

Abstract
The dopamine D4 receptor (D4R) is predominantly expressed in the frontal cortex (FC), a brain region that receives dense input from midbrain dopamine (DA) neurons and is associated with cognitive and emotional processes. However, the physiological significance of this dopamine receptor subtype has been difficult to explore because of the slow development of D4R agonists and antagonists the selectivity and efficacy of which have been rigorously demonstrated in vivo. We have attempted to overcome this limitation by taking a multidimensional approach to the characterization of mice completely deficient in this receptor subtype. Electrophysiological current and voltage-clamp recordings were performed in cortical pyramidal neurons from wild-type and D4R-deficient mice. The frequency of spontaneous synaptic activity and the frequency and duration of paroxysmal discharges induced by epileptogenic agents were increased in mutant mice. Enhanced synaptic activity was also observed in brain slices of wild-type mice incubated in the presence of the selective D4R antagonist PNU-101387G. Consistent with greater electrophysiological activity, nerve terminal glutamate density associated with asymmetrical synaptic contacts within layer VI of the motor cortex was reduced in mutant neurons. Taken together, these results suggest that the D4R can function as an inhibitory modulator of glutamate activity in the FC.
Dopamine (DA) modulates multiple neuronal circuits, including those responsible for the initiation of locomotor activity and the integration of goal-oriented behaviors, through the stimulation of five distinct G-protein-coupled receptors (Civelli et al., 1993). These receptors have been classified as either dopamine D1-like (D1R and D5R) or D2-like (D2R, D3R, and D4R) according to similarities in their pharmacological profiles, coupling to second messenger systems, nucleotide sequence, and genomic organization.
One of the major ascending DAergic pathways originates in the ventral tegmental area and innervates the frontal cortex (FC) in which DA is thought to play an important role with respect to the integration of diverse neuronal signals, especially those produced in response to novel environmental stimuli (Tassin et al., 1980; Roth et al., 1988;Bardo et al., 1996). It is also in the FC, where the modulation of neuronal excitability by DA is thought to contribute to working memory and the establishment of memory fields (Williams and Goldman-Rakic, 1995). Although there is considerable evidence suggesting that the D1R plays an important role in mediating the effects of DA on cognition (Lidow et al., 1998), little is known about the functional consequences of D2-like receptor-mediated signaling in the FC.
Among the members of the D2-like receptor family, the D4R has been of interest since its discovery, in part because it is densely expressed in the FC (Meador-Woodruff et al., 1996; Mrzljak et al., 1996; Ariano et al., 1997) and has high affinity for various atypical antipsychotics (Van Tol et al., 1991; Roth et al., 1995). Consequently, it has been suggested that the D4R may play an important role in cognitive functions associated with the cortex and as such may be involved in the etiology or pathophysiology of diseases such as schizophrenia that are thought to involve altered cortical function. This latter notion is supported by the demonstration that striatal D4R binding is six times higher in schizophrenic patients than in normal individuals (Seeman et al., 1993). At the molecular–genetic level, some alleles of the highly polymorphic human D4R gene (DRD4) have been associated with human diseases, social habits, and personality traits, including attention deficit hyperactivity disorder (ADHD) (Barkley, 1998; Swanson et al., 1998), opiate and alcohol use (Muramatsu et al., 1996; Geijer et al., 1997; Kotler et al., 1997), and novelty-seeking behavior (Benjamin et al., 1996; Ebstein et al., 1996).
In vitro electrophysiological studies have shown that D4Rs can couple to hyperpolarizing, inwardly rectifying potassium channels (Werner et al., 1996). Therefore, the activation of the D4R could play a fundamental role in the overall inhibitory modulation of FC neuronal activity (Starr, 1996; Gulledge and Jaffe, 1998) and may explain, in part, how increased cortical excitability can be elicited by atypical neuroleptics (D2-like receptor blockers) (Menza et al., 1993; Lidow et al., 1998).
Given the demonstration that D4Rs are expressed in the FC of primates and rats (Meador-Woodruff et al., 1996; Mrzljak et al., 1996; Ariano et al., 1997), it was of interest to determine whether the D4R is involved in the modulation of cortical excitability. To this end we have examined a strain of genetically engineered mutant mice that lack D4Rs as well as a novel D4R selective antagonist, PNU-101387G (Merchant et al., 1996). Previously, we reported that these mutant mice are supersensitive to the psychomotor stimulant effects of ethanol, methamphetamine, and cocaine (Rubinstein et al., 1997) and that DA synthesis and turnover are significantly increased in the striatum of these mutants. More recently it has been shown that mice lacking D4Rs exhibit reduced exploration of novel stimuli (Dulawa et al., 1999). Herein we present electrophysiological, pharmacological, immunohistochemical, and ultrastructural evidence that the D4R-deficient mice display cortical hyperexcitability, consistent with the interpretation that in wild-type mice the activation of D4Rs may have an inhibitory influence on pyramidal neurons of the FC.
