as assayed in the warm water tail-flick assay, in NET-knock-out (-KO) mice than in wild-type (WT) mice. As anticipated, yohimbine, an inhibitor of α2ARs, blocked this potentiation. Moreover, a warm water swim-stress paradigm, which is known to induce the release of endogenous opioids, produced greater antinociception in NET-KO than in the WT mice. Naloxone, an inhibitor of opioid receptors, blocked the development of the swim-evoked analgesia in both WT and NET-KO mice, confirming the involvement of the endogenous opioid system. In the NET-KO mice, DMI did not further enhance analgesia but was still able to produce inhibitory effects on the locomotor activity of these mutants, suggesting that the effects of this TCA are not exclusively via interactions with the NET. In summary, these results demonstrate in a genetic model that both endogenous and exogenous opiate-mediated analgesia can be enhanced by elimination of the NET, indicating that the interaction of TCAs with NET mediates these effects.
- adrenergic
- monoamine transporters
- opiates
- opioid receptors
- antinociception
- tricyclic antidepressants
- desipramine
In humans, as well as in mice, treatment with tricyclic antidepressants (TCAs) produces mild analgesia (Ward et al., 1979; Tura and Tura, 1990; Gray et al., 1999). TCAs that block norepinephrine transporters (NETs), such as desipramine (DMI), prevent the presynaptic reuptake of norepinephrine (NE) and lead to increased postsynaptic NE levels. The resulting increase of NE and the subsequent activation of α2-adrenergic receptors (α2ARs) in spinal cord neurons are thought to mediate TCA-induced antinociception (Howe and Yaksh, 1982; Howe et al., 1983; Fleetwood-Walker et al., 1985;Yaksh, 1985; Solomon et al., 1989; Takano and Yaksh, 1992). This is consistent with the clinically proven efficacy of α2AR agonists in the treatment of pain associated with cancer in humans (Davis et al., 1991; Eisenach et al., 1995, 1996).
Direct activation of α2ARs has also been shown to potentiate morphine-induced spinal analgesia (Howe et al., 1983;Ossipov et al., 1990a,b; Roerig et al., 1992; Fairbanks and Wilcox, 1999). Interestingly, in addition to the mild analgesia reported after TCA treatment, a potentiation of opioid antinociception also occurs after coadministration of TCAs and morphine in both mice and humans (Kellstein et al., 1984, 1988; Hwang and Wilcox, 1987; Gray et al., 1998, 1999; Reimann et al., 1999). Although the limited availability of pharmacologically selective ligands has prevented delineation of the particular α2AR subtypes involved, many studies have implicated the α2AAR as the target for adrenergic antinociception (Millan et al., 1994; Graham et al., 1997). In addition, studies using transgenic mice expressing a functionally compromised α2AAR support the idea that the α2AAR is specifically involved in modulating spinal nociception (Lakhlani et al., 1997) and that this receptor is principally involved in the synergistic potentiation of morphine analgesia (Stone et al., 1998).
Recently, we generated by homologous recombination a knock-out (KO) mouse lacking the NET (Xu et al., 2000). The NET-KO mice resemble mice treated with antidepressants in a number of tests classically used to assess the actions of antidepressants (Xu et al., 2000). In addition,Xu et al. (2000) showed that although brain tissue storage of NE is decreased, extracellular levels of NE are increased, indicating a potential for chronic activation of α2ARs. These mice provide a genetic model in which we can assess the effect of the loss of NE reuptake on adrenergic-signaling systems without involving the administration of TCAs. Moreover, the contribution of TCAs, such as DMI, to behavioral parameters can be assessed in these mice that genetically lack the proposed target of such drugs.
MATERIALS AND METHODS
Animals. Mice were generated as described previously (Wang et al., 1999; Xu et al., 2000). Wild-type (WT) mice were littermates from the NET-KO heterozygous cross and were used as controls in these assays. Both males and females were tested in each assay. Initially, the results from males and females were analyzed separately, but results were ultimately combined because no significant differences were found between the genders. All experiments were conducted in accordance with the National Institutes of Health guidelines for the care and use of animals and with an approved animal protocol from the Duke University Animal Care and Use Committee.
Materials. Morphine sulfate (Research Biochemicals, Natick, MA), desipramine, yohimbine, naloxone (Sigma, St. Louis, MO), and guanfacine (Tocris Cookson, Inc., Ballwin, MO) were prepared and administered as described in Nociceptive testing.
