

Abstract
To investigate the tissue distribution and subcellular localization of ST3GalV (CMP-NeuAc:lactosylceramide α2,3 sialyltransferase/GM3 synthase) in the adult mouse, we generated two antisera against mouse ST3GalV that were designated CS2 (directed against amino acids K227–I272) and CS14 (directed against amino acids D308–H359). We previously reported that CS2 antiserum stains medial andtrans-Golgi cisternae in all cell types investigated. In neural tissue, however, CS14 antiserum reveals a subpopulation of ST3GalV with a subcellular distribution complementary to CS2 antiserum. CS14 antiserum strongly stains axons in cortical, cerebellar, brainstem, and spinal cord tissue sections. The subcellular localization of neuronal ST3GalV is maintained in primary cultures of rat hippocampal neurons and in PC12 cells. In PC12 cells, ST3GalV localization evolves during NGF-induced differentiation such that a pool of enzyme leaves the Golgi for a distal compartment in conjunction with neurite outgrowth. In PC12 cells transfected with an epitope-tagged form of ST3GalV, staining for the epitope tag coincides with expression of endogenous enzyme. The non-Golgi pool of ST3GalV does not colocalize with markers for the trans-Golgi network, endosome, or synaptic vesicles, nor is it detected on the cell surface. Distinct subpopulations of ST3GalV imply that ganglioside synthesis can occur outside of the Golgi or, alternatively, that a portion of the total ST3GalV pool subserves a nonenzymatic function. Significantly fewer transfected cells were found in PC12 cultures treated with plasmid encoding ST3GalV than in cultures treated with control plasmid, indicating that the expression of ST3GalV in excess of endogenous levels results in either cell death or a decreased rate of cell division.
The glycosyltransferase ST3GalV (CMP-NeuAc:lactosylceramide α2,3 sialyltransferase or GM3 synthase) catalyzes the addition of sialic acid to a neutral glycosphingolipid acceptor, lactosylceramide, thereby forming ganglioside GM3, the simplest glycosphingolipid bearing a ganglio-series oligosaccharide (van Echten and Sandoff, 1993; Tsuji et al., 1996; Ishii et al., 1998; Kono et al., 1998; Fukumoto et al., 1999). In turn, GM3 (NeuAcα3Galβ4GlcCer) [nomenclature based onSvennerholm (1964)] serves as the precursor for almost all other gangliosides, a class of glycosphingolipids implicated in transmembrane signaling, cell–cell interactions, specialized membrane domain formation, and synaptic transmission (Goldenring et al., 1985;Blackburn et al., 1986; Bremer et al., 1986; Kreutter et al., 1987;Nojiri et al., 1988; Tsuji et al., 1988; Hakomori and Igarashi, 1993;Yamamura et al., 1997). Because GM3 synthesis occurs early in ganglioside biosynthesis, ST3GalV was predicted to be localized to thecis-Golgi (van Echten et al., 1990; Young et al., 1990; Iber et al., 1992; Maccioni et al., 1999). However, biochemical and immunohistochemical localization studies have demonstrated that ST3GalV is found in both medial and trans-Golgi cisternae (Lannert et al., 1998; Stern et al., 2000). The exclusion of ganglioside synthetic enzymes from the early Golgi, combined with the broad distribution of ST3GalV in the remaining Golgi, indicates that the topographic distribution of glycolipid-directed glycosyltransferases differs from the restrictive spatial hierarchy characteristic of glycoprotein oligosaccharide-processing enzymes. Furthermore, it implies that mechanisms governing subcellular localization and compartmental retention of glycolipid-directed glycosyltransferases may be specific for this class of enzymes and are deserving therefore of direct investigation.
Definition of the topography of glycolipid synthetic enzymes in neurons is of particular importance because many proposed functions for gangliosides require the presence and maintenance of glycosphingolipid along the axon or at the nerve terminal. Glycosphingolipids constitute ∼15% of total neuronal membrane (Futerman and Banker, 1996). Although Golgi synthesis and vesicular transport can provide a constitutive supply of glycolipid, dynamic local synthesis would offer a more immediately responsive pool of ganglioside for the modulation of growth factor signaling and membrane domain formation (Hakomori and Igarashi, 1995; Mutoh et al., 1995; Grimes et al., 1997; Hakomori et al., 1998). In fact, the existence of extra-Golgi glycosyltransferase activities in neurons has been suggested previously by the detection of two distinct sialyltransferase activities, one protein-directed and one lipid-directed, in synaptosomal membranes prepared from calf cortex (Preti et al., 1980) and by the detection of an additional glycolipid synthase in rat brain (Durrie et al., 1987, 1988). The difficulty inherent in preparing synaptosomal membranes free of Golgi contamination has limited, however, the interpretation of results derived from subcellular fractionation (Ng and Dain, 1977; Durrie et al., 1988). Alternatively, immunohistochemical probes provide a powerful complementary approach to investigating the subcellular distribution of glycosyltransferases (Taatjes et al., 1988; Daniotti and Maccioni, 1997; Maccioni et al., 1999; Stern et al., 2000).
We have investigated the subcellular distribution of mouse ST3GalV (mST3GalV) using two different antisera raised against nonoverlapping peptide regions. Our immunolocalization reveals two distinct pools of ST3GalV in neurons. As expected, mST3GalV is localized to the Golgi apparatus but is also found in axons throughout the adult neuraxis. Similarly, dual localization is seen in primary cultures of rat hippocampal neurons and in PC12 cells. Epitope-tagged ST3GalV (C-terminal myc) expressed in PC12 cells demonstrates the same distribution as endogenous enzyme. By confocal microscopy, ST3GalV fails to colocalize with markers for known post-Golgi vesicular pools, placing the non-Golgi associated pool of ST3GalV in a novel compartment.