MATERIALS AND METHODS
D4R-deficient mice. All animals tested were originally derived from F1 Drd4+/− mice (C57BL/6J,129/Ola) back-crossed for five successive generations to produce N5-incipient congenic C57BL/6J mice. Details regarding the targeted mutagenesis and generation of these mice have been described elsewhere (Rubinstein et al., 1997). Heterozygous N5 mice were bred to obtain wild-type and homozygous mutant mice. Animals were housed in same-sex groups of five or six with ad libitum access to food and water. In all experiments, D4R-deficient mice and their wild-type controls were age and sex matched. The vivarium was maintained at 20–22°C on a 12 hr light/dark cycle (lights on at 7:00 A.M.). All experimental procedures were performed in accordance with the National Institutes of Health guidelines for animal research by individuals that were blind to the animal's genotype.
Immunohistochemistry. A polyclonal, anti-peptide antiserum was produced in rabbits against a unique 20-residue sequence (MGNSSATGDGGLLAGAGPEC) that is part of the N terminus of the rat D4R and is predicted to be extracellular. This peptide was chosen with the aid of IBI-Pustell MacVector DNA-Protein sequence analysis software that identifies regions of potential antigenicity. The peptide was synthesized by Peninsula Laboratories (Belmont, CA), purified by HPLC, and conjugated to keyhole limpet hemocyanin (Sigma, St. Louis, MO) via the terminal cysteine residue using the cross-linking reagent, m-maleimido-benzoyl-N-hydroxysuccinimide (Sigma) as described previously (Ariano et al., 1997). The specificity of the antiserum was assessed using transfected Chinese hamster ovary cells that stably express full-length, functionally active rat D4Rs (Ariano et al., 1997). Immunofluorescent staining in mouse brain sections was performed as described previously (Ariano et al., 1997). The primary antiserum was diluted 1:1000 in PBS and applied to 10-μm-thick, frozen-fixed sections of mouse brain and incubated overnight in a moist environment. Unbound anti-D4R antibodies were removed by rinsing in PBS and then applying fluorescently conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Labs, West Grove, PA; Molecular Probes, Eugene, OR). After a further 1 hr incubation in a moist environment at 4°C, the sections were rinsed again in PBS and examined using routine epifluorescence. Controls for the immunofluorescence staining reaction included omission of the primary antisera, substitution of preimmune sera, or adsorption challenge of the primary antisera with peptide before incubation. Examination of the tissues under conditions that produce epifluorescence was performed by a trained investigator (M.A.A.) using a Zeiss Photomicroscope 3 that was equipped with a 100 W Hg burner as the ultraviolet light source. Data were captured using HP-5 plus black and white film (Ilford, Ciba-Geigy, Paramus, NJ) and developed using D-19 (Kodak, Rochester, NY) to increase the contrast of the film negatives. All scoring was done without previous knowledge of the genotype of an individual mouse.
Electrophysiological recordings. Mice were killed for rapid brain extraction, the brains were blocked, and rostral cortical tissue was sectioned coronally (∼350 μm thick). Throughout the sectioning process, slices were bathed in an oxygenated (95% O2, 5% CO2), low-Ca2+, artificial CSF (ACSF) composed of (in mm): NaCl 130, KCl 3, NaH2PO4 1.25, MgCl2 5, NaHCO3 26, CaCl2 1, glucose 10. Slices were incubated in lactated bicarbonate ACSF (as above except CaCl22, MgCl2 2, lactate 4) at 33°C for at least 1 hr before being placed in a Haas-type recording chamber. In the recording chamber, slices were superfused (1.4 ml/min) with standard ACSF of the following concentrations (in mm): NaCl 124, KCl 5, NaH2PO4 1.25, MgSO4 2, NaHCO3 26, CaCl2 2, glucose 10, for a minimum of 30 min before electrophysiological recording began. Glass micropipettes filled with 3 m KAc (60–150 MΩ) were used for recording. Signals were amplified (Axoclamp-2A; Axon Instruments, Foster City, CA), displayed on an oscilloscope, and digitized for subsequent computer analysis (pCLAMP6.0.1; Axon Instruments). After a neuron was impaled, a baseline recording (20–30 min) was obtained to ensure stability of membrane properties. Only data obtained from neurons with resting membrane potentials of at least −60 mV and action potentials exceeding 55 mV (measured from the start of the rapid rising phase to the peak of the depolarization) were used. All data were obtained from well impaled neurons in which recordings were made for 1–2 hr. Membrane properties (membrane potential and input resistance) and action potential amplitude were measured during the baseline recording period. Current–voltage relationships were obtained by injection of depolarizing and hyperpolarizing pulses, and the input resistance of the cell was determined from hyperpolarizing pulses in the linear portion of the current–voltage plots.
Paroxysmal activity in cortical slices was produced by bath applying the convulsant agents 4-aminopyridine (4-AP; 100 μm) and bicuculline methiodide. In cortical slices, bath application of 4-AP induces paroxysmal discharges (Dube et al., 1988). In an effort to eliminate the confounding effects of inhibitory amino acid release, bicuculline, also a convulsant agent, was applied in conjunction with the 4-AP. All quantitative measurements were taken 20–40 min after drug application. Cell identification was achieved by filling electrodes with 2% biocytin (Sigma) and injecting it with hyperpolarizing current pulses (0.5 Hz, 0.3–0.6 nA). Slices were then fixed overnight in 4% paraformaldehyde.