Nociceptive testing. Morphine sulfate (Research Biochemicals) was prepared in saline and administered subcutaneously. Desipramine and yohimbine or naloxone (Sigma) were prepared in water or saline, respectively, and administered via intraperitoneal injection. Guanfacine (Tocris Cookson, Inc.) was prepared in water and administered subcutaneously (Millan et al., 1994). Morphine-induced antinociception was evaluated by measuring response latencies in the warm water tail-flick and hot plate assays. Response latencies were measured as the amount of time the animal took to respond to the thermal stimuli (Bohn et al., 1999; Gainetdinov et al., 1999b). The warm water (54°C) tail-flick test was performed by the use of a method similar to that described by Stone et al. (1997), and the response was defined as the removal of the tail from the warm water. In the hot plate (56°C) test, the response was manifested as either paw licking or flicking. For both tests, the mice were not permitted to exceed 30 sec of exposure to the thermal source to prevent prolonged painful stimulation. The reported data account for this artificial ceiling as well as for the basal responsiveness of each mouse to the test and are presented as the percent maximum possible effect (% MPE) that is calculated by the following formula: 100% × [(drug response time − basal response time)/(30 sec − basal response time)] = % maximum possible effect (% MPE).
Norepinephrine tissue content. The norepinephrine content in spinal cord tissue was determined by HPLC with electrochemical detection (HPLC-EC) as described previously (Wang et al., 1997). Dissected spinal cords of adult mice were homogenized in 0.1m HClO4 containing 100 ng/ml 3,4-dihydroxybenzylamine as an internal standard. Homogenates were centrifuged for 10 min at 10,000 × g. Supernatants were filtered through 0.22 μm filters and analyzed for levels of NE using HPLC-EC. Monoamines and metabolites were separated on a microbore reverse-phase column (C-18, 5 μm, 1 × 150 mm; Unijet; Bioanalytical Systems) with a mobile phase consisting of 50 mm monobasic sodium phosphate, 0.2 mm octyl sodium sulfate, 0.1 mm EDTA, 10 mm NaCl, and 10% methanol, pH 2.6, at a flow rate of 90 μl/min and were detected by a 3 mm glass carbon electrode (Unijet; Bioanalytical Systems) set at +0.65 V. The volume of the injection was 5 μl.
Binding assays. Radioligand-binding assays were performed on membranes from mouse spinal cords prepared by Polytron homogenization in 50 mm Tris-HCl, pH 7.4, and centrifugation (20,000 × g). Homogenates were prepared by Dounce homogenization in 50 mm Tris-HCl buffer, and concentrations of 50 μg per tube were used in each assay. Saturation binding assays for the α2AAR, α1AR, and μ-opioid receptor (μOR) were performed with the respective antagonists [3H]RX821002 (49 Ci/mmol; Amersham, Piscataway, NJ), [3H]prazosin (77 Ci/mmol; NEN, Boston, MA), and [3H]naloxone (52 Ci/mmol; Amersham). Increasing concentrations of each radioligand were incubated with membranes for 1 hr at 25°C. Nonspecific binding was determined with 10 μm unlabeled naloxone or phentolamine as indicated. Membranes were collected by rapid filtration via a Brandel (Gaithersburg, MD) cell harvester onto GF/B filters and washed three times with cold 10 mm Tris buffer, pH 7.4 (Bohn et al., 1999).
Locomotor testing. Spontaneous open field locomotion and rearing behaviors of littermate wild-type and knock-out mice were measured in an Omnitech digiscan activity monitor (42 cm2). Activity studies were performed between the hours of 10 A.M. and 2 P.M. Locomotor activity was measured at 5 min intervals, and cumulative counts were taken for data analysis. To evaluate the effects of DMI on locomotor behavior, DMI or vehicle was injected intraperitoneally 30 min before testing, and different parameters of locomotor activity were monitored for the following 60 min as described previously (Gainetdinov et al., 1999a).
Swim-induced analgesia. The warm water swim-stress-induced analgesia paradigm was followed as described previously (Mogil et al., 1996; Rubinstein et al., 1996). Thirty minutes after basal nociception was measured, mice were placed in warm water (33°C) and allowed to swim for 3 min. They were then taken from the water and dried in a soft terrycloth towel and allowed to rest for 2 min before they were subjected to the tail-flick assay.
