

Abstract
Cyclin-dependent kinase 5 (Cdk5) is a critical regulator of neuronal migration in the developing CNS, and recent studies have revealed a role for Cdk5 in synaptogenesis and regulation of synaptic transmission. Deregulation of Cdk5 has been linked to the pathology of neurodegenerative diseases such as Alzheimer's disease. Activation of Cdk5 requires its association with a regulatory subunit, and two Cdk5 activators, p35 and p39, have been identified. To gain further insight into the functions of Cdk5, we identified proteins that interact with p39 in a yeast two-hybrid screen. In this study we report that α-actinin-1 and the α-subunit of Ca2+/calmodulin-dependent protein kinase II (CaMKIIα), two proteins localized at the postsynaptic density, interact with Cdk5 via their association with p35 and p39. CaMKIIα and α-actinin-1 bind to distinct regions of p35 and p39 and also can interact with each other. The association of CaMKIIα and α-actinin-1 to the Cdk5 activators, as well as to each other, is stimulated by calcium. Further, the activation of glutamate receptors increases the association of p35 and p39 with CaMKIIα, and the inhibition of CaMKII activation diminishes this effect. The glutamate-mediated increase in association of p35 and CaMKIIα is mediated in large part by NMDA receptors, suggesting that cross talk between the Cdk5 and CaMKII signal transduction pathways may be a component of the complex molecular mechanisms contributing to synaptic plasticity, memory, and learning.
Cyclin-dependent kinase 5 (Cdk5) is a proline-directed serine/threonine kinase that has emerged as a crucial regulator of neuronal migration in the developing brain (Dhavan and Tsai, 2001). Cdk5-deficient mice die at birth and display widespread disruptions in neuronal layering of many brain structures, indicating impairment in directing neuronal positioning (Ohshima et al., 1996). Activation of Cdk5 requires binding to one of two regulatory subunits, p35 or p39 (Lew et al., 1994; Tsai et al., 1994;Tang et al., 1995). The phenotype of the p35/p39 double knock-out mice is identical to the Cdk5-deficient mice, suggesting that together the two activators of Cdk5 are necessary and sufficient for Cdk5 function (Ko et al., 2001). Over 20 functionally diverse proteins involved in cytoskeleton dynamics, cell adhesion, transport, and membrane trafficking have been identified as Cdk5 substrates, elucidating the molecular mechanisms of Cdk5 function (Dhavan and Tsai, 2001; Smith et al., 2001).
Recently, synaptic functions of Cdk5 have come to light. Cdk5 and its activators are localized at synapses and are present in presynaptic and postsynaptic subcellular fractions, including postsynaptic density (PSD) fractions (Humbert et al., 2000b; Niethammer et al., 2000). Cdk5 and p35 also are enriched at the neuromuscular junction, where Cdk5 activity is required for the neuregulin-induced transcription of acetylcholine receptors (Fu et al., 2001). Cdk5-mediated phosphorylation of Synapsin 1, Munc18, Amphiphysin 1, and the α-subunit (1A) of P/Q-type voltage-dependent calcium (Ca2+) channels has revealed a role for this kinase in synaptic vesicle trafficking and neurotransmitter release (Dhavan and Tsai, 2001; Tomizawa et al., 2002). Cdk5 also appears to play a role in postsynaptic transmission, and phosphorylation of DARPP32 by Cdk5 modulates dopamine signaling in striatal neurons (Bibb et al., 1999). Stimulation of metabotropic glutamate receptors (mGluRs) in neostriatal slices causes a transient increase in Cdk5 activity, and Cdk5 activity has been implicated in the mGluR-mediated enhancement of voltage-dependent Ca2+ currents (Liu et al., 2001). Finally, Cdk5 phosphorylates the NR2A subunit of the NMDA receptor, and pharmacological inhibition of Cdk5 modulates NMDA receptor conductance (Li et al., 2001).
To gain further insight into the functions of Cdk5, we identified proteins that interact with its activator p39 in a yeast two-hybrid screen. Two of the proteins identified in this screen, α-actinin-1 and Ca2+/calmodulin-dependent protein kinase II (CaMKIIα), are enriched at the PSD and interact with the NMDA receptor (Wyszynski et al., 1997; Walikonis et al., 2000; Lisman et al., 2002). We report that α-actinin-1 and CaMKIIα interact with p35, p39, Cdk5, and each other in a Ca2+-dependent manner. Furthermore, the activation of glutamate receptors increases the association of p35 and p39 with CaMKIIα, suggesting that synaptic activity may influence the cross talk between Cdk5 and CaMKIIα.
MATERIALS AND METHODS
Constructs. Mouse p39 cDNA was amplified by PCR, and the fragment was inserted into the SalI and NotI sites of the pPC97 vector (pDBLeu; Invitrogen, Carlsbad, CA) for expression as a GAL4 DNA binding domain fusion protein (p39-pPC97). Identical cloning strategy was used to generate p35-pPC97, p25-pPC97 (residues 99–307 of human p35), p29-pPC97 (residues 115–367 of mouse p39), p10-pPC97 (residues 1–98 of human p35), and p19-pPC97 (residues 1–113 of mouse p39).
The cDNAs encoding the N-terminal (residues 1–316) and C-terminal (residues 314–478) domains of CaMKIIα were generated by PCR from a CaMKIIα-containing plasmid (a gift from Dr. H. Schulman, Stanford University, CA) and subcloned into the SalI andNotI sites of pPC97 and pPC86 vectors (Invitrogen) for expression as GAL4 DNA binding or activation domain fusion proteins, respectively. A cDNA encoding full-length α-actinin-1 (I.M.A.G.E. Consortium) was used as a template to generate truncation mutants of α-actinin-1 by PCR (residues are indicated in Fig. 1) and cloned into the SalI and NotI sites of pPC97 and pPC86 vectors. The cDNAs encoding p39, p29, p35, p25, Cdk5, and the α-actinin-1 cDNA clone identified in the screen were subcloned into the SalI and NotI sites of PGEX4T2 (Amersham Pharmacia, Uppsala, Sweden) for the expression of GST fusion proteins.