MATERIALS AND METHODS
Materials. DMEM high-glucose media, horse serum, fetal calf serum, glutamine, penicillin, and streptomycin were obtained from Life Technologies (Gaithersburg, MD). Nerve growth factor (NGF) (2.5S, type II) was obtained from Boehringer Mannheim (Indianapolis, IN). Secondary antibodies (HRP-conjugated and fluorescein-conjugated goat anti-rabbit, Texas Red-conjugated goat anti-mouse and unconjugated goat anti-rabbit IgG) were from Jackson Laboratories (West Grove, PA). pcDNA 3.1 (−)myc/His expression vector, Protein A Sepharose, laminin, collagen type I from rat tail, and poly-l-lysine were supplied by Sigma (St. Louis, MO). pATH11 vector was from American Type Culture Collection (Manassas, VA). Affigel 15 resin was from Bio-Rad (Hercules, CA). ECL reagent was from Amersham Pharmacia Biotech (Arlington Heights, IL). Swiss Webster-CD1 mice were obtained from Charles River Laboratories (Wilmington, MA). Monoclonal antibody against MAP2 (clone HM2) was obtained from Sigma. Monoclonal antibody against c-myc epitope (9E10) was provided by Pocono Rabbit Farm and Laboratory (Canadensis, PA), monoclonal antibody against transferrin receptor by I. Mellman (Yale University, New Haven, CT), antiserum against mannosidase II by P. Cresswell (Yale University), monoclonal antibody against synaptobrevin by P. DeCamilli (Yale University), and monoclonal antibody against TGN-38 by K. Howell (University of Colorado School of Medicine, Denver, CO). All other chemicals and reagents were obtained from standard commercial sources.
mST3GalV antibody production. The preparation of anti-mouse ST3GalV antiserum, designated CS2, has been described previously (Stern et al., 2000). Another antiserum to mouse ST3GalV was raised against a TrpE fusion protein in rabbit. Nucleotides 922–1077 (D308–H359) were subcloned into pATH11 vector, and the fusion protein was expressed after induction in Escherichia coli XL-1 Blue (Koerner et al., 1991). Fusion protein was prepared and injected into rabbits as described previously (Stern et al., 2000). The harvested serum was designated CS14, and a portion was affinity purified by binding to and eluting from an Affigel 15 column coupled with mST3GalV-TrpE fusion protein. All protein determinations were made by the bicinchoninic acid assay (Smith et al., 1985).
Western blot and immunodepletion. Enriched Golgi membrane fractions were prepared from adult mouse brains as described previously (Fleischer and Kervina, 1974; Ma and Colley, 1996). Western blot analysis was performed on aliquots of membrane preparations as described previously (Stern et al., 2000). The primary antiserum dilution for CS14 was 1:5000. No difference was detected between blots probed with affinity-purified or nonpurified serum.
For immunodepletion with anti-mouse ST3GalV antisera, CS2 or CS14 antiserum was bound to Protein A Sepharose for 2 hr at ambient temperature. Mouse brain extract (4 μg of protein) was solubilized in 50 mm Tris, pH 7.6, 100 mm NaCl, and 1% Triton X-100 and precleared by incubation with Protein A Sepharose for 45 min at 4°C. Precleared extract was incubated with CS2- or CS14-loaded Protein A Sepharose for 2 hr at 4°C and then centrifuged to pellet the Protein A Sepharose; the supernatant was recovered. An aliquot of the supernatant (5%) was prepared for SDS-PAGE by reduction with 50 mm dithiothreitol followed by sulfhydryl alkylation with 250 mm iodoacetamide (Stern et al., 2000). Samples were then electrophoresed and transferred to nitrocellulose (Harlow and Lane, 1988). The blots were blocked in 5% dry milk and 0.2% Tween 20 in PBS and incubated with primary antiserum (CS2 or CS14) diluted 1:5000 and then with secondary antibody (HRP-conjugated goat anti-rabbit) diluted 1:5000. Antibody binding was detected by ECL reagent using the manufacturer's recommended conditions.
Immunofluorescence staining of mouse tissue sections. Cortical, cerebellar, brainstem, and spinal cord tissue sections from four adult mice were prepared as described previously (Stern et al., 2000). Primary antibody (CS14) was diluted 1:100, and secondary antibody (fluorescein-conjugated goat anti-rabbit) was diluted 1:200. Confocal images were acquired on a Zeiss Axiovert 100 microscope equipped with an argon–krypton laser. Microscope operation and image acquisition were performed with the Zeiss LSM510 version 2.3 software package. Optical sections were taken every 1.0 μm parallel to the coverslip. Acquired images were prepared with Adobe software by minimally adjusting background fluorescence levels equally across all color channels; the relative fluorescence between red and green channels was unaltered from the original confocal scan.
Cell culture. PC12 rat pheochromocytoma cells were obtained from P. DeCamilli and cultured in DMEM with 10% horse serum, 5% fetal bovine serum, 0.3 mg/ml glutamine, 12.5 U of penicillin, and 12.5 μg/ml streptomycin (Greene and Tischler, 1976). For differentiation assays, PC12 cells were cultured in low-serum DMEM (1% horse serum plus glutamine, penicillin, and streptomycin).
PC12 cells were exposed to 1 or 50 ng/ml NGF in low-serum media for 3 d to induce neurite formation and cellular differentiation (Greene and Tischler, 1976; Campbell and Neet, 1995). Cells were plated on coverslips coated with a laminin/poly-l-lysine substrate (1:6; 33 μg/ml laminin and 0.2 mg/ml poly-l-lysine) for double-label immunofluorescence staining or with a collagen/poly-l-lysine substrate (25:1; 250 μg/ml collagen and 10 μg/ml poly-l-lysine) for quantitation of cell survival and proliferation. Transfected cells were quantitated from images acquired using IP Lab digital imaging software (Scanalytics, Fairfax, VA) and a Zeiss axiophot microscope. For each experiment, at least 10 fields were counted to give >100 cells per experiment. In total, >4000 cells were counted at 0 ng/ml NGF, and >1900 cells were counted at 50 ng/ml NGF.
Primary cultures of postnatal day 5–13 rat hippocampal neurons were obtained from I. Mellman (Brewer et al., 1993; Winckler et al., 1999) and P. DeCamilli (Banker and Cowan, 1977; Bartlett and Banker, 1984;Chilcote et al., 1995). Cultured neurons were fixed and stained as described for PC12 cells.