RESULTS
Basal spinal nociceptive threshold is augmented in NET-KO mice
Mice of each genotype were assessed for their responsiveness to pain using two classic antinociceptive tests: the “hot plate” and the warm water tail-flick assay. Although both genotypes respond equally to the hot plate test (Fig.1A), the NET-KO mice display a significantly, although modestly, elevated pain threshold in the tail-flick test (Fig. 1B). These observations suggest that there is a slightly enhanced antinociceptive predisposition in mice that lack the NET.

Basal antinociceptive thresholds in WT and NET-KO mice. Mice were subjected to the indicated test before any drug administrations. The average of their basal responses to the painful stimuli is presented here. A, Hot plate (56°C) paw withdrawal latency. B, Warm water tail-flick (54°C) tail withdrawal latency. Data are presented as the mean ± SEM (***p < 0.01; Student's ttest; n = 40–50 mice).
NE tissue content in spinal cord of WT and NET-KO mice
The increase in basal antinociception revealed in the NET-KO mice may reflect changes in NE levels, because it has been shown that increasing NE (by blocking reuptake or by direct administration) can induce mild antinociception (Howe and Yaksh, 1982; Howe et al., 1983; Fleetwood-Walker et al., 1985; Yaksh, 1985; Solomon et al., 1989; Takano and Yaksh, 1992). Because these mutant mice lack the NE reuptake system, there exists the potential for elevated NE. Previously, we reported that NE levels in brain tissue were decreased by ∼50–70% and that this decrease in NE storage was accompanied by a greater than twofold increase in extracellular NE levels (Xu et al., 2000). Similar results were described previously for the homeostatic control of dopamine and serotonin in dopamine transporter- and serotonin transporter (SERT)-KO mice, respectively (Jones et al., 1998; Murphy et al., 1998). Altogether, these observations support a generalized principle that decreased storage and a corresponding increase in extracellular transmitter levels occur in neuronal systems without active transport (Gainetdinov et al., 1998; Jones et al., 1998; Xu et al., 2000). Using the same approach, we evaluated the NE content in spinal cord tissue from WT and NET-KO mice by HPLC-EC analysis (Fig. 2). Although technical limitations prevent reliable assessment of extracellular NE levels in the spinal cord of mice, the 50% decrease of tissue NE levels detected in NET-KO mice strongly suggests altered NE homeostasis in a manner similar to that seen in brain tissues. Thus, increased extracellular NE levels in the spinal cord can reasonably be inferred in NET-KO mice.

NE content in spinal cord tissue. NE levels were determined by HPLC-EC analysis of spinal cords of WT and NET-KO mice (***p < 0.001; Student's t test;n = 6 of each genotype).
Enhanced effects of morphine analgesia revealed in NET-KO mice
To assess the effects of opiate treatment in mice that genetically lack the NET, increasing doses of morphine were administered, and analgesia was assessed after 20 min by both the hot plate and tail-flick methods. In the hot plate test, each genotype responded to morphine in a dose-dependent manner, revealing no difference between the two genotypes (Fig. 3A). In the tail-flick test, however, morphine analgesia was significantly enhanced in the NET-KO mice (Fig. 3B). This striking difference in the potency of morphine between the genotypes in the tail-flick test may reflect enhanced spinal antinociception. Similar enhancement of spinal antinociception has been described previously in the synergistic actions of morphine and adrenergic agonists (Howe and Yaksh, 1982; Howe et al., 1983; Fleetwood-Walker et al., 1985;Yaksh, 1985; Solomon et al., 1989; Takano and Yaksh, 1992).

Morphine-induced analgesia in the tail-flick assay. WT and NET-KO mice were injected subcutaneously with the indicated dose of morphine, and antinociception was measured after 20 min. A, Hot plate (56°C) response latencies.B, Tail-flick (54°C) response latencies. NET-KO mice experienced significantly greater analgesia than did WT mice at 10 and 20 mg/kg morphine. Data are presented as the mean ± SEM (***p < 0.001; Student's t test;n = 9–19 mice/dose).