Yeast two-hybrid screen. A yeast two-hybrid screen was performed in yeast strain MaV203 containing HIS3,LacZ, and URA reporter genes under the control of the GAL4-activating sequences as described previously (Vidal et al., 1996a,b; Niethammer and Sheng, 1998). Briefly, >10 million cotransformants of p39-pPC97 and a human fetal (third trimester) brain cDNA library (Invitrogen) were screened for interacting proteins by growth on synthetic dropout Leu−Trp− His−plates containing 20 mm 3-amino-1,2,4-triazole. Positive colonies were tested further for expression of theLacZ reporter gene by β-galactosidase assay (Clontech yeast protocol handbook, Palo Alto, CA) and for induction of theURA reporter gene that allowed growth on Leu− Trp−Ura− plates and conferred sensitivity to 5-fluoroorotic acid (Vidal et al., 1996a,b). Plasmids were isolated from positive yeast colonies as described previously (Niethammer and Sheng, 1998), and the interacting proteins were identified by sequencing. The p35 yeast two-hybrid screen has been described previously (Kwon et al., 2000).
Mapping domains of interaction. pPC86-α-actinin-1 or pPC86-CaMKIIα identified in the screen was cotransformed into MaV203 yeast strain with pPC97-p39, p35-pPC97, p29-pPC97, p25-pPC97, p19-pPC97, or p10-pPC97. β-Galactosidase assay was used to compare the strength of interaction among the proteins encoded by cotransformed cDNA. We judged colonies that turned blue within 1 hr to contain strongly interacting proteins (++++). The +++, ++, and + symbols were used to designate colonies that turned blue within 1–4 hr, 4–8 hr, and 8–24 hr, respectively. The − symbol was used to indicate colonies that did not turn blue within 24 hr. To map the binding region of CaMKIIα, we cotransformed the CaMKIIα truncation mutants in pPC86 vector with p39-pPC97, p35-pPC97, or α-actinin-1-pPC97. Deletion constructs of α-actinin-1 in pPC86 vector were cotransformed with p39-pPC97, p35-pPC97, or CaMKIIα-pPC97 and were assayed as described above.
GST pulldown assay. GST fusion proteins were expressed, purified, and cross-linked to glutathione-Sepharose 4B (Amersham Pharmacia) as described previously (Tarricone et al., 2001). Adult rat brain was lysed in RIPA buffer (150 mm NaCl, 50 mm Tris, pH 8.0, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with protease and phosphatase inhibitors [containing (in mm) 1 PMSF, 1 DTT, 1 Na3VO4, 5 NaF, 2 sodium pyrophosphate, and 50 β-glycerophosphate plus 1 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 nm okadaic acid] in a homogenizing Dounce and then was spun at 13,000 rpm for 15 min at 4°C. Two milligrams of lysate were incubated with 5 μg of cross-linked GST fusion proteins in RIPA buffer for 2 hr at 4°C. The beads were washed three times with RIPA buffer, and the associated proteins were resolved by SDS-PAGE and analyzed by immunoblotting.
Antibodies for immunoblotting. For Western blot analysis we used an affinity-purified p35 polyclonal antibody (1:1000) (Ko et al., 2001), an affinity-purified p39 polyclonal antibody (1:1000) (Humbert et al., 2000a), a monoclonal Cdk5 antibody DC-17 (1:10) (Tsai et al., 1993), a monoclonal CaMKIIα antibody (1:2000; Chemicon, Temecula, CA), a monoclonal α-actinin-1 antibody (1:1000; Sigma-Aldrich, St. Louis, MO), an affinity-purified polyclonal Lis1 antibody (1:1000) (Smith et al., 2000), or a polyclonal actin antibody (1:1000; Sigma-Aldrich).
Coimmunoprecipitation. Two milligrams of adult rat brain lysate (described above) were incubated with 1 μg of p35, p39, Cdk5 (C8; Santa Cruz Biotechnology, Santa Cruz, CA), or HA (Santa Cruz Biotechnology) polyclonal antibodies or α-actinin-1 or HA (Santa Cruz Biotechnology) monoclonal antibodies for 1 hr at 4°C in RIPA buffer. Then 100 μl of a 10% slurry of protein A-Sepharose (Amersham Pharmacia), or IgM-Sepharose (Sigma-Aldrich) for α-actinin-1 IPs, was added for 1 hr at 4°C. The beads were washed three times with RIPA buffer, and bound proteins were analyzed by SDS-PAGE and Western blotting. In some cases the indicated concentration of CaCl2, MgCl2, EDTA, or EGTA was added to adult rat brain lysate and incubated for 20 min before coimmunoprecipitation.