Transfection of PC12 cells. Myc-tagged fusion constructs of mouse ST3GalV were generated by directional cloning of mouse ST3GalV at nucleotides −7 and 1077 using EcoRI and KpnI restriction sites in pcDNA 3.1 (−)myc/His expression vector downstream of the CMV promoter. Constructs contain the mouse ST3GalV translational start site. PC12 cells (2.5 × 105) were transfected with 20 μg of myc-tagged mouse ST3GalV or control pcDNA 3.1 vector DNA by electroporation with a Bio-Rad electroporator. After electroporation, cells were recovered in low-serum media and plated at a density of 1.0 × 105cells per 3.5 cm well. After 18 hr, we added fresh low-serum media containing 0, 1, or 50 ng/ml NGF. During the 3 d differentiation period, NGF was added in fresh media every 24 hr. Before fixation, cells were washed with PBS that was warmed previously to 37°C. Then PC12 cells were fixed with 4% paraformaldehyde and 4% sucrose in PBS, prewarmed to 37°C for 15 min, and washed three times with 120 mm sodium phosphate, pH 7.4. Alternatively, PC12 cells were fixed for 8 min with methanol prechilled to −20°C and then washed three times with 120 mm sodium phosphate, pH 7.4.
Immunofluorescence staining of PC12 cells. Fixed PC12 cells were sequentially washed twice for 5 min in 120 mm sodium phosphate, pH 7.4, then in low-salt buffer (150 mm NaCl, 10 mm sodium phosphate, pH 7.4), and finally in high-salt buffer (500 mm NaCl, 20 mm sodium phosphate, pH 7.4). Coverslips were blocked for 30 min in 15% normal goat serum, 0.3% Triton X-100, 0.3m NaCl in 0.04 m sodium phosphate, pH 7.4. Primary antibody dilutions were 1:100 for CS14, 1:200 for anti-mannosidase II antiserum, 1:500 for anti-myc antiserum and anti-synaptobrevin antiserum, and 1:10 for anti-TGN38 and anti-transferrin receptor antibodies. Incubation with primary antibody was performed for 2 hr in a humidified chamber. Coverslips were then washed in high-salt buffer and incubated in secondary antibody, fluorescein-conjugated goat anti-rabbit and/or Texas Red-conjugated goat anti-mouse at 1:200. After two washes in high-salt buffer and one wash in 120 mm sodium phosphate buffer, pH 7.4, all coverslips were mounted in 4% w/v 1,4-diazabi-cyclo[2,2,2]octane in 50% glycerol in PBS. Confocal images were acquired as described above.
For PC12 cell surface staining, PC12 cells were treated with either 0 or 50 ng/ml NGF for 3 d. Before staining, cells were washed briefly in cold PBS and chilled on ice for 15 min in 0.02% sodium azide in PBS. Probes were diluted 1:100 (CS14 antiserum) or 1:500 (anti-c-myc antiserum or Texas Red-conjugated wheat germ agglutinin) in 5% normal goat serum, 0.02% sodium azide in PBS. Primary antibody incubation was performed for 15 min on ice. Cells were then washed in PBS and blocked in 15% normal goat serum, 0.3% Triton X-100 in PBS for 15 min before the addition of secondary antibody. Either fluorescein-conjugated goat anti-rabbit or goat anti-mouse was used at a dilution of 1:200 in 15% normal goat serum, 0.3% Triton X-100 in PBS; incubation was performed for 90 min. Cells were washed in PBS and mounted as described above.
RESULTS
Antiserum CS14 reveals a non-Golgi pool of mST3GalV in neurons
Immunostaining with an anti-mST3GalV antiserum designated CS2 (raised against amino acids K227–I272) has shown that a pool of GM3 synthase is localized to medial and trans-Golgi compartments (Fig. 1A) (Stern et al., 2000). However, another anti-mST3GalV antiserum, designated CS14 (directed against the most C-terminal 52 amino acids, D308–H359), reveals a non-Golgi pool of this key ganglioside synthetic enzyme. In cerebellar tissue sections, for instance, axons coursing through the granule and Purkinje cell layers are stained with CS14 antiserum. Parallel processes are seen in the molecular layer, and multiple axon profiles are visible in the white matter of the cerebellum (Fig.1B). Furthermore, CS14 immunostaining is complementary to CS2 immunostaining in neurons. An elaborate Golgi apparatus is apparent in Purkinje cells stained with CS2 antiserum, but Purkinje cells are completely devoid of Golgi staining when stained with CS14 antiserum (Fig. 1A,B). However, the adjacent molecular layer shows parallel fiber staining (Fig. 1C), and axons reactive with CS14 antiserum are seen weaving between granule cells (Fig. 1D,arrow) or are visualized in cross section (arrowhead) within the granule layer. The white matter tracts of the cerebellum, consisting primarily of oligodendrocytes and axonal processes, show concentrated CS14 axonal staining.