For studies involving voltage clamp, cortical neurons were visualized in mouse brain slices using infrared video microscopy and differential interference contrast optics (IR-DIC). Excitatory spontaneous synaptic activity was measured from visually identified cortical neurons using the whole-cell patch-clamp technique. Briefly, the membrane potential was held at −70 mV, and the membrane current was recorded continuously for a period of 3–6 min in each of two conditions per cell (19 of 22 recordings were at least 5 min in duration). The membrane current was filtered at 1 kHz and digitized at 200 μsec using Fetchex (Axon Instruments). Spontaneous synaptic activity was first recorded in a control bath solution composed of the following (in mm): NaHCO3 26, NaH2PO4 1.25, NaCl 130, KCl 3, MgCl2 2, CaCl2 2, glucose 10, and bicuculline (10 μm). The potassium channel blocker 4-AP (50 μm) was then added to the bath solution for ∼5 min before a second measurement of spontaneous synaptic activity from the same cell but in the presence of 4-AP. Spontaneous synaptic events were detected off-line using the Mini Analysis Program (Jaejin Software, Leonia, NJ); the minimum amplitude required for the detection of an event was set to 5 pA. The internal solution in all experiments was composed of the following (in mm): Cs-methanesulfonate 130, TEA-Cl 5, BaCl2 5, NaCl 4, MgCl2 1, CaCl2 0.5, EGTA 5, ATP 3, GTP 0.3, HEPES 10; biocytin, 0.2%; pH 7.2.
Convulsant effects of bicuculline. To establish a dose–response curve for bicuculline-induced seizures, (+)-bicuculline (Sigma), prepared fresh daily in 0.2N HCl and neutralized with 0.4N NaOH to pH 5.0, was administered intraperitoneally to mice of both genotypes. After bicuculline administration, mice were placed individually in a transparent acrylic box (30 × 21 × 16 cm) where they were monitored by a trained observer blind to the genotype of the animal. All sessions were also videotaped to permit analysis at a later time. Latencies to clonus, “wild” running, and tonus were recorded. Each observation period was 15 min in duration.
Electron microscopy. Mice were anesthetized (10 ml/kg of 5% ketamine, 1% acepromazine, and 2% xylazine) and then perfused transcardially with a fixative containing 2.5% glutaraldehyde, 0.5% paraformaldehyde, and 0.1% picric acid in 0.1 mHEPES, pH 7.3. Whole brains were immediately harvested and soaked in cold fixative at 4°C overnight. The following day, 200 μm coronal sections containing the FC were cut with a Vibratome (Ted Pella, Redding, CA) and washed in buffer. The tissue was then incubated in a solution of 1% osmium tetroxide, 1.5% potassium ferricyanide for 30 min at room temperature, stained en bloc with aqueous 0.5% uranyl acetate, dehydrated in alcohol, cleared in propylene oxide, and embedded in Epon/Spurrs at 60°C overnight. Ultrathin sections of tissue were cut on an ultramicrotome (RMC, Phoenix, AZ) and processed for post-embedding glutamate immunocytochemistry. The glutamate antibody (Arnel, NY), the specificity of which has been described previously (Phend et al., 1992; Meshul et al., 1999), was diluted 1:250,000 before use. Sections were viewed and photographed using a JEOL 1200 EX TEMSCAN electron microscope. Photographs were taken randomly throughout the neuropil of asymmetrical nerve terminals, most of which synapse on dendritic spines within layer VI of the FC. Glutamatergic nerve terminals were identified by the accumulation of gold particles within the presynaptic ending and a prominent postsynaptic density. Only those 10 nm gold particles within presynaptic nerve terminals and associated with the vesicular pool were counted by an experienced investigator (L.B.K.), without previous knowledge of the genotype of a given animal. The area of the nerve terminal was determined using Image Pro Plus software (Media Cybernetics, Silver Spring, MD). The mean density of gold particles (number of particles per square micrometers) and the area of the presynaptic terminals (square micrometers) were determined for each animal. The data were analyzed using a one-way ANOVA with the significance level set at p < 0.05.
Electrochemical detection ofl-3,4-dihydroxyphenylalanine, DA, and DOPAC. HPLC-coupled electrochemical detection ofl-3,4-dihydroxyphenylalanine (l-DOPA), DA, and DOPAC was performed using a Varian 5000 liquid chromatograph coupled to an electrochemical detector (BAS LC-4C) by an investigator who was blind with respect to the genotype of the mouse under study. l-DOPA accumulation in the FC was measured after blocking its conversion to DA using the DOPA decarboxylase inhibitor m-hydroxybenzylhydrazine (NSD-1015; Aldrich, Milwaukee, WI). To determine the slope of DOPA accumulation, mice from both sexes and genotypes received two intraperitoneal injections separated by 15 min after three different schemes. One group of animals received saline in both injections, a second group of mice received saline first and NSD-1015 (100 mg/kg) 15 min later, and a third group of mice received first NSD-1015 (100 mg/kg) and then 15 min later a second injection of NSD-1015 (50 mg/kg). The number of mice of each group was 6. Mice were killed by cervical dislocation 15 min after the second injection; the FC was immediately removed, weighed, frozen in dry ice, and kept at −70°C until electrochemical determination of l-DOPA levels. In the group of mice receiving only saline, DOPAC and DA were also determined using the same procedure to calculate the DOPAC/DA ratio as an index of DA turnover.