Immunostaining. Dissociated hippocampal neurons from embryonic day 18 (E18) rat brains were plated on coverslips coated with poly-l-lysine (Sigma-Aldrich) at a density of 5 × 104/coverslips and maintained in culture in Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) and l-glutamine (Sigma-Aldrich) as described previously (Brewer et al., 1993) for 3 weeks. Neurons were fixed in 4% paraformaldehyde/5% sucrose in PBS for 30 min at room temperature (RT), followed by 5 min in cold methanol at –20°C. Cells were permeabilized in blocking buffer [10% normal goat serum (NGS), 0.2% Triton X-100 in PBS] for 1 hr at RT. Primary antibodies in staining buffer (3% NGS, 0.1% Triton X-100 in PBS) were applied for 16 hr at 4°C. The CaMKIIα, p35, p39, Cdk5 (C8; Santa Cruz Biotechnology), and α-actinin-1 antibodies described above were used at a dilution of 1:250 for immunostaining. Oregon green-conjugated anti-rabbit and Texas Red-conjugated anti-mouse secondary antibodies (Molecular Probes, Eugene, OR) were used at a dilution of 1:500 in staining buffer for 1 hr at RT. For triple staining, a Texas Red-conjugated IgM-specific anti-mouse secondary antibody was used with Oregon green-conjugated anti-rabbit and Cy5-conjugated IgG-specific anti-mouse secondary antibodies (Molecular Probes) to visualize α-actinin-1, p35 (or p39 or Cdk5), and CaMKIIα, respectively. All images were acquired by using a 60× or 100× oil-immersion objective on a Nikon inverted microscope linked to a Delta vision deconvolution imaging system (Applied Precision, Issaquah, WA). Images of double- and triple-labeled cells were combined and colorized with Adobe Photoshop software.
Glutamate treatment of hippocampal neurons. As described above, 3 × 106 hippocampal neurons were plated on poly-l-lysine-coated 10 cm plates. After 2 weeks in culture the medium was replaced with KRH buffer [containing (in mm) 128 NaCl, 5 KCl, 1 NaH2PO4, 2.7 CaCl2, 1.2 MgSO4, 10 glucose, and 20 HEPES, pH 7.4] for 30 min at 37°C. Neurons were treated with 100 μm glutamate/10 μm glycine in Mg2+-free KRH buffer for 5 min and then were Dounce homogenized in ice-cold sucrose buffer [0.32 msucrose plus (in mm) 4 HEPES, pH 7.4, 1 MgCl2, 1 NaHCO3, and 0.5 CaCl2] containing all of the protease and phosphatase inhibitors listed above. The homogenized cells were centrifuged at 1000 × g for 10 min. The resulting supernatant was centrifuged at 3000 × g for 15 min. The pellet was lysed in RIPA buffer (described above) by 10 strokes in a glass-glass Dounce homogenizer and centrifuged at 13,000 rpm for 15 min at 4°C; then the supernatant was collected. One milligram of protein was used per immunoprecipitation, as described above.
Treatment of hippocampal slices. Hippocampal slices were prepared from 3-week-old Sprague Dawley rats with a McIlwain tissue chopper and placed in KRH buffer aerated with 95% O2/5% CO2. After equilibration at 37°C for 30 min the slices were stimulated in Mg2+-free KRH buffer for 5 min with 100 μm glutamate/10 μm glycine, 100 μm NMDA/10 μm glycine, or 100 μm AMPA. In some cases the slices were pretreated with 10 μm each KN-62, KN-93, KN-92, and MK801, 1 μm H-89, 100 μm CNQX, 100 μmAIDA, or 5 mm EDTA for 10 min before the stimulations. Treatments were stopped on ice, and the drug was removed. Slices were homogenized in ice-cold sucrose buffer. Lysates were prepared and immunoprecipitations were performed as described above.
Cdk5 kinase activity. One milligram of neuronal lysate was immunoprecipitated with p35 antibody as described above. The Sepharose-bound complexes were washed two times with kinase buffer (20 mm MOPS, pH 7.2, 5 mmMgCl2). Kinase buffer (30 μl) containing 500 μm Histone H1 peptide (PKTPAKAKKL), 100 μmATP, and 5 μCi [γ-32P]ATP was incubated with the immunocomplexes for 30 min at RT. Samples were centrifuged at 13,000 rpm for 2 min, and 10 μl of the supernatant was spotted on phosphocellulose disc paper. The discs were washed four times with 0.3% phosphoric acid and counted in 10 ml of scintillation fluid.
CaMKII activity from hippocampal slices. CaMKII-dependent activity was measured by using a Ca2+/calmodulin-dependent protein kinase assay system (Invitrogen) according to the manufacturer's instructions. Briefly, slices were Dounce homogenized in cold extraction buffer [containing (in mm) 20 PIPES, pH 7.0, 0.5 EGTA, 1 EDTA, 10 sodium pyrophosphate, 2 DTT, and 0.4 ammonium molydate plus 10 μg/ml leupeptin and 0.1% Triton X-100]. Then 20 μl of the extract was incubated in a buffer containing (in mm) 50 PIPES, pH 7.0, 20 MgCl2, 0.05 ATP, and 15 μm autocamtide-3 plus 200 mg/ml BSA and 50 μCi/ml [γ-32P]ATP in a final volume of 50 μl either in the presence of 1 mm EGTA (Ca2+/CaM-independent activity) or 1 mm CaCl2/2.4 μmcalmodulin (total activity). The reaction was incubated for 2 min at 30°C and stopped by the addition of 10 μl of ice-cold 20% TCA. Reactions were centrifuged at 13,000 rpm for 2 min. Then 30 μl of supernatant was spotted on phosphocellulose discs and counted as described above. Autonomous CaMKII activity is expressed as the percentage of total activity that is Ca2+/CaM-independent.
Statistic analysis. The Western blot images on film were digitized by a calibrated ImageScanner scanner and LabScan 3.0 software (Amersham Pharmacia). The volumes of image spots were quantified by using the ImageMaster 3 software (Amersham Pharmacia). Data were analyzed by paired Student's t test or by one-way ANOVA, followed by Newman–Keuls multiple comparison tests that used GraphPad Prism (version 3.02; GraphPad, San Diego, CA). Differences were considered significant at p < 0.05.
RESULTS
p39 interacts with α-actinin-1 and CaMKIIα in a yeast two-hybrid screen
Full-length p39 was used as bait in a yeast two-hybrid screen of a human brain cDNA library. Two clones identified in this screen were identical cDNA inserts encoding the full-length α-subunit of CaMKII (Fig. 1A). Both CaMKIIα cDNAs contained 120 bp of 5′-untranslated region (UTR), the 1434 bp coding region, and a 1495 bp 3′-UTR.