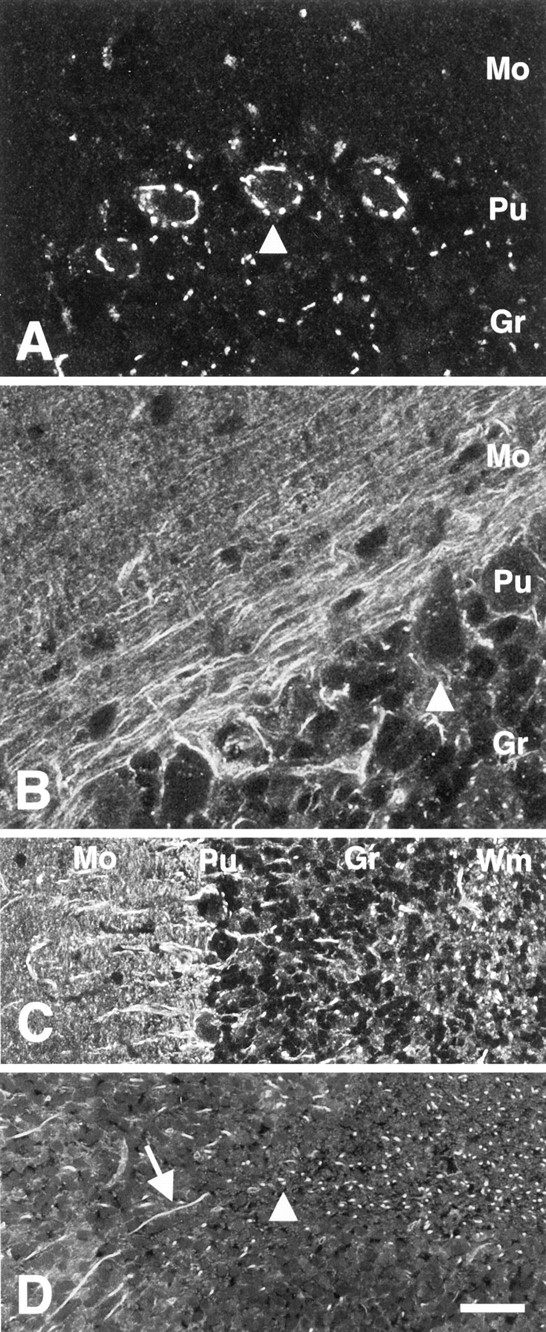
Anti-mST3GalV antisera reveal distinct subpopulations of ST3GalV in cerebellar tissue sections.A, CS2 antiserum stains large perinuclear puncta identified as medial trans-Golgi (Stern et al., 2000) in cerebellar Purkinje cells (Pu,arrowhead). In granule cells (Gr), mST3GalV is restricted to a single locus in each cell.B, Cerebellar molecular and Purkinje cell layers show processes staining with CS14 antiserum extending around the Purkinje cells, with no CS14 immunore activity observed in Purkinje cell bodies (arrowhead). In the molecular layer (Mo) immediately adjacent to the Purkinje cell layer (Pu), parallel fibers strongly stain with CS14 antiserum. A decreasing gradient of parallel fiber staining exists toward the pial surface.C, Axons stained with CS14 antiserum are observed extending in the molecular layer (Mo) through the Purkinje cell layer (Pu) and within the granule cell layer (Gr). In this section, axonal processes are oriented primarily in cross section throughout the cerebellar white matter (Wm). D, Higher magnification of granule cell layer. CS14-immunoreactive neuronal processes are seen both in longitudinal section (arrow) and cross section (arrowhead) within the granule cell layer. CS14 antiserum staining is also observed in the white matter. Scale bars:A, B, 17 μm; C, 41 μm;D, 34 μm.
CS14 antiserum reveals a non-Golgi distribution of mST3GalV in all neurons. Within the spinal cord, CS14 staining is seen in axon fascicles coursing through the gray matter and in ascending and descending tracts within the white matter (Fig.2A). In cross section (Fig. 2B), profiles of small-caliber axons (arrow) are uniformly stained. In large-caliber axons (thin arrows), however, a peri-axonal ring of staining is evident, consistent with the presence of the enzyme in close association with the axolemma or with the innermost myelin lamellae. CS14-reactive ST3GalV was not apparent in layers of compacted myelin (small arrowhead). Punctate staining within the axon profile was also observed in large-caliber axons (arrowhead). CS14 antiserum also reveals a non-Golgi pool of ST3GalV in all brainstem and cortical regions examined (data not shown).

mST3GalV localizes to axons in spinal cord sections. A, A montage of the ventrolateral region of the spinal white matter of the cervical spinal cord (Wm, white matter; Vh, ventral horn). CS14 stains axons cut in cross section within white matter, and a fine meshwork of axons is seen in the ventral horn. B, Higher magnification of spinal white matter reveals that CS14 antiserum stains axons differentially depending on their caliber. Large axons exhibit a peri-axolemmal ring of staining (thin arrows) within which punctate staining is also observed (arrowhead). Dark regions surrounding large-caliber axons are the nonstaining regions of compact myelin (small arrowhead). Smaller caliber axons (arrow) stain throughout the diameter of the process. Scale bars: A, 56 μm; B, 15 μm.
The non-Golgi pool of mST3GalV is preserved in cultured cells of neural origin but is undetectable in non-neural cells
Primary cultures of rat hippocampal neurons display CS14 staining similar to intact tissue. CS14 reactivity is found both in the cell body and confined to a single neuronal process (Fig.3A). MAP2 staining, which reveals the dendritic arbor of the neuron (Caceres et al., 1984), fails to colocalize with CS14 immunoreactivity, indicating that ST3GalV is preferentially localized to the axon. In the NGF-inducible cell line PC12, CS14 antiserum stains all neuritic processes in addition to a portion of the cell body corresponding to the Golgi apparatus (Fig.3B). The non-Golgi distribution of ST3GalV was equally apparent when cells were fixed with cold methanol or aldehyde-based fixatives.

CS14 antiserum stains axons in primary cultures of rat hippocampal neurons and neurites of PC12 cells. A, CS14 (green) immunoreactivity fails to colocalize with the MAP2 antibody (red) in primary culture rat hippocampal neurons. B, CS14 antiserum stains both the cell body and neurites (arrow) of NGF-induced PC12 cells. Scale bars: A, 60 μm; B, 34 μm.
In non-neural cells, CS14 antiserum only stains the Golgi, implying that the complementary distribution of CS2 and CS14 epitopes detected in nervous tissue arises from a neural-specific modification of the polypeptide. For instance, CS14 antiserum stains Golgi in numerous cell types of the seminiferous tubules, including germ cells (spermatogonia and primary spermatocytes), Leydig cells (Fig.4A), and Kupffer cells in the liver (data not shown); identical staining of seminiferous tubule cell types and Kupffer cells was observed with the Golgi-specific anti-ST3GalV antiserum, CS2 (Stern et al., 2000). The ability of CS14 antiserum to stain ST3GalV in neural tissue sections was abolished by the immunoadsorption of antiserum with immobilized fusion peptide (Fig. 4B,C).