RESULTS
D4R immunoreactivity is expressed in neurons of the mouse FC
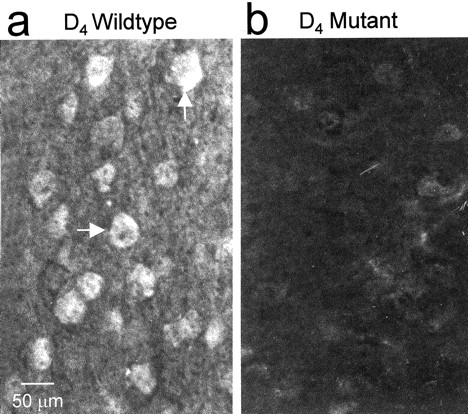
In both the monkey (Mrzljak et al., 1996) and the rat (Ariano et al., 1997), the majority of D4R immunoreactivity (D4R-ir) is found in the FC. Using a polyclonal antiserum, we determined that the neuroanatomical distribution of D4R-ir in the mouse brain parallels that reported previously for the rat (Ariano et al., 1997), with particularly dense staining throughout the prefrontal, frontal, and sensorimotor cortices (Fig. 1a). No labeling was detected when either the primary antibody was omitted (data not shown) or the brain sections were taken from D4R-deficient mice (Fig. 1b). Within the cortical regions, D4R-ir was found associated with large-diameter pyramidal neurons in laminas III and V, as well as smaller, more rounded cells with morphology consistent with that of interneurons but not glia. At the subcellular level, D4R-ir appeared along the proximal portions of apical and basilar dendrites of pyramidal cells. In addition, relatively dense staining was observed in the neuropil of the cortex and was probably caused by receptor expression on dendrites as well as local axon collaterals in the cortex.

D4R immunoreactivity is detectable in frontal cortical pyramidal neurons in wild-type, but not mutant, mice.a, The arrows point to two cortical pyramidal neurons in a coronal section of wild-type mouse brain that reacted with the polyclonal antiserum directed against the D4R. b, Brain section prepared from a D4R-deficient mouse and incubated with the D4R antiserum.
Cortical glutamatergic neurons from D4R-deficient mice display increased excitability
Because D4Rs have been localized to glutamatergic pyramidal neurons and GABA-containing interneurons of the FC, and D2-like receptor agonists are known to act as anticonvulsant agents (Starr, 1996), we investigated the contribution of D4Rs to the modulation of pyramidal neuron excitability.
Recordings were made from a total of 20 pyramidal neurons (10 obtained from eight different wild-type mice and 10 from nine different D4R-deficient mice) located in layers III–V of the FC. The average membrane resting potentials (−74 ± 0.7 mV, wild type vs −75.8 ± 1.3 mV, mutant) and action potential amplitudes (76.5 ± 1.8 mV, wild-type vs 78.0 ± 1.8 mV, mutant) were similar. In contrast, the input resistance was significantly higher in the wild-type compared with the mutant neurons (37.7 ± 2.4 MΩ, wild-type vs 31.4 ± 1.6 MΩ, mutant; t = 2.218; df = 19; p = 0.032).
After baseline recordings were made, slices were exposed to 100 μm 4-AP, a convulsant agent that can induce paroxysmal discharges in cortical slices (Mattia et al., 1993). The GABAA receptor antagonist bicuculline (10 μm) was applied in conjunction with 4-AP to eliminate the confounding effects of inhibitory amino acid release. The application of both drugs consistently evoked paroxysmal discharges in pyramidal neurons from both the mutant and wild-type mice (Fig.2a). In most cells, paroxysmal discharges occurred after 7 min exposure to 4-AP and bicuculline and consisted of an abrupt membrane depolarization and a burst of action potentials, sometimes followed by an afterhyperpolarization. The frequency of paroxysmal discharges increased over time and reached a plateau ∼15 min after application of 4-AP and bicuculline. All subsequent measurements were made after this plateau was reached.