CaMKIIα and α-actinin-1 interact with the Cdk5 activators p39 and p35, and with each other, in a yeast two-hybrid system. A, A schematic of full-length CaMKIIα and α-actinin-1 proteins is presented above a diagram of the protein encoded by the cDNA clones identified in the screen. Thenumbers in parentheses indicate the number of copies of CaMKIIα and α-actinin-1 cDNAs isolated in the screen. CaMKIIα contains an N-terminal kinase domain, a Ca2+/calmodulin binding domain (CBD), and an association domain (AD). Both CaMKIIα cDNAs identified in the screen contain the entire open reading frame (amino acid residues 1–478). α-Actinin-1 is composed of an N-terminal actin binding domain, a central region containing four spectrin repeats (I–IV), and a C terminus with two EF-hand motifs. The cDNA identified in the screen encodes amino acid residues 363–892 of α-actinin-1. B, cDNAs encoding full-length p39 (residues 1–369) and p35 (residues 1–307) or N- and C-terminal truncation mutants of p39 and p35 were tested for association with the CaMKIIα and α-actinin-1 clones identified in the yeast two-hybrid screen. Strength of an interaction is indicated by ++++, +++, ++, or + on the basis of β-galactosidase assays, whereas – denotes no interaction (see Materials and Methods). C, Full-length or truncation mutants of CaMKIIα were tested for their ability to interact with full-length p35 and p39 in a yeast two-hybrid β-galactosidase assay. A schematic of the proteins encoded by the CaMKIIα cDNAs is indicated. D, Full-length or truncation mutants of α-actinin-1 were tested for their interaction with p35 and p39. A schematic representation of the proteins encoded by α-actinin-1 cDNAs is presented. The N terminus contains an actin binding domain (ABD); the central region contains four spectrin-like repeats (SRI–IV). The C terminus contains a pair of EF-hand motifs (EF-H) depicted by two vertical lines. E, Full-length or truncation mutants of CaMKII were tested for their ability to interact with full-length α-actinin-1 in a yeast two-hybrid assay.F, Full-length or truncation mutants of α-actinin-1 were tested for their ability to interact with full-length CaMKIIα in the yeast two-hybrid assay.
An additional cDNA clone identified in the screen represented the C-terminal two-thirds of human α-actinin-1 (also called nonskeletal muscle α-actinin). The α-actinins are a family of closely related proteins that are composed of three domains: an N-terminal actin binding domain, a central region composed of four spectrin-like repeats, and a C-terminal region containing two Ca2+-binding EF-hand motifs (Blanchard et al., 1989). Our cDNA insert began at nucleotide 1087 (amino acid 363) and extended for 1.8 kb. The encoded protein contains the last three spectrin-like repeats and continues to the C-terminal end of the protein (Fig. 1A).
α-Actinin-1 and CaMKIIα bind distinct regions of p39 and p35
Seven cDNA clones of α-actinin-1 also had been identified previously in a yeast two-hybrid screen by using full-length p35 as bait and an embryonic mouse (E12.5) cDNA library (M. Nikolic and L.-H. Tsai, unpublished observations). The α-actinin-1 protein encoded by all seven clones contained the last three spectrin-like repeats and continued to the C-terminal end of the protein. Moreover, p35 is 46% identical to p39, with >70% identity in the C-terminal Cdk5 binding region (Tang et al., 1995). p35 could bind to α-actinin-1, and with greater affinity than p39, in the yeast two-hybrid assay (Fig.1B). p35 also could interact with CaMKIIα in the yeast two-hybrid system, although the association was weaker than with p39 (Fig. 1B).
The domains of p35 and p39 required for association with full-length α-actinin-1 and CaMKIIα were investigated in a yeast two-hybrid system by using N- and C-terminal truncation mutants of p35 and p39. The N terminus of p35 and p39 contains a myristoylation signal and is thought to be critical for the membrane localization of the two activators (Patrick et al., 1999; Patzke and Tsai, 2002). The C-terminal two-thirds of p35 and p39 adopt a cyclin-box tertiary structure that is sufficient for binding and activating Cdk5 (Qi et al., 1995; Tarricone et al., 2001). The N-terminal regions of p39 and p35 could interact with α-actinin-1 with equivalent or greater efficiency than the respective full-length proteins, whereas the Cdk5 binding domains did not display any association with α-actinin-1 in the yeast two-hybrid assay (Fig. 1B). This suggests that the N terminus of p35 and p39 is necessary and sufficient for association with α-actinin-1. In contrast, CaMKIIα could bind to the C terminus of p35 and p39 but did not interact with the N-terminal domains of these two proteins (Fig. 1B). Thus CaMKIIα and α-actinin-1 bind to different regions of both p35 and p39.
The N terminus of CaMKIIα is required for the association with p35 and p39
All CaMKII subunits contain an N-terminal catalytic domain, a central regulatory region, and a C-terminal association domain (Fig.1A) (Soderling et al., 2001). The regulatory region contains an overlapping autoinhibitory domain and a Ca2+/calmodulin binding motif, whereas individual subunits interact via their association domains to form a multimeric holoenzyme. The N-terminal 316 residues of CaMKIIα, containing the catalytic domain and the regulatory region, were necessary and sufficient for binding to the Cdk5 activators (Fig.1C).