Adrenergic and opioid receptor-binding profiles
To ascertain whether receptor-binding profiles were altered in the NET-KO mice, radioligand-binding analyses were preformed. Because previous work implicates the μOR as well as the α2AAR subtypes in the opioidergic–adrenergic-potentiated antinociception, levels of these receptors were determined in preparations of spinal cord membranes. Binding of the α2AAR-preferring antagonist [3H]RX821002 did not vary between the genotypes, nor did the binding of [3H]naloxone, indicating that the α2AAR and μOR levels in the spinal cord of WT and NET-KO mice are not significantly different (Table1). Therefore, changes in morphine sensitivity in the mutant mice are probably not caused by increases in receptor numbers. In addition, α1AR-binding parameters were also measured in spinal cord membrane preparations using [3H]prazosin. Although theKD for the ligand showed no difference, a >50% decrease in α1AR number was observed similar to that seen in hippocampal membranes prepared from the NET-KO mice (Xu et al., 2000). This downregulation of α1AR is consistent with increased levels of NE in these mice.
Saturation binding of α2AAR and μOR in WT and NET-KO mouse spinal cord
Enhanced morphine analgesia in NET-KO mice is caused by activation of α2ARs
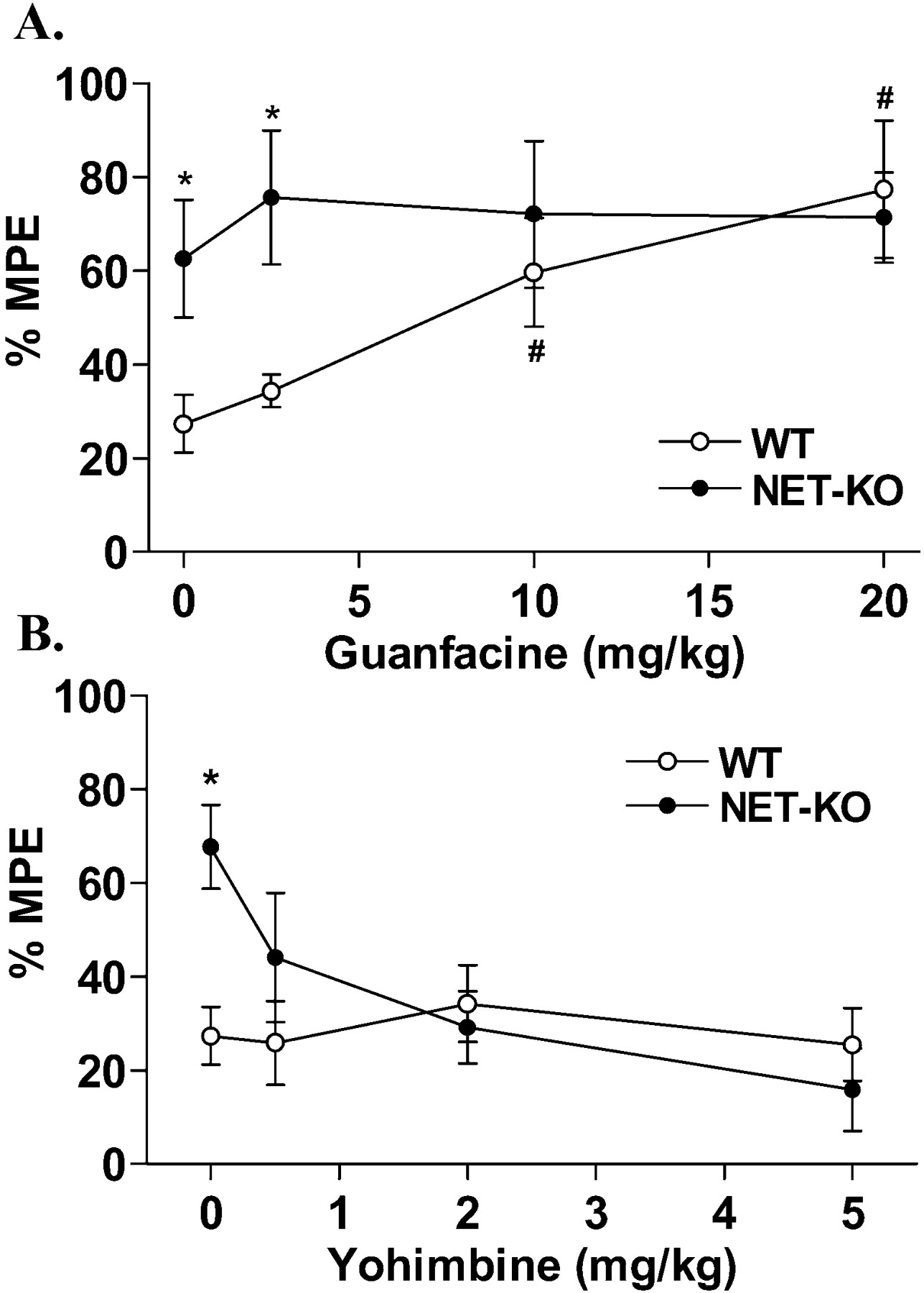
The blockade (via TCAs) or loss (via genetic deletion) of the NET may lead to increased NE in the spinal cord, resulting in increased activation of the α2AR. Because the α2AAR subtype has been implicated in the opioid–adrenergic synergy of spinal antinociception (Millan et al., 1994; Graham et al., 1997; Stone et al., 1998), guanfacine, a preferential agonist at the α2AAR (Millan et al., 1994), was administered before morphine in both NET-KO and their WT controls. Guanfacine enhanced morphine analgesia in WT mice in a dose-dependent manner, resulting in the same degree of antinociception as that observed for morphine alone in NET-KO mice (Fig.4A). Interestingly, guanfacine had no significant further effect on morphine analgesia when given to the NET-KO mice (Fig. 4A). Guanfacine, when give alone, produced mild analgesia (11.8 ± 1% MPE) in WT mice and had a similar effect on NET-KO mice (10.6 ± 3.1% MPE). To test further the involvement of α2AR signaling in the apparent enhancement of morphine analgesia, yohimbine, an α2AR antagonist, was used (Fig.4B). Whereas yohimbine