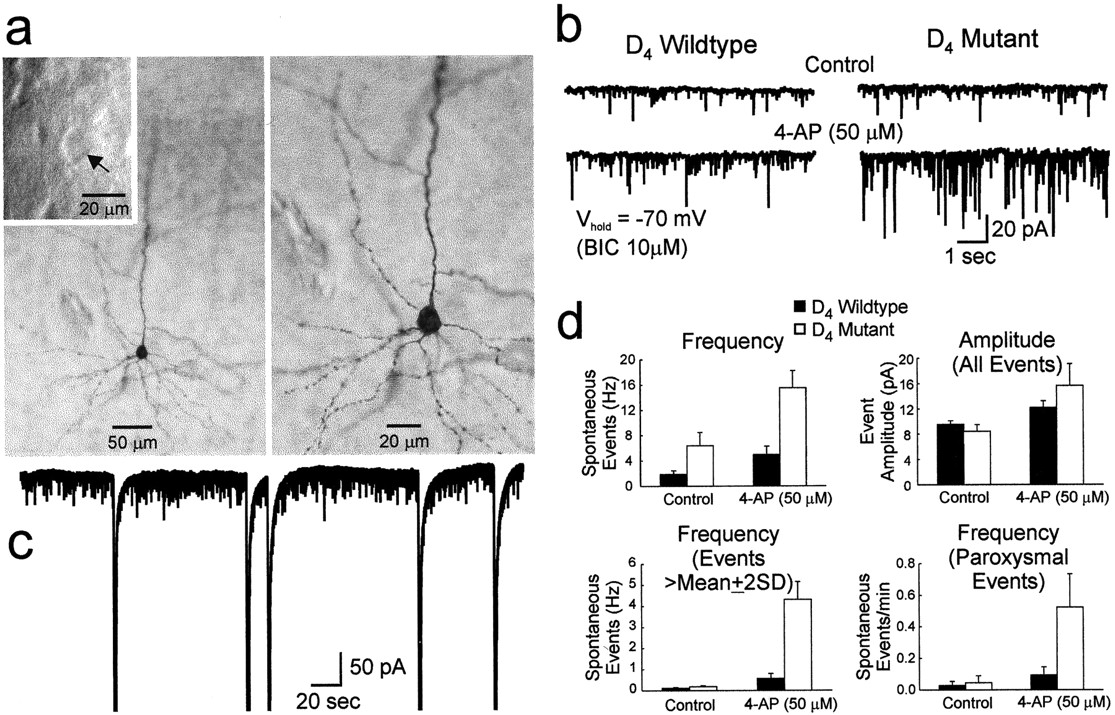
Intracellular recordings reveal increased excitability of cortical pyramidal neurons in D4R-deficient mice. a, Effects of 4-AP (100 μm) and bicuculline (10 μm) on cortical pyramidal neurons from wild-type (left four traces) and mutant (right four traces) mice. b, Representative single, faster-speed traces of paroxysmal depolarizations from the neurons shown in a. Theleft trace is from a wild-type neuron, whereas thetrace on the right is from a mutant neuron. c, The effect of the non-NMDA receptor antagonist CNQX on paroxysmal discharges evoked by 4-AP and bicuculline in a neuron from a mutant mouse is shown. The hyperpolarizing pulses (downward deflections) used to measure input resistance are shown in the trace on the right. d, Biocytin-filled cortical pyramidal cells from wild-type and mutant mice. The top panel displays three different magnifications of a wild-type pyramidal neuron. The bottom three panels show similar magnifications of a pyramidal neuron taken from a mutant mouse. The calibrations refer to both thetop and bottom panels.
Not only did this treatment significantly increase the frequency of spontaneous discharges in the mutant neurons (0.066 ± 0.009/sec) compared with wild-type neurons (0.039 ± 0.009/sec;t = −2.182; df = 19; p = 0.042) (Fig. 2a), but the average duration of their bursting was consistently longer (2247 ± 499 msec) (Fig. 2b,right trace) than the wild-type response (982 ± 109 msec; t = −2.245; df = 18; p = 0.038) (Fig. 2b, left trace).
Evidence for the participation of glutamate in the production of these paroxysmal discharges was provided by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μm), a non-NMDA receptor antagonist that abolished all spontaneous activity when added to the bath (Fig. 2c). At the conclusion of the recording session, individual neurons were labeled with biocytin. Subsequent light microscopic analysis failed to reveal any gross morphological differences in the appearance of mutant pyramidal cells compared with their wild-type counterparts (Fig. 2d).
A total of 11 whole-cell voltage-clamp recordings were made from adult pyramidal neurons (7 obtained from three wild-type mice and 4 taken from three mutant mice) visualized by infrared videomicroscopy before recording (Fig. 3a,inset). Biocytin was routinely included in the patch pipette to further verify the identity of the cell at a later time (Fig.3a). Both wild-type and mutant neurons displayed spontaneous inward currents of varying amplitudes and frequencies (Fig.3b) that were mediated by the activation of non-NMDA glutamate receptors because they could be blocked by CNQX (data not shown). The neurons from D4R-deficient mice displayed a small but statistically significant increase in the frequency of spontaneous events compared with the wild-type neurons (Fig.3d, top left panel, left set ofbars) (t = 2.66; df = 9;p = 0.026). In the presence of 50 μm 4-AP, burst frequency increased in neurons from both wild-type and mutant mice, but the effect was most pronounced in the latter (Fig. 3d, top left panel,right set of bars) (t = 3.45; df = 9; p = 0.007).

Whole-cell voltage-clamp recordings reveal increased excitation of cortical pyramidal neurons from D4R-deficient mice. a, An example of a biocytin-filled pyramidal neuron from a mutant mouse from which voltage-clamp data were obtained. The inset shows an IR-DIC image of the cell (arrow) before the patch electrode was attached. Panels show two different magnifications of the same pyramidal neuron. b, Recordings of inward membrane currents showing excitatory events in a cortical neuron isolated from a wild-type (left traces) and a mutant mouse (right traces). Currents were recorded either in control conditions (top traces) or in the presence of 50 μm 4-AP (bottom traces). Bicuculline (10 μm) was present throughout the experiment, and the membrane was held at −70 mV.c, Approximately 5.5 min of membrane current recorded from a neuron prepared from a D4R-deficient mouse in the presence of 4-AP (50 μm). The five large downward deflections reflect paroxysmal activity evoked by the addition of 4-AP to the bath solution. d, Summary plots of quantified spontaneous synaptic activity. Columns indicate the results obtained from wild-type (black) and D4R-deficient mice (white), respectively.d (top left panel), Plot of the frequency (in Hertz) of all the detected spontaneously occurring events. d (top right panel), Plot of the mean amplitude of all the detected spontaneous events.d (bottom left panel), Plot of the frequency of the events greater in amplitude than the sum of the mean amplitude and twice the SD for any given data set (see Results).d (bottom right panel), Plot of the frequency (events per minute) of paroxysmal activity.