p35 and p39 recognize two independent binding sites on α-actinin-1
The α-actinin-1 clone identified in our yeast two-hybrid screen contains the last three spectrin repeats and the EF-hand-containing C-terminal domain. To narrow further the residues of α-actinin-1 that are required for association with the Cdk5 activators, we assessed the ability of truncation mutants of α-actinin-1 to bind to full-length p39 and p35 in a yeast two-hybrid assay (Fig. 1D). The N-terminal actin binding domain of α-actinin-1 did not interact with p39 or p35 (Fig. 1D). A C-terminal truncation mutant of α-actinin-1, lacking the C-terminal EF-hand-containing domain, demonstrated reduced binding to p35 and p39 compared with the full-length protein (Fig. 1D). This suggests that, although the C-terminal region of α-actinin-1 is not necessary for association, residues in this domain may contribute to the interaction with p35 and p39. In fact, the C-terminal region of α-actinin-1 alone was sufficient for association with p35 and p39 (Fig.1D). A truncation mutant of α-actinin-1 lacking both the C-terminal domain and the N-terminal actin binding domain could also interact with p39 and p35 (Fig. 1D). Larger N-terminal deletions did not reduce binding further, and residues encoding just the spectrin-like repeat IV domain were sufficient for association with both Cdk5 activators (Fig.1D). Thus p35 and p39 can recognize at least two distinct regions of α-actinin-1, one contained in the spectrin-like repeat IV and the other in the C-terminal region of the protein.
α-Actinin-1 interacts with CaMKIIα
Walikonis and colleagues have shown that α-actinin-4, another member of the α-actinin family, interacts with both the α- and β-subunits of CaMKII at a site just downstream of its EF-hands, between residues 806 and 871 (Walikonis et al., 2001). α-Actinin-1 and α-actinin-4 are 88% identical and 97% similar in this region (Honda et al., 1998). The authors suggest that α-actinin-1, which has been localized to the PSD (Walikonis et al., 2000), also may be a physiologically relevant binding partner of CaMKII. We therefore investigated this possibility directly. In a yeast two-hybrid assay the full-length CaMKIIα and α-actinin-1 indeed did display a very strong interaction (Fig. 1E,F).
The N terminus of CaMKIIα, encompassing the kinase domain and the regulatory region, was sufficient for interaction with α-actinin-1, whereas the C-terminal association domain of CaMKIIα did not display any binding (Fig. 1E). Thus, consistent with the observations for α-actinin-4 (Walikonis et al., 2001), the N terminus of CaMKIIα is necessary and sufficient for association with α-actinin-1. Deletion of the C-terminal domain of α-actinin-1 completely abolished binding to CaMKIIα (Fig. 1F). Further, the C-terminal domain of α-actinin-1 alone could interact with CaMKIIα with the same strength as the full-length protein (Fig.1F). As is the case for α-actinin-4, the EF-hand-containing C terminus of α-actinin-1 is therefore necessary and sufficient for binding to CaMKIIα (Walikonis et al., 2001).
CaMKIIα and α-actinin-1 interact with p35/Cdk5 and p39/Cdk5 complexes
We used GST pulldown assays to confirm the association of α-actinin-1 and CaMKIIα with p39 and p35 independently of the yeast two-hybrid system. For these experiments we expressed recombinant p39, p25, and Cdk5 as GST fusion proteins in bacteria. Full-length p35 cannot be expressed as a GST fusion protein because of severe degradation (Qi et al., 1995). Therefore, GST-p25, the C-terminal Cdk5 binding and activating region of p35, was used in these assays. All GST fusion proteins were used to pull down interacting proteins from adult rat brain lysate, and associated proteins were identified by immunoblotting. As expected, GST-p39 and GST-p25 both could bind to Cdk5 in this assay (Fig.2A). GST-p39 and GST-p25, but not GST alone, also could precipitate CaMKIIα from adult rat brain lysate (Fig. 2A). GST-p29, containing the C terminus of p39, also can interact with CaMKIIα (data not shown). These results are consistent with the yeast two-hybrid data that indicate that the Cdk5-activating domain of p35 and p39 is necessary and sufficient for interaction with CaMKIIα. The specificity of the GST pulldowns is demonstrated by showing that Lis1, a protein expressed at high levels in the adult brain (Smith et al., 2000), does not interact with the GST fusion proteins under these conditions.

CaMKIIα, α-actinin-1, and Cdk5 and its activators interact in adult rat brain. A, GST fusion proteins of p39 (GST-p39), p25 (GST-p25), Cdk5 (GST-Cdk5), or GST alone were incubated with adult rat brain lysate. Then 10% of the lysates used for the pulldowns (10% Input) and the associated proteins were resolved by SDS-PAGE and detected by immunoblotting with CaMKIIα, α-actinin-1, Cdk5, and Lis1 antibodies, as indicated on theright. B, GST alone and a GST fusion protein encoding residues 363–892 of actinin-1 (GST-α-actinin-1) were incubated with adult rat brain lysate, and the associated proteins were detected by Western blotting with p39, p35, Cdk5, and CaMKIIα antibodies, as indicated on the right. The p35 antibody recognizes both full-length p35 and the N-terminal truncation product termed p25, as indicated by the arrows on the left.C, CaMKIIα and α-actinin-1 coimmunoprecipitate with Cdk5 and its activators. p35, p39, Cdk5, and HA antibodies (indicated at the top of the lanes) were used for immunoprecipitations from adult rat brain lysate. Then 5% of the lysate used for the immunoprecipitation (5% Input) and the immunoprecipitated proteins were resolved by SDS-PAGE and detected by immunoblotting with the antibodies indicated on theright. D, α-Actinin-1 or HA antibody was used for immunoprecipitations from adult rat brain lysate. Then 10% of the proteins used for the immunoprecipitations (10% Input) and the associated proteins were resolved by SDS-PAGE and detected by immunoblotting with the antibodies indicated on theright.