CS14 antiserum stains Golgi in non-neural cell types, and neural staining is inhibited by relevant peptide.A, CS14 antiserum stains Golgi in testis cell types, including spermatogonia (arrows), primary spermatocytes (thin arrow), and Leydig cells (arrowheads). B, C, Immunoadsorption of CS14 antiserum with peptide abolishes axonal staining in spinal cord tissue sections. Scale bars: A, 43 μm; B, C, 34 μm.
The anti-ST3GalV antisera CS2 and CS14 recognize a single polypeptide and cross-immunodeplete the same protein
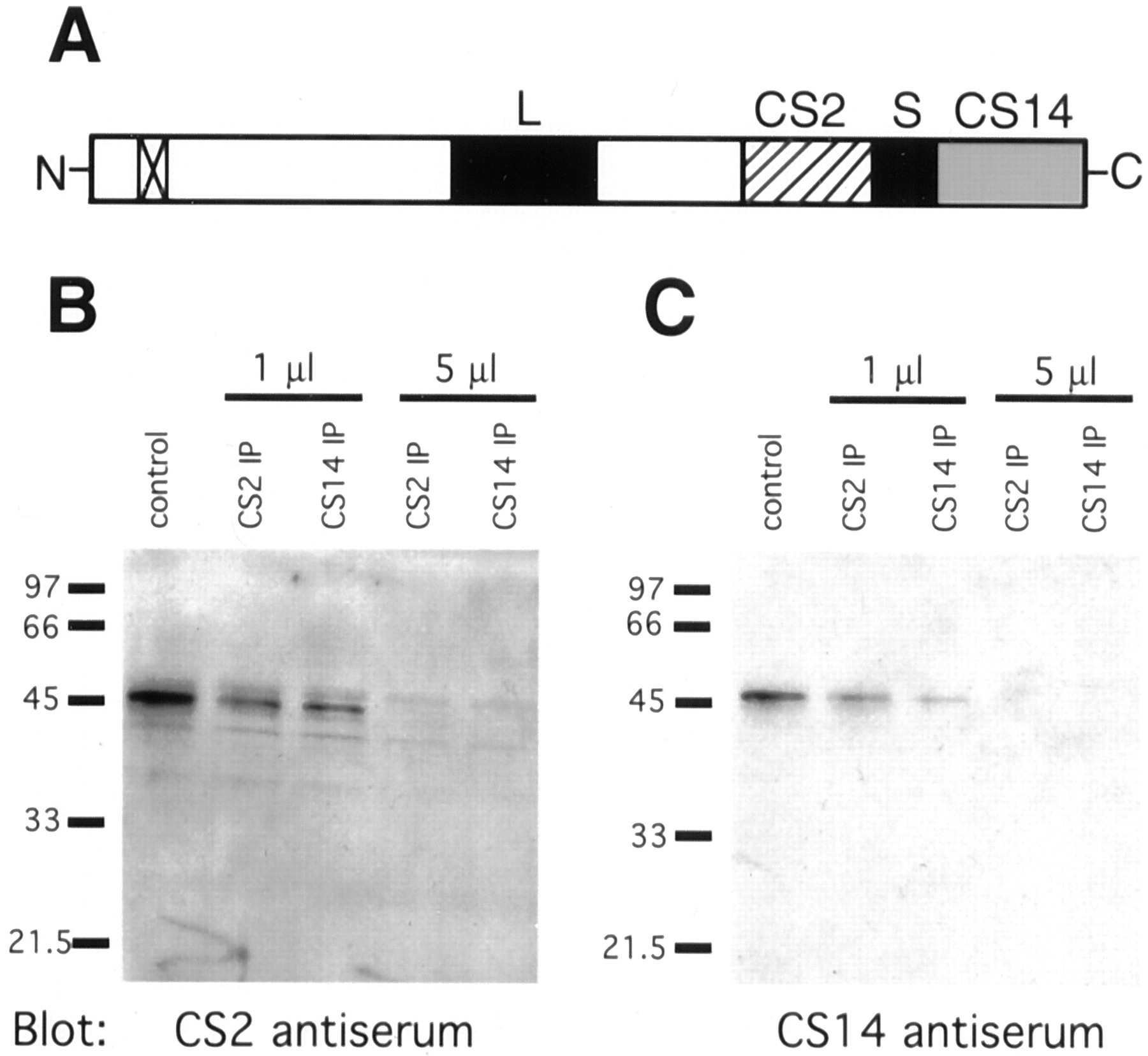
By Western blot analysis, CS2 and CS14 antisera recognize a single protein of 45 kDa in membrane preparations from mouse brain (Fig.5), liver, and testis (Stern et al., 2000) (data not shown), consistent with the predicted molecular weight of mouse ST3GalV (41,263 Da with between one and three N-linked oligosaccharides). Preimmune serum does not exhibit reactivity. Furthermore, CS2 antiserum depletes a band recognized by CS14 antiserum from tissue extracts (Fig.6B), and, likewise, CS14 antiserum depletes a band recognized by CS2 antiserum (Fig.6C). These results are consistent with both CS2 and CS14 antisera recognizing mST3GalV in adult mouse brain membrane preparations.

Both anti-mST3GalV antisera, CS2 and CS14, recognize a single protein band by Western blot. A single polypeptide of 45 kDa apparent molecular weight is recognized by both CS2 (A) and CS14 (B) antisera in Golgi-enriched membrane preparations from adult mouse brain.