Although the average amplitude of inward currents was not significantly different between wild-type and mutant neurons, in either the presence or absence of 4-AP (Fig. 3d, top right panel), closer inspection of the data revealed two classes of large inward currents, as defined by amplitude criteria. The first event group comprised simple inward currents that had amplitudes greater than the sum of the mean amplitude and twice the SD for any given data set. Under control conditions the frequency of this type of current was the same in both genotypes (Fig. 3d,bottom left panel, left set of bars) but in the presence of 4-AP was significantly increased only in the mutant neurons (Fig. 3d, bottom left panel,right set of bars) (t = 5.42; df = 9; p < 0.001). The second large-amplitude event was characterized by a robust inward current (Fig. 3c) that probably corresponded to the paroxysmal depolarizations observed in the current-clamp experiments described above. Again, under control conditions the frequency of this type of current was the same in both genotypes (Fig. 3d, bottom right panel,left set of bars) but was significantly increased by 4-AP only in the mutant neurons (Fig. 3d, bottom right panel, right set of bars) (t = 2.57; df = 9; p = 0.03). The reduced frequency of these events in the voltage-clamp experiments compared with the current-clamp experiments may be a reflection of the lower concentration of 4-AP used in the voltage-clamp studies.
When taken together, these electrophysiological results indicate that cortical excitability is increased in the D4R-deficient mice, a finding consistent with the hypothesis that in vivothe D4R acts as an inhibitory modulator of neuronal activity in the FC. To test this hypothesis, we performed three types of experiments. In the first, the effect of PNU-101383G (a novel D4R antagonist) on the electrophysiological activity of cortical pyramidal neurons from wild-type and D4R-deficient mouse brain slices was evaluated. In the second set of experiments, the D4R-deficient mice were analyzed with respect to their sensitivity to the convulsant effects of the GABAA blocker bicuculline. Third, we investigated whether the cortical output to cortical layer VI was modified in D4R-deficient mice by performing an ultrastructural analysis of glutamate terminal immunolabeling.
The D4R antagonist PNU-101387G increases cortical excitability in cortical slices of wild-type mice
Several putative D4R antagonists exhibiting different ratios of binding selectivity in comparison to other DA, 5-HT, and noradrenaline receptors have emerged during the last several years, although their efficacy in vivo is still a matter of controversy (Tarazi and Baldessarini, 1999). One of these compounds is PNU-101387G. This D4R antagonist displays moderately high affinity (Ki = 3.6 nm) and selectivity for the D4R (Merchant et al., 1996). More recently PNU-101387G has been shown to prevent amphetamine sensitization in rats (Feldpausch et al., 1998) and stress-induced memory deficits in monkeys (Arnsten et al., 2000). We used PNU-101387G to determine the possible participation of the D4R in the modulation of the activity of pyramidal neurons of the FC in normal mouse brain slices. Whole-cell voltage-clamp recordings were made from nine cortical pyramidal neurons obtained from four wild-type mice. In standard ACSF at a holding potential of −60 mV, all neurons displayed spontaneous inward currents of varying frequencies and amplitudes (Fig.4a). In the presence of PNU-101387G (10 μm; 3 min exposure duration), six of nine neurons displayed an increased frequency of spontaneous inward currents (Fig. 4a). Two neurons did not display changes, and one neuron showed a decreased frequency of spontaneous inward currents. As a group, the increase in frequency was statistically significant (87 ± 15.4%; t = −2.48; df = 8; p < 0.05) (Fig. 4b). In the neurons that displayed increases in the frequency of spontaneous inward currents, we examined normalized amplitude–frequency histograms to determine whether changes in frequency were associated with changes in amplitude. There were no consistent changes in the normalized amplitude–frequency histograms in the presence of PNU-101387G (Fig.4c). In the two neurons that did not change frequency of spontaneous inward currents in the presence of PNU-101387G, after washing we subsequently added 4-AP to increase the frequency of spontaneous events and then re-exposed the neurons to PNU-101387G 10 μm for 3 min. Both neurons now displayed an increase in frequency (317 and 54%). To provide a test of pharmacological specificity of the D4R antagonist, we examined the effects of applying the same concentration of PNU-101387G in five neurons from two D4R-deficient mice. PNU-101387G (10 μm) had no significant net effect (Fig.4b): two neurons displayed slight increases in the frequency of inward currents, and three displayed decreases. These data indicate that blockade of the D4R can increase the frequency of spontaneous inward currents, thereby providing additional support for the hypothesis that this receptor subtype mediates at least some of inhibitory actions of DA in the cortex. Furthermore, the observation that a change in frequency did not appear to be associated with a change in amplitude suggests that some of these inhibitory actions may be presynaptic.