Although full-length p39 could bind to α-actinin-1 in our pulldown assay, GST-p25 and GST alone did not interact with α-actinin-1 (Fig.2A). GST-p29 also did not pull down α-actinin-1 in these assays (data not shown), confirming that the N terminus of p35 and p39 is necessary for interaction with α-actinin-1. GST-Cdk5 could pull down both CaMKIIα and α-actinin-1, but not Lis1, from brain lysate, presumably via its interaction with p35 and p39 (Fig.2A).
The α-actinin-1 clone obtained in the screen (residues 363–892) was expressed as a recombinant GST fusion protein in bacteria and was used to pull down interacting proteins from adult rat brain lysate. In this assay GST-α-actinin-1, but not GST alone, could interact with p39, p35, and Cdk5 (Fig. 2B). Immunoreactivity representative of p25 was not detected in the α-actinin-1 pulldowns, consistent with our previous GST pulldown and yeast two-hybrid results that the N terminus of p35 and p39 is necessary for association with α-actinin-1 (Fig. 2B). Together, the results from the GST pulldown assays suggest that CaMKIIα and α-actinin-1 can interact with the p35/Cdk5 and p39/Cdk5 complexes in brain. Consistent with the yeast two-hybrid interaction results, GST-α-actinin-1 also bound CaMKIIα from adult rat brain lysate (Fig.2B).
CaMKIIα, α-actinin-1, and the Cdk5 activators interactin vivo
To determine whether p39 and p35 interact with CaMKIIα and α-actinin-1 in vivo, we performed immunoprecipitations from adult rat brain lysate by using antibodies against p35, p39, and Cdk5, and we detected associated proteins via immunoblotting (Fig.2C). Both CaMKIIα and α-actinin-1 were detected in p35, p39, and Cdk5 immunoprecipitates, but not in control immunoprecipitations with HA antibody (Fig. 2C). Under these conditions Lis1 did not interact with p35, p39, or Cdk5, demonstrating the specificity of the coimmunoprecipitations. Reverse immunoprecipitations with the α-actinin-1 antibody also precipitated p35, p39, Cdk5, and CaMKIIα from adult rat brain lysate (Fig.2D). Together, these results suggest that CaMKIIα and α-actinin-1 interact with each other and also form a complex with p35/Cdk5 and p39/Cdk5 in vivo. p39, p35, Cdk5, or α-actinin-1 was not detected in CaMKIIα immunoprecipitations (data not shown). This could be attributable to the fact that CaMKIIα is a very abundant protein in brain, and only a small portion of it interacts with the Cdk5 activators and α-actinin-1.
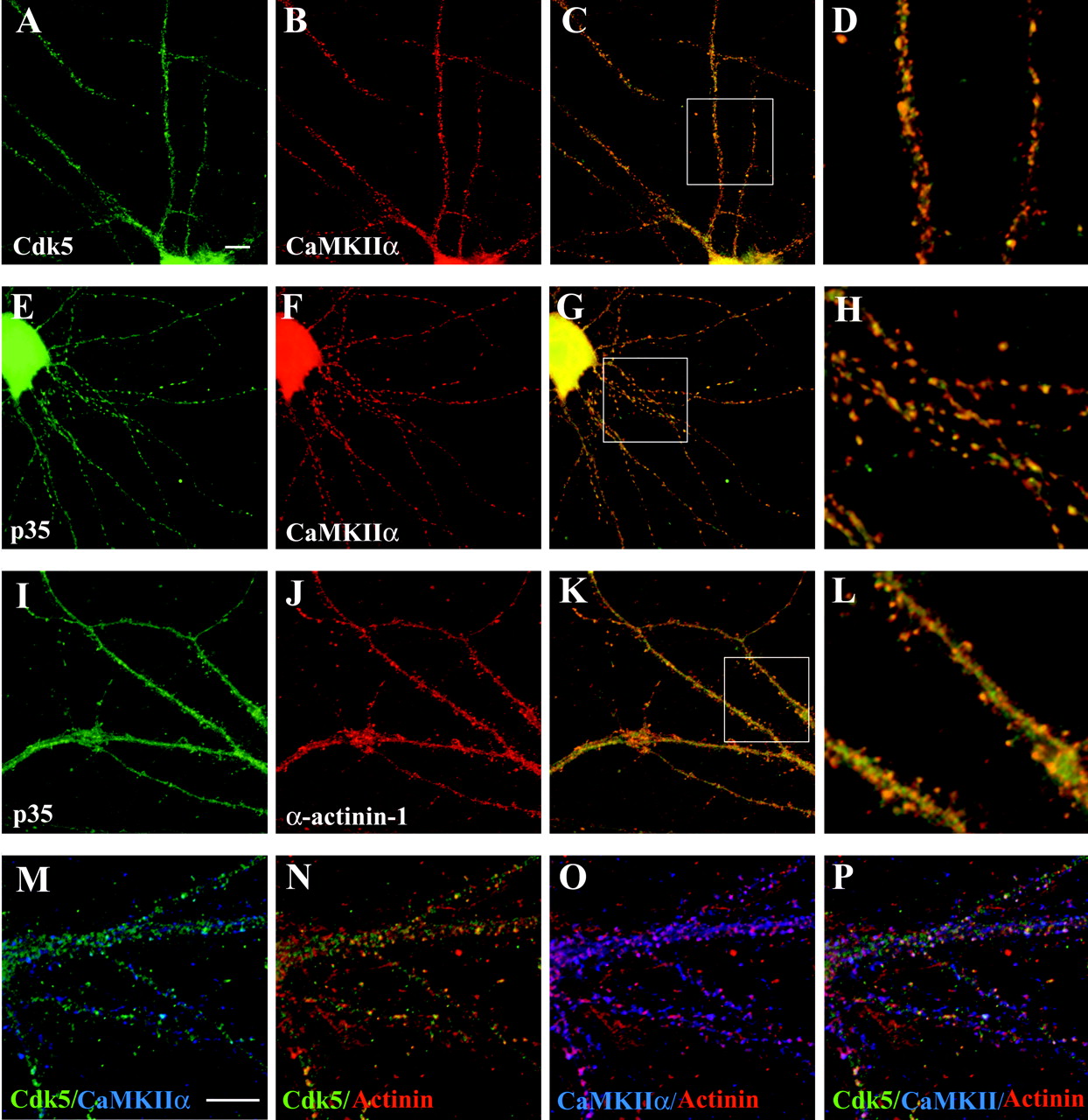
CaMKIIα, α-actinin-1, and Cdk5 and its activators colocalize in hippocampal neurons
CaMKIIα is a major component of excitatory synapses and is enriched particularly at the PSD (Lisman et al., 2002). α-Actinin also has been localized at the PSD and in dendritic spines (Wyszynski et al., 1997, 1998; Walikonis et al., 2000). We used immunocytochemistry to determine whether Cdk5 and its activators colocalize with CaMKIIα and α-actinin-1 at synaptic sites in neurons (Fig. 3). Dissociated hippocampal neurons were maintained in culture for 3 weeks, during which time they develop extensive processes and synaptic junctions and then were used for immunocytochemistry.