Anti-mST3GalV antisera, CS2 and CS14, cross-immunodeplete mST3GalV from mouse brain extracts.A, ST3GalV schematic diagrams the peptides used as immunogens to generate CS2 (hatched box) and CS14 (stippled box) antisera. Also shown are the amino-terminal transmembrane domain (box withx) and the highly conserved L- and S-sialyl motifs (black boxes). B, C, Mouse brain extracts were incubated with Protein A Sepharose previously loaded with the indicated volume of antiserum (CS2 IP or CS14 IP). After centrifugation to pellet the Sepharose beads, aliquots of the supernatants were subjected to SDS-PAGE and Western blot analysis with the indicated antisera. Both antisera were able to quantitatively deplete the extract of ST3GalV, whether recognized by CS2 or CS14.
mST3GalV localizes to PC12 cell neurites after induction with NGF
In undifferentiated PC12 cells, mST3GalV localizes to the Golgi apparatus (Fig.7A,C,E,G). On induction with NGF, the Golgi increases in size (Rhodes et al., 1989), and a pool of ST3GalV appears within the extending PC12 cell neurite (Fig.7B,D,F,H). Myc-tagged ST3GalV is detected in the same subcellular distribution as endogenous ST3GalV (Fig. 7C,D); CS14 and anti-myc antisera staining colocalize throughout the Golgi and neurites in PC12 cells transfected to express myc-mST3GalV (Fig.7E,F). Because only a portion of the total ST3GalV population colocalizes with mannosidase II (a medial trans-Golgi marker), the pool of ST3GalV distributed into neurites is not a result of an aberrant elaboration of the Golgi apparatus in NGF-induced PC12 cells (Fig.7G,H) (Stieber et al., 1987; Rabouille et al., 1995).

ST3GalV localizes to neurites after induction with NGF in PC12 cells. A, C,E, G, ST3GalV localizes to Golgi in undifferentiated PC12 cells. B, D,F, H, After induction with 50 ng/ml NGF for 3 d, ST3GalV is also found in PC12 neurites. A,B, Endogenous ST3GalV stained with CS14 antiserum.C, D, Myc-tagged mST3GalV detected by anti-c-myc antiserum after transfection of the tagged construct into PC12. E, F, Colocalization of CS14 antiserum (green) and anti-c-myc antiserum (red). G, H, Colocalization of medial trans-Golgi marker, anti-mannosidase II (green), and anti-c-myc (red). Scale bar, 34 μm.
The non-Golgi pool of mST3GalV is not at the cell surface, nor does it colocalize with markers for known post-Golgi vesicular compartments
Staining of nonpermeabilized PC12 cells with anti-ST3GalV antisera failed to detect the presence of the enzyme at the cell surface. Both endogenous ST3GalV (visualized by CS14 antiserum) and myc-tagged ST3GalV (visualized by anti-myc antiserum) failed to stain the cell surface as defined by colocalization with wheat germ agglutinin (data not shown). Double-labeling experiments with CS14 antiserum and markers for the trans-Golgi network, endosomal membranes, or synaptic vesicles were performed to identify other subcellular compartments in which the axonal pool of ST3GalV might reside. Endogenous mST3GalV does not colocalize with TGN38, atrans-Golgi network marker (Luzio et al., 1990). Although partial colocalization in the differentiated PC12 cell suggests that ST3GalV passes through the trans-Golgi network (Fig.8B), the Golgi-associated pool of ST3GalV predominantly remains distinct from the trans-Golgi network. The distal ST3GalV pool is distributed away from the cell body and TGN38 (Fig. 8C).

ST3GalV does not colocalize withtrans-Golgi network, endosomal, or synaptic vesicle markers. A–C, Endogenous ST3GalV (green) and TGN38 (red) maintain distinct sub-Golgi compartmentalization in both undifferentiated (A) and differentiated PC12 cells (B, C). B, Some partial colocalization is observed, suggesting that ST3GalV passes through thetrans-Golgi network as it moves to a more distal neurite location. D–F, ST3GalV (green) also fails to colocalize with endosomes marked by transferrin receptor (red). D, In undifferentiated PC12 cells, the anti-transferrin receptor staining surrounds the CS14 antiserum Golgi staining. E, In the fully differentiated PC12 cell, both CS14 and transferrin receptor staining become more diffuse throughout the cell body, but both maintain distinct subcellular locations. F, CS14 antiserum stains ST3GalV along the neurite process but is not located at the neurite terminus that stains red with anti-transferrin receptor antiserum (arrow, neurite terminus). G–I, Colocalization of ST3GalV (green) with the synaptic vesicle marker, synaptobrevin (red).G, H, Either without NGF treatment or in the cell body of the fully differentiated PC12 cell, there is some partial colocalization between ST3GalV and synaptobrevin, reflecting the processing of synaptobrevin. H, I, At the nerve terminus, synaptobrevin and ST3GalV do not colocalize. Scale bar, 34 μm.
Between the trans-Golgi network and the cell surface, two potential post-Golgi locations for ST3GalV are the endosome and the synaptic vesicles at the nerve terminus. Immunostaining with CS14 antiserum and an anti-transferrin receptor monoclonal antibody demonstrates that ST3GalV is not localized to the endosome (Fig.8D,E) (Mellman, 1996). Both markers remain in distinct cellular compartments throughout differentiation. In PC12 cell neurites, for instance, ST3GalV is present for most of the length of the extension, whereas transferrin receptor is clustered at the neurite terminus (Fig. 8F, arrow). Double immunostaining with CS14 and a synaptic vesicle marker (anti-synaptobrevin) shows that ST3GalV is not directly associated with this pool of recycling membrane either (Fig.8H,I) (Baumert et al., 1989;Chilcote et al., 1995). Synaptobrevin and CS14 staining are complementary; CS14 staining was never observed extending into the neurite tip. Some colocalization between CS14 and synaptobrevin is observed within the cell body and is likely attributable to synaptobrevin staining in the Golgi during processing (Fig.8G,H).
Expression of mST3GalV in PC12 cells decreases cell survival or rate of division
Quantitation of the number of transfected cells in a treated PC12 cell population revealed that significantly fewer cells were found expressing plasmid containing mST3GalV than control plasmid (GFP plus pcDNA). In undifferentiated PC12 cultures (0 ng/ml NGF), there is at least a threefold difference between the number of cells expressing control plasmid and those expressing ST3GalV-encoding plasmids (p ≤ 0.0015) (Fig.9). In the fully differentiated PC12 cell population, the effect of ST3GalV overexpression is less dramatic, with a twofold or less difference between ST3GalV and control plasmids (p ≤ 0.025).