The D4R antagonist PNU-101387G increases the frequency of spontaneous inward currents in cortical pyramidal neurons in the FC of wild-type mice but has no effect in D4R-deficient mice. a, Whole-cell voltage-clamp recordings. Top trace shows spontaneous activity before application of PNU-101387G. Bottom traceshows that during application of the D4 antagonist (10 μm, 3 min application) spontaneous inward currents increased in frequency. b, Bar graphshows increase in frequency of spontaneous inward currents in wild-type mice and return to baseline after wash. Asteriskindicates that increase was statistically significant (p < 0.05). In D4R-deficient mice there was no net change in spontaneous inward currents.c, Amplitude–frequency histograms before (Control), during, and after (Wash) exposure to the D4 antagonist in the FC neuron shown in a. d, Amplitude–frequency histograms were first normalized to the total number of events in each of the three histograms shown inc, and then the cumulative frequencies were plotted before (Control), during, and after (Wash) exposure to the D4antagonist. The curves superimpose, indicating that although the D4 antagonist markedly increased the frequency of spontaneous inward currents, their amplitudes were not altered.
D4R-deficient mice are supersensitive to the convulsant effects of bicuculline
On the basis of the electrophysiological data, we hypothesized that pyramidal neurons in the FC of D4R-deficient mice might become hyperexcitable under proconvulsive conditions. To test this hypothesis, wild-type and mutant mice were exposed to increasing concentrations of the convulsant bicuculline. At doses up to 3 mg/kg, bicuculline produced only mild to medium signs of epileptogenic activity in mice of both genotypes. However, at 4 mg/kg, bicuculline displayed pronounced convulsant effects that differed as a function of genotype (Fig. 5). After the administration of 4 mg/kg bicuculline, all of the wild-type mice entered a tonic–clonic seizure phase that was characterized by a loss of postural control, barrel rolling, spontaneous jumping, and convulsions. At this dose, 60% of the animals died by the end of the 15 min observation period. In contrast, the mice lacking D4Rs displayed more pronounced epileptic-like seizure behavior and a significantly earlier onset of tonus, wild running, and clonus (Fig. 5). In addition, all of the D4R-deficient mice experienced severe seizures and died within 15 min of administration of the bicuculline.

D4R-deficient mice are hypersensitive to the convulsant effects of the GABAA blocker bicuculline. Shown is the onset of the different convulsive phases induced by bicuculline (4 mg/kg, i.p.) in wild-type (black columns) and D4R-deficient (white columns) mice. Bars represent the mean ± SEM time values of wild-type (n = 10) and D4R-deficient mice (n = 9), respectively. Data were analyzed by Student's t test. *p < 0.05 and **p < 0.005 compared with wild-type mice. Each animal was observed for 15 min.
Glutamate immunoreactivity is decreased in D4R-deficient mice
Recent in vivo microdialysis and electron microscopic studies have demonstrated that enhanced extracellular levels of glutamate are correlated with a decrease in the density of terminal glutamate immunolabeling as a consequence of reduced accumulation of neurotransmitter (Meshul et al., 1999). Given the apparent cortical hyperexcitability that we observed in our mutant mice, we hypothesized that glutamate immunolabeling of FC pyramidal neurons might be reduced in the D4R-deficient mice compared with wild-type cells. To test this hypothesis we performed an ultrastructural analysis of glutamate immunolabeling in 349 FC terminals, 165 of which came from wild-type mice (n = 6) (Fig.6a) and the remaining 184 from D4R-deficient mice (n = 6) (Fig.6b). This study revealed significantly less glutamate-ir in FC layer VI of mutant (91 ± 3 SEM gold particles/μm2), compared with wild-type, mice (114 ± 4 SEM gold particles/μm2).

Ultrastructural analysis of frontal cortex from wild-type and D4R-deficient mice. Electron photomicrographs of nerve terminals within the frontal cortex prepared from wild-type (a) and D4R-deficient (b) mice. In a andb, the small arrows identify gold particles that reveal the location of the glutamate antibody; thelarge arrows indicate asymmetrical synaptic contacts. Scale bar (shown in b for a andb): 0.25 μm.
DA synthesis and turnover in the FC is unaffected in mice lacking D4Rs
Because glutamatergic pyramidal neurons of the FC appeared to be hyperactive in the D4R-deficient mice, we investigated whether this condition might be associated with altered DA synthesis and release in this brain region. l-DOPA accumulation was assessed after treatment with the l-aromatic decarboxylase inhibitor NSD-1015 to estimate the rate of DA synthesis. The accumulation of l-DOPA in the FC of wild-type and mutant mice was similar, with a calculated DA synthesis rate of 2.627 ± 0.046 pg/min and 3.010 ± 0.345 pg/min in each respective genotype. The DOPAC/DA ratio was also calculated and found to be similar between the two genotypes (wild type: 3.26 ± 0.71; mutant: 2.71 ± 0.14).
DISCUSSION
The absence of D4Rs leads to increased cortical excitability and glutamate synaptic activity
On the basis of anatomical studies, it has been proposed that D4Rs may modulate the activity of cortical neurons (Mrzljak et al., 1996), and now our electrophysiological analyses, including current- and voltage-clamp experiments, have demonstrated that cortical neuronal activity is enhanced in D4R-deficient mice. There is considerable evidence that DA, acting on multiple receptor subtypes (Cepeda et al., 1992; Goldman-Rakic, 1996), can have both excitatory and inhibitory influences on the activity of neurons in the FC. Furthermore, D1-like agonists are known to promote seizure activity, whereas D2-like agonists can act as anticonvulsants, presumably by elevating the excitatory threshold of the FC (Starr, 1996). In the present study, we found that the total absence or pharmacological blockade of D4Rs increases excitability in the FC and that mice lacking D4Rs also have an increased susceptibility to the tonic–clonic seizures produced by bicuculline. Therefore, the ability of typical and atypical neuroleptics, including clozapine (Denney and Stevens, 1995), to induce paroxysmal discharges may be explained, in part, by their ability to block D4Rs in the FC. On the basis of these observations, it is conceivable that selective stimulation of D4Rs may constitute a rational pharmacotherapy for certain types of epileptic disorders.