had no effect on analgesia in WT mice after morphine treatment, in the NET-KO mice it reversed the enhanced morphine response to the same level observed in WT mice after morphine treatment alone (Fig. 4B). Taken together, these results suggest that the enhanced effects of morphine in the NET-KO mice are caused by activation of α2ARs and that this phenomenon can be recapitulated in WT mice after coadministration of morphine and an α2AAR-preferring agonist.

Role of α2AR in the tail-flick assay. A, The α2AAR agonist guanfacine (2.5, 10, and 20 mg/kg, s.c.), when given 10 min before morphine, enhances morphine (10 mg/kg, s.c.; 20 min) analgesia in WT mice to the same extent that morphine alone enhances analgesia in the NET-KO mice. Guanfacine (10 mg/kg, s.c.) alone induced mild analgesia in WT mice (11.8 ± 1.0% MPE) and NET-KO mice (10.6 ± 3.1% MPE).B, The α2AR antagonist yohimbine (0.5, 2, and 5 mg/kg, s.c.) reverses potentiated morphine analgesia in NET-KO mice when given 10 min before morphine (10 mg/kg, s.c.; 20 min). Data are presented as the mean ± SEM [*p < 0.01, vs WT; p < 0.01, vs WT (0, 2.5 mg/kg guanfacine); Student's t test; n = 5–9 mice/dose].
Desipramine enhances morphine analgesia in WT but not NET-KO mice
The degree of enhanced morphine analgesia in the NET-KO mice is similar to that seen in previous studies in which normal mice were coadministered morphine and DMI or morphine and α2AR agonists (Kellstein et al., 1984;Fairbanks and Wilcox, 1999; Reimann et al., 1999), suggesting that the loss of NET results in greater tonic activation of the α2AR system in the NET-KO mice. After a 2 hr pretreatment with DMI (2.5, 10, or 20 mg/kg, i.p.), we found that morphine analgesia was enhanced in WT littermates to the same extent as that seen in NET-KO mice receiving only morphine (Fig.5). Interestingly, DMI at this dose had no apparent effect on morphine analgesia in NET-KO mice, suggesting that the antinociceptive contributions of this TCA are primarily caused by blockade of the NET.

Effects of DMI on antinociception in the tail-flick assay. WT and NET-KO mice were first injected with DMI (2.5, 10, and 20 mg/kg, i.p.); 2 hr later, mice were treated with morphine (10 mg/kg, s.c.), and tail-flick latencies were measured after 20 min. Data are presented as the mean ± SEM [*p < 0.01, vs WT; p < 0.01, vs WT (0, 2.5 mg/kg desipramine); Student's t test; n = 8–18 mice/dose].