CaMKIIα, α-actinin-1, and Cdk5 and its activators colocalize in hippocampal neurons. Hippocampal neurons maintained in culture for 3 weeks were immunostained as described in Materials and Methods. A–L, Images were acquired by using a 60× oil-immersion objective. Scale bar in A, 10 μm (also applies to A–C,E–G, and I–K).M–P, Image was acquired by using a 100× oil-immersion objective. Scale bar in M, 10 μm (also applies to N–P). A–D, Colocalization of Cdk5 and CaMKIIα. Hippocampal neurons were double stained for Cdk5 (A, green) and CaMKII (B,red). C, The images shown inA and B are combined to reveal the colocalization of Cdk5 (green) and CaMKII (red) in yellow. The boxed area in C is presented in D at 3.5-fold magnification. E–H, Colocalization of p35 and CaMKIIα. Hippocampal neurons were double stained for p35 (E, green) and CaMKIIα (F, red). G, The images were merged to indicate colocalization between p35 (green) and CaMKIIα (red) inyellow. The boxed region of the image inG is presented at 3.5-fold magnification inH. I–L, Colocalization of p35 and α-actinin-1. Hippocampal neurons were double stained for p35 (I, green) and α-actinin-1 (J, red). L, The images were merged to indicate colocalization of p35 (green) and α-actinin-1 (red) inyellow. The boxed region of the image inK is presented at 3.5-fold magnification inL. M–P, Triple colocalization of Cdk5, CaMKIIα, and α-actinin-1. IgG- and IgM-specific secondary antibodies were used to triple-stain hippocampal neurons with Cdk5, CaMKIIα, and α-actinin-1 antibodies (see Materials and Methods).M, Colocalization of Cdk5 (green) and CaMKII (blue) in triple-stained neurons is revealed by the blue-green color. N, Colocalization of Cdk5 (green) and α-actinin-1 (red) in triple-stained hippocampal neurons is visible as yellow. O, Colocalization of CaMKIIα (blue) and α-actinin-1 (red) in triple-stained hippocampal neurons is visible as pink.P, Triple colocalization of Cdk5 (green), α-actinin-1 (red), and CaMKIIα (blue) in triple-stained neurons is visible aswhite.
In these hippocampal neurons the punctate staining of CaMKIIα along processes was readily detectable (Fig. 3B,F). Discrete spots of Cdk5 immunostaining were visible along dendrites (Fig. 3A), and an overlay of these images revealed punctate colocalization of Cdk5 and CaMKIIα immunoreactivity along dendrites (Fig. 3C,D). Like Cdk5, p35 immunoreactivity was visible in neuronal shafts as well as in discrete puncta along processes (Fig.3E,I). Merged images of p35 and CaMKIIα immunoreactivity revealed a punctate pattern of colocalization of these proteins (Fig. 3G,H). p39 also colocalized with CaMKII in these neurons (data not shown).
α-Actinin-1 immunoreactivity was detected primarily in dendritic spines, with lower levels in the cytoplasm of neuronal processes (Fig.3J). In addition to staining along the length of neurites, p35 also was present in very fine spiny protrusions, and these p35 positive spines also were immunoreactive for α-actinin-1 (Fig. 3I–L). Similar colocalization with α-actinin-1 also was observed for Cdk5 (Fig. 3N) and p39 in dendritic shafts and spines (data not shown).
We used IgG- and IgM-specific secondary antibodies to determine whether triple colocalization of CaMKIIα, α-actinin-1, and Cdk5 was detectable in mature hippocampal neurons. High-magnification images confirm the colocalization of Cdk5 with CaMKIIα (Fig.3M) and α-actinin-1 (Fig. 3N). Overlap of CaMKIIα and α-actinin-1 immunoreactivity indicates that these two proteins also colocalize in discrete spots along dendrites (Fig. 3O). Triple labeling indicates that a subset of α-actinin-1/CaMKII sites also contains Cdk5 immunostaining, as indicated by the white color in the merged images (Fig. 3P). Triple colocalization of CaMKIIα and α-actinin-1 with either p35 or p39 also was detectable in these neurons (data not shown). This suggests that CaMKIIα, α-actinin-1, and Cdk5 along with its activators all can colocalize in mature hippocampal neurons, supporting the possibility that they can exist in a complex. The partial overlap between Cdk5/α-actinin-1 and Cdk5/CaMKIIα coimmunostaining suggests that, although a complex of Cdk5, CaMKIIα, and α-actinin-1 may exist at synapses, exclusive complexes of Cdk5/CaMKIIα, Cdk5/α-actinin-1, and CaMKIIα/α-actinin-1 are also probably present.