Increased cortical excitability and reduced glutamate immunogold labeling in mutant mice are consistent with the interpretation that D4Rs may normally exert an inhibitory influence on FC glutamatergic pyramidal neurons in wild-type mice. Alternatively, our findings could be explained by a significant increase in the activity of ascending dopaminergic mesocortical neurons that would elevate cortical DA levels and possibly prolong the activation of D1-like receptors in the D4R-deficient mice. To address the possibility that the presynaptic component of mesocortical DA transmission was altered in the D4R mutant mice, we assessed the rate of DA synthesis and turnover in the FC of wild-type and receptor-deficient mice. The results of these studies indicated that in the mutant mice the presynaptic components of DA neurotransmission are not altered in the FC, similar to what we observed in the nucleus accumbens of the mutant mice (Rubinstein et al., 1997), but in marked contrast to what was observed in the dorsal striatum of these animals. The unaltered synthesis and turnover of cortical DA in the D4R-deficient mice support the hypothesis that DA can have an inhibitory influence on cortical excitability via its postsynaptic actions on D4R-expressing cortical neurons, some of which may project to the dorsal striatum. This interpretation may help to explain how some atypical antipsychotics that are also D4R antagonists (Van Tol et al., 1991; Roth et al., 1995), such as clozapine, can increase neuronal activity in the FC and ameliorate some cognitive and affective deficits that afflict schizophrenics without producing the unwanted side effects associated with the typical antipsychotics (Lidow et al., 1998).
Recent genetic studies have associated certain alleles of the human DRD4 gene with the occurrence of ADHD (LaHoste et al., 1996; Swanson et al., 1998). ADHD, characterized by difficulty with concentration, has been associated with FC malfunction (Barkley, 1998) and has shown a heritability of 50–90% (Gillis et al., 1992). Neuroanatomical imaging studies performed on normal individuals as well as individuals with ADHD have revealed that the FC, dorsal striatum, and globus pallidus, the three areas richest in D4Rs (Mrzljak et al., 1996;Ariano et al., 1997), are significantly smaller in affected individuals compared with the corresponding structures in control subjects (Castellanos et al., 1996). Given that individuals suffering from ADHD are frequently medicated with catecholamine uptake inhibitors such as methylphenidate, our results are consistent with the interpretation that by indirectly increasing DA levels in the FC, methylphenidate may derive its clinical benefit, in part, by prolonging the hyperpolarization of pyramidal neurons, perhaps via the sustained activation of D4Rs.
In conclusion, we have provided behavioral, electrophysiological, immunocytochemical, and ultrastructural evidence that cortical activity is elevated in mice lacking D4Rs. We have also presented pharmacological data that D4R blockade increases excitability in wild-type pyramidal neurons of the FC. Knowing that D4Rs are expressed in cortical neurons and that their activation can result in cellular hyperpolarization, we have concluded that the abnormal hyperexcitability displayed by cortical neurons in slices of brain taken from D4R-deficient mice is a consequence of the inability of DA to exert its normal inhibitory influence over these neurons. Consequently, we anticipate that future studies involving these receptor-deficient mutant mice and their wild-type counterparts will serve to further illuminate the contributions that this DA receptor subtype makes to normal brain function.
Footnotes
This work was supported, in part, by an International Research Scholar Grant of the Howard Hughes Medical Institute (M.R.), Universidad de Buenos Aires (M.R.), Agencia Nacional de Promoción Cientifica y Tecnológica (M.R.), National Institute on Drug Abuse (D.K.G., L.B.K., M.J.L.), National Alliance for Research on Schizophrenia and Depression (M.S.L), National Institute of Neurological Disorders and Stroke (M.S.L.), the Portland Alcohol Research Center (D.K.G., M.J.L.), the Department of Veterans Affairs Merit Review Program (C.K.M.), and the Smokeless Tobacco Research Council (C.K.M.). Tomás Falzone is the recipient of a predoctoral fellowship from the Consejo Nacional de Investigaciones Cientı́ficas y Tecnológicas, Argentina. We thank Marta Barontini, Sylvia Gill, Ahrin Koppel, Jennifer Larson, Elizabeth Lutz, Norberto Malarini, and Lidia Parodi for excellent technical assistance. We also acknowledge Kalpana Merchant of Pharmacia-Upjohn Pharmaceuticals for supplying PNU-101387G.
Correspondence should be addressed to Dr. Marcelo Rubinstein, Instituto de Investigaciones en Ingenierı́a Genética y Biologia Molecular (Universidad de Buenos Aires–Consejo Nacional de Investigaciones Cientificas y Técnicas), Vuelta de Obligado 2490, Buenos Aires, 1428 Argentina, E-mail:mrubins{at}dna.uba.ar, or Dr. David K. Grandy, Department of Physiology and Pharmacology, L334 Oregon Health Sciences University, 3181 SW Sam Jackson Park Road, Portland, OR 97201. E-mail:grandyd{at}ohsu.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}