Desipramine retains some effects in the NET-KO mice
In rodents, acute administration of DMI produces an inhibitory effect on locomotor activity (Tucker and File, 1986). To assess whether DMI elicits this inhibitory effect exclusively via interaction with the NET, we monitored WT and NET-KO mice for horizontal and vertical locomotor activity after acute drug administration. As we described previously, the NET-KO mice demonstrate markedly lower basal levels of both horizontal and vertical activities when exposed to a new environment [Fig. 6, vehicle (saline) treated] (Xu et al., 2000). Pretreatment with DMI (10 mg/kg, i.p.) induces significant inhibitory effects in WT mice (Fig. 6). Despite lower levels of spontaneous activity, the NET-KO mice retained some degree of sensitivity to the inhibitory effects of DMI on locomotor behavior (two-way ANOVA, p < 0.01). Thus, desipramine still elicits some behavioral effects in mice lacking the NET.

Effect of DMI on locomotor activity. The effect of DMI (10 mg/kg, i.p.) on the horizontal (top) and vertical (bottom) activity of WT (left) and NET-KO (right) mice is shown. DMI significantly depresses horizontal and vertical activities in both genotypes (p < 0.01 vs vehicle-treated controls; two-way ANOVA). Numbers of animal per group are depicted on the figure.
Endogenous opioid effects are enhanced in NET-KO mice
Because the effects of an exogenous opioid such as morphine could be so dramatically enhanced in mice lacking the NET, we evaluated the effects of endogenous opioids in these mice. A 3 min swim in warm (30–33°C) water has been shown previously to induce mild endogenous opioid-mediated analgesia in mice (Mogil et al., 1996; Rubinstein et al., 1996). After this swim-stress paradigm, low levels of antinociception could be detected by the warm water tail-flick assay in the WT mice; however, antinociception in NET-KO mice was dramatically induced (Fig. 7). To demonstrate that this induction of antinociception was mediated via the activation of opioidergic systems, naloxone, the OR antagonist, was used to block completely the analgesic effects of the swim stress (Fig. 7). Naloxone alone had no significant effect on the tail withdrawal latency of either genotype (data not shown).

Warm water swim-stress-induced antinociception. Antinociception was assessed by the tail-flick assay after the warm water swim. The endogenous opioid-mediated analgesia could be reversed by pretreating mice with the opioid receptor antagonist naloxone (2 mg/kg, s.c.) 20 min before the swim. Data are the mean ± SEM (*p < 0.1, vs WT basal; **p < 0.001, vs NET-KO basal; #p < 0.001, vs NET-KO after the swim; one-way ANOVA followed by a Tukey post test).
DISCUSSION
These studies demonstrate that the NET-KO mice experience enhanced opioid analgesia as compared with WT mice as well as a modest increase in basal nociceptive thresholds. We have provided evidence that the loss of the NET results in increased extracellular NE (Xu et al., 2000) (Fig. 2) and that this increase in the extracellular concentrations of the neurotransmitter contributes to the increasing pain thresholds by acting at the α2AAR. The increase in basal nociceptive thresholds seen in the NET-KO mice in the tail-flick but not the hot plate test suggests that these effects may predominantly involve spinal noradrenergic systems, a suggestion that is in agreement with previous observations (Hylden and Wilcox, 1983; Wilcox et al., 1987; Ossipov et al., 1990a,b). Furthermore, α2AARs are robustly present in spinal cord neurons (Stone et al., 1998), and agonists at these receptors are known to prolong tail-flick latencies (Reddy et al., 1980;Yasuoka and Yaksh, 1983; Milne et al., 1985; Solomon et al., 1989; Lakhlani et al., 1997).
Although basal antinociception is only modestly enhanced in the NET-KO mice, greater differences between the genotypes can be clearly seen after administration of morphine. Many strains of mice, as well as the WT and the parental lines used to generate this transgenic line, reach moderate analgesia (∼30–50% MPE as measured in this assay) 20–30 min after a dose of morphine (10 mg/kg, s.c.; data not shown). The NET-KO mice display significantly enhanced morphine analgesia at this dose as compared with the WT controls (Fig. 3).