Expression of ST3GalV reduces cell survival or rate of division. At 0 ng/ml NGF, at least threefold fewer PC12 cells express ST3GalV-containing plasmid than control plasmid (p ≥ 0.0015). At 50 ng/ml NGF, the effect of overexpressing ST3GalV is less dramatic. Solid black bars, Cells cotransfected with control plasmid, pcDNA3.1, and GFP encoding plasmids (n ≥ 3); hatched bars, cells cotransfected with mST3GalV and GFP encoding plasmids (n ≥ 4); stippled bars, cells transfected with C-terminal myc-tagged mST3GalV (n = 4).
DISCUSSION
By raising antisera to nonoverlapping mST3GalV peptides, we have generated immunological probes that define two distinct subcellular pools of this ganglioside synthetic enzyme. Previously, we reported the Golgi localization of ST3GalV in adult mouse tissue sections using an antiserum designated CS2 (Stern et al., 2000). Localization of this pool of ST3GalV is consistent with its synthetic function, the generation of GM3 ganglioside from a lactosylceramide acceptor (Ishii et al., 1998; Kono et al., 1998; Fukumoto et al., 1999). In this study, however, we describe a non-Golgi localization for ST3GalV identified with a second antiserum, designated CS14. The non-Golgi-associated pool of the enzyme is only observed in neurons in which CS14 staining is seen along axonal processes in cortical, cerebellar, brainstem, and spinal cord sections. Similarly, in primary cultures of rat hippocampal neurons, CS14 immunoreactivity is distributed in an axon-like (MAP2-negative) process.
CS14 also reveals a pool of non-Golgi-localized ST3GalV in differentiated PC12 cells. Although ST3GalV is localized to the Golgi in undifferentiated PC12 cells, the enzyme distributes into extending neurites after NGF treatment. Whether the distribution of endogenous enzyme is visualized by CS14 antiserum or by epitope-tagged mST3GalV in transfected cells, staining exhibits a punctate appearance consistent with localization to a vesicular structure. These vesicles may transiently pass through the trans-Golgi network, but colocalization with compartment-specific markers demonstrates that ST3GalV does not accumulate in the trans-Golgi network, in endosomes, or in synaptic vesicles, nor is staining detected at the cell surface.
Non-Golgi distributions have been described for other glycosyltransferases. In particular, a glycoprotein-directed β1,4-galactosyltransferase has been detected at the cell surface, and several glycosyltransferases are secreted as a result of cleavage from their membrane anchor within the secretory pathway (Nelson et al., 1973; Shur, 1989; Jaskiewicz et al., 1996a; Allende et al., 1997; Cho and Cummings, 1997; Zhu et al., 1998; Kitazume-Kawaguchi et al., 1999). Although the expression of epitope-tagged glycosyltransferases in cultured cells has furthered greatly our understanding of the subcellular localization of other glycan synthetic enzymes, this study is the first to investigate the endogenous distribution of a ganglioside-directed glycosyltransferase in neurons located within their normal tissue environment. As additional immunological probes directed against other glycosyltransferases become available, the commonality of non-Golgi enzyme localization can be evaluated. Neurons, with their highly polarized and spatially distributed morphology, provide unique opportunities to visualize the distribution and transport of such molecules.
The specificity of the CS14 antiserum for ST3GalV is demonstrated by several findings. First, both CS2 and CS14 antisera stain the Golgi apparatus in some non-neural cell types. Second, both antisera recognize a single, reduced, denatured polypeptide of the same apparent molecular weight. Third, by immunoprecipitation from brain extracts, either antiserum can deplete the epitope recognized by the other antibody. And finally, a myc-tagged form of ST3GalV expressed in PC12 cells is found both in the Golgi and in neurites, independently verifying that the enzyme possesses targeting information for localization beyond the Golgi.
The exclusion of the CS14 epitope from the Golgi apparatus of neurons in tissue is of particular interest because CS14 antiserum does stain the Golgi in other cell types. It is possible that both neural and non-neural cells produce a mixed population of ST3GalV, but neurons effectively segregate CS14-recognized forms from CS2-recognized forms. Alternatively, a neuron-specific structural or post-translational modification may simultaneously reveal the CS14 epitope while masking the CS2 epitope in the axonal pool of ST3GalV. Among the leading candidates for such a structural modification are alternate disulfide bond formation, differential glycosylation, and phosphorylation, all of which have the potential to mask or generate a peptide epitope. Multiple mature disulfide-bonded and phosphorylated forms of other glycosyltransferases, most notably of another sialyltranferase, have been suggested to modulate catalytic activity and participate in Golgi retention (Jaskiewicz et al., 1996b; Ma and Colley, 1996; Ma et al., 1997, 1999; Zhu et al., 1997; Chen et al., 2000). However, in preliminary experiments, treatment of PC12 cells with DTT before fixation had no effect on anti-ST3GalV staining. Although the diversity of ST3GalV glycoforms remains to be determined, the sharptrans-Golgi boundary that delimits CS2 from CS14 staining in nervous tissue is most consistent with a processing event, such as sialylation, inducing differential epitope display (Nagai et al., 1997;Martina et al., 1998). Regardless of the nature of the structural difference, we predict that changes in ST3GalV conformation or modification generate differences in compartmental localization and retention.
The endoplasmic reticulum and Golgi apparatus have generally been considered the center for glycosphingolipid synthesis. Therefore, the identification of an axonal distribution for ST3GalV, the activity of which is required early in ganglioside biosynthesis, implies either that axonal ST3GalV subserves a nonenzymatic function or that ganglioside biosynthesis, like phospholipid biosynthesis, is more broadly distributed than previously thought (Vance et al., 1991, 1994;de Chaves et al., 1995; Futerman and Banker, 1996). Efficient recycling and repair of axolemmal glycosphingolipids might partially rely on a pool of synthetic capacity located closer to the sight of insertion than the Golgi apparatus.
Ganglioside recycling occurs via endocytosis, with internalized lipid assuming one of three fates: lysosomal degradation, return to the cell surface, or glycan remodeling before return to the cell surface (Trinchera et al., 1990; Riboni and Tettamanti, 1991; Riboni et al., 1991, 1997; Sofer et al., 1996). However, the subcellular distribution and kinetics of glycosphingolipid recycling in neurons are poorly defined. Attempts to measure the bulk turnover rate of gangliosides in cultured cells have yielded wide variations in lipid half-lives (Suzuki, 1970; Van Meer, 1989; Riboni et al., 1996, 1997; Sofer et al., 1996), but studies of plasma membrane turnover, growth cone retraction, or extension after axotomy imply the existence of a rapid mechanism, such as local synthesis of ganglioside, that would liberate axonal glycosphingolipid recycling from complete reliance on anterograde axonal transport (Bray et al., 1978; Wessells et al., 1978; Steinman et al., 1983; Ashery et al., 1996; Diefenbach et al., 1999).
Although the Golgi is still predicted to be the major site of ganglioside synthesis (van Echten and Sandoff, 1993), axonal compartments containing ST3GalV are poised for direct reglycosylation of glycosphingolipid. Previous reports (Schengrund and Rosenberg, 1970;Tettamanti et al., 1972; Schengrund and Nelson, 1975; Chigorno et al., 1986; Miyagi et al., 1990; Varki, 1993; Riboni et al., 1997; Monti et al., 2000) have described the vital importance of cell surface sialylation and have identified sialidase activity at the plasma membrane of multiple cell types, including neurons. To maintain lipid sialylation levels, cells must either synthesize and transport new ganglioside or locally sialylate the pool of desialylated substrates. Candidate lipid-directed sialyltransferase activities have been biochemically identified in nerve terminal preparations, but the interpretation of such subcellular fractionation studies has been limited by the difficulty inherent in preparing material free of Golgi membrane contamination (Schengrund and Nelson, 1975; Ng and Dain, 1977;Preti et al., 1980; Durrie et al., 1987, 1988). Immunostaining with CS14 antiserum provides independent evidence that a sialyltransferase is distributed in axons and nerve termini, but primarily in sub-plasmalemmal vesicles.
Comprehensive local ganglioside synthesis, sialylation, or remodeling requires that additional transferase activities, transporters, and substrates be present in the same compartments. For ST3GalV to function within the axon or nerve terminal as a sialyltransferase, both acceptor (lactosylceramide) and donor sugar-nucleotide (CMP-NeuAc) are necessary. CMP-NeuAc is synthesized in the nucleus (Kean, 1991). It is currently unknown whether CMP-NeuAc or the required antiport machinery for lumenal transport also exists in axons and, if so, whether they colocalize with sialyltransferase. Perhaps vesicles containing multiple glycolipid biosynthetic components are segregated in the Golgi and transported to their axonal destination as a preloaded ganglioside synthesome. A complete analysis of the localization of all ganglioside-directed glycosyltransferases and sugar-nucleotide transporters must await the generation of appropriate antibodies and epitope-tagged constructs but promises to provide new insight into the regulation and importance of membrane turnover in neurons.
Gangliosides have been implicated in the modulation of signaling through the epidermal growth factor (EGF), NGF (trkA), and PDGF receptors (Igarashi et al., 1989; Hakomori and Igarashi, 1993). The relevant lipid modulators range from a single glycolipid species (lyso-GM3 downregulates EGF receptor signaling) to the overall cellular glycolipid profile (Bremer et al., 1984; Nores et al., 1988; Hakomori and Igarashi, 1993; Zhou et al., 1994; Yates et al., 1995; Rebbaa et al., 1996; Sachinidis et al., 1996). In PC12 cells, overexpression of GD3 synthase, an α2,8-sialyltransferase necessary for synthesis of complex (highly sialylated) gangliosides, leads to depletion of gangliosides like GM3 and GM1 but increases the presentation of the complex gangliosides GD1b and GT1b at the surface of PC12 cells (Fukumoto et al., 2000). In parallel with the shift in ganglioside sialylation profile, overexpressing PC12 cells no longer extend neurites in response to NGF; rather the cells continue to divide as undifferentiated cells (Fukumoto et al., 2000). However, when we overexpress ST3GalV (GM3 synthase) in PC12 cells, we observe no effect on neurite outgrowth but a significantly smaller than expected population of transfected cells. Although this may suggest that transfection with ST3GalV leads to higher levels of cell death, it is more likely that cell division is slowed because transfected cells are fully capable of extending neurites and exhibit no outward signs of impending demise. A model suggesting that less sialylated gangliosides decrease cell division and complex gangliosides increase cell division is a gross oversimplification, but it is clear that cells must regulate their ganglioside expression to optimize environmental responsiveness.
Finally, it is possible that the non-Golgi pool of ST3GalV has no synthetic capacity, either because it is structurally altered or because other components necessary for enzyme activity are not present (CMP-NeuAc, etc.). However, if the non-Golgi pool of the enzyme retains substrate (lactosylceramide)-binding activity, it may function as a lectin, chaperoning lipids through the secretory pathway or participating in the formation or stabilization of membrane microdomains (Fiedler et al., 1994; Fiedler and Simons, 1996; Simons and Ikonen, 1997; Chigorno et al., 2000; Prinetti et al., 2000). Such a binding activity could also impact the efficacy of transmembrane signaling by sequestering or presenting glycosphingolipid in the appropriate context. Additional characterization of ST3GalV function, either through immunological perturbation or the transgenic generation of null mutants, should provide important insights into ganglioside function in the nervous system.
Footnotes
This work was supported by National Institutes of Health–National Institute on Child Health and Human Development Grant HD33878, by a Basil O'Connor Award from the March of Dimes, and by The Patrick and Catherine Weldon Donaghue Foundation (all to M.T.). C.S. received support from National Institutes of Health–National Institute of General Medical Services Grant GM07223. We thank members of the Pietro DeCamilli and Ira Mellman laboratories for immunological probes and neuronal primary cultures. We also thank Katherine Howell for immunological probes.
Correspondence should be addressed to Michael Tiemeyer, Glyko, Inc., 11 Pimentel Court, Novato, CA 94949. E-mail:mtiemeyer{at}glyko.com.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}