Previous studies have demonstrated consistently that morphine analgesia is potentiated after coadministration of α2AR agonists. Both μ and δ opioid receptors have been implicated as targets mediating the effects of morphine in the antinociception potentiated by α2AR agonists (Ossipov et al., 1990b; Roerig et al., 1992; Stone et al., 1997; Grabow et al., 1999). Moreover, because of a lack of pharmacologically selective agents, it had remained uncertain which subtype of α2ARs is targeted by agonists, such as clonidine and norepinephrine, in modulating spinal transmission of pain, although many studies now suggest that the α2AAR plays a prominent role (Millan et al., 1994; Graham et al., 1997; Lakhlani et al., 1997). This observation is recapitulated in the WT littermates of the NET-KO mice (Fig. 4A) using the selective α2AAR agonist guanfacine (Millan, 1992). Interestingly, the WT mice treated with both guanfacine and morphine experience the same extent of analgesia as do the NET-KO mice treated only with morphine. This suggests that in the NET-KO mice, the α2AAR component is already stimulated and that additional agonist treatment has no effect. This supposition was confirmed by using the α2AR antagonist yohimbine (Howe et al., 1983; Takano and Yaksh, 1992), which eliminates the enhanced effects of morphine analgesia, causing the NET-KO mice to respond to morphine to the same extent as do WT mice (Fig.4B). Interestingly, these data suggest that the tonically elevated NE does not seem to lead to functional desensitization of the adrenergic response. In addition, although we see a downregulation of α1AR, the number of α2AARs does not change in the NET-KO mice. This is in agreement with previous observations that the α2AAR resists downregulation in the presence of chronic agonist (Daunt et al., 1997; Saunders and Limbird, 1999).
Additionally, we were able to recreate the effect of potentiated morphine analgesia in the presence of elevated levels of α2AR agonists or TCAs in the absence of any drug treatment. By inducing opioid release by the swim-stress-induced analgesia paradigm, the nociceptive threshold was raised in the NET-KO mice to the same extent that it was raised in WT mice treated with morphine alone. This elevation in nociceptive thresholds in the NET-KO mice indicates that endogenous opioid-mediated analgesia can be significantly potentiated by elevation of NE in the mouse.
In studies in both humans and mice, TCAs have proven to be effective in inducing mild analgesia. However, such transporter blockers appear to be nonselective in vivo, making it hard to discriminate between their actions at other monoamine uptake sites (for review, seeChen and Reith, 1997). Additionally, there are reports in the literature that suggest that DMI may bind directly to opioid receptors, thereby contributing to their analgesic properties (Biegon and Samuel, 1980). However, it is generally accepted that the DMI provides moderate analgesia because of the blockade of NE reuptake and the consequent activation of the α2AR. The generation of transgenic mice lacking the NET has made it possible to assess directly the contribution of blocking the specific transporter to the induction of antinociception.
The results of the present study directly demonstrate that analgesic effects of DMI can be attributed to interactions with the NET. However this is not the case for the inhibitory effect of DMI on behavior. Previously, we have shown that NET-KO mice are hypoactive in a new environment, and this hypoactivity was correlated with lower levels of extracellular dopamine (DA) in the striatum (Xu et al., 2000). Despite this fact, DMI was able to induce further inhibitory effects on the activity of these mice. Thus it is likely that DMI may have two independent effects on locomotor activity; one may be related to secondary alterations in the midbrain DA system, and the other may be via interactions of the drug with targets other than the NET. The most likely candidate for this additional effect of DMI is the SERT, which is consistent with the established inhibitory effect of serotonin on novelty-provoked locomotor activity (Lucki, 1998; Gainetdinov et al., 1999a).
In conclusion, these data demonstrate that genetic elimination of the NET results in enhanced antinociception and that this is attributable to increased activation of the target of NE, the α2AR. Moreover, these data illustrate that the NET is required for the analgesic but not the locomotor inhibitory properties mediated by DMI in mice. Finally, these observations support the idea that an elevation of NE by blockade of the transporter results in potentiated morphine-induced spinal antinociception.
Footnotes
This work was supported in part by National Institutes of Health Grants NS-19576 and MH-40159 and a neuroscience unrestricted award from Bristol Myers Squibb to M.G.C. M.G.C. is an Investigator of the Howard Hughes Medical Institute, L.M.B is supported by National Institutes of Health Grant DA-06023, and R.R.G. is a visiting researcher from the Institute of Pharmacology, Russian Academy of Medical Sciences (Baltiyskaya 8, 125315 Moscow, Russia). We thank J. Holt and S. Suter for excellent technical assistance.
Correspondence should be addressed to Dr. Marc G. Caron, Howard Hughes Medical Institute, Departments of Cell Biology and Medicine, Duke University Medical Center, Box 3287, Durham, NC 27710. E-mail:caron002{at}mc.duke.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}