Ca2+ stimulates the association of p35 with CaMKIIα and α-actinin-1
Changes in intracellular Ca2+ levels have been shown to regulate the binding and signaling properties of both α-actinin-1 and CaMKII (Blanchard et al., 1989; Krupp et al., 1999; Soderling et al., 2001; Lisman et al., 2002). To determine whether Ca2+ affects the association of CaMKIIα or α-actinin-1 with the Cdk5 activators, we tested the ability of p35, p39, or α-actinin-1 antibodies to coimmunoprecipitate associated proteins from adult rat brain lysate in the presence of increasing Ca2+ concentrations (Fig.4).

Ca2+-dependent association of CaMKIIα, α-actinin-1, and the Cdk5 activators. A, Ca2+ enhances the coimmunoprecipitation of CaMKIIα and α-actinin-1 with p35. EDTA (1 mm) or CaCl2 was added to adult rat brain lysate to a final concentration indicated above the lanes (0–5 mm). p35 antibody (p35 IP) or no antibody (no Ab) was used for immunoprecipitations from these lysates, and the immunoprecipitated samples were resolved by SDS-PAGE. The amounts of associated α-actinin-1 (top), CaMKIIα (middle), and Cdk5 (bottom) were analyzed by Western blots. Then 5% of the lysates used for the immunoprecipitations (Lysate) was analyzed by Western blotting for the levels of p35, α-actinin-1, CaMKIIα, and Cdk5. B, Ca2+ enhances the coimmunoprecipitation of CaMKIIα and α-actinin-1 with p39. EGTA (1 mm) or CaCl2 was added to adult rat brain lysate to a final concentration indicated above the lanes. p39 antibody (p39 IP) was used for immunoprecipitations from these lysates; the amounts of associated α-actinin-1 (top), CaMKIIα (middle), and Cdk5 (bottom) were analyzed by Western blots.C, Ca2+ enhances the coimmunoprecipitation of p35 and CaMKIIα with α-actinin-1. The same samples described in A were used for immunoprecipitations with the α-actinin-1 antibody (α-actinin-1 IP) or no antibody (no Ab). The amounts of associated p35 (top), CaMKIIα (middle), and α-actinin-1 (bottom) were analyzed by Western blots.D, Magnesium does not enhance the coimmunoprecipitation of CaMKIIα and α-actinin-1 with p35. CaCl2(Ca2+) or MgCl2(Mg2+ ) was added to adult rat brain lysate to a final concentration indicated above the lanes.p35 antibody was used for immunoprecipitations from these lysates. Then 10% of the lysates used for the immunoprecipitations (10% Input) and the immunoprecipitated samples (p35 IP) were resolved by SDS-PAGE. The amounts of associated α-actinin-1 (top), CaMKIIα (middle), and Cdk5 (bottom) were analyzed by Western blots.
In the absence of exogenously added Ca2+, we could detect coimmunoprecipitation of both α-actinin-1 and CaMKIIα with p35 (Figs. 2C, 4A). The addition of 10 μmCa2+ to brain lysates stimulated a detectable increase in the coimmunoprecipitation of both α-actinin-1 and CaMKIIα with p35 (Fig. 4A). The addition of 10–50 μm Ca2+ to brain lysates resulted in a twofold increase in the coimmunoprecipitation of α-actinin-1 with p35 and a threefold to fourfold increase in the association of CaMKII with p35. The α-actinin-1 and CaMKIIα that coimmunoprecipitated with p35 at high Ca2+ concentrations was not attributable to nonspecific precipitation of protein aggregates because no protein was detected in the non-IgG controls (Fig. 4A,C). The addition of 1 mm EDTA reduced the coimmunoprecipitation of α-actinin-1 and CaMKIIα with p35 (Fig.4A). The addition of calcium to brain lysates similarly induced coimmunoprecipitation of actinin-1 and CaMKII with p39, whereas the addition of EGTA inhibited the associations (Fig.4B). In contrast, the coimmunoprecipitation of Cdk5 with p35 or p39 was not affected by changing calcium, EDTA, or EGTA concentration in brain lysates (Fig. 4A,B).
Reverse immunoprecipitations with α-actinin-1 antibody also revealed the increased coimmunoprecipitation of p35 with α-actinin-1 after the elevation of Ca2+ concentrations in brain lysates (Fig. 4C). Interestingly, increasing the Ca2+ concentration in brain lysates also enhanced the coimmunoprecipitation of CaMKIIα with α-actinin-1, with a twofold increase in the coimmunoprecipitation of these proteins after the addition of 10 μmCa2+ (Fig. 4C). The addition of MgCl2 to brain lysates did not increase the coimmunoprecipitation of CaMKIIα or α-actinin-1 with p35 (Fig.4D), indicating that this effect is specific for Ca2+ and is not the result of increasing concentrations of general divalent cations. Our results suggest that Ca2+ can stimulate the association of CaMKIIα and α-actinin-1 with p35 as well as with each other.
Glutamate treatment of neurons can regulate the association of p35 with CaMKIIα
The stimulation of glutamate receptors in neurons can lead to increases in cytosolic Ca2+ (Nakanishi et al., 1998). We therefore investigated whether the treatment of neurons with glutamate could modulate the association of the Cdk5 activators with CaMKIIα and α-actinin-1. Dissociated hippocampal neurons were maintained in culture for 2 weeks and then were treated with 100 μm glutamate/10 μm glycine in a Mg2+-free buffer for 5 min. After treatment, p35 and α-actinin-1 were immunoprecipitated from lysates of these neurons, and the associated proteins were detected by Western blotting (Fig. 5A,B). Both CaMKIIα and α-actinin-1 coimmunoprecipitated with p35 under basal conditions, and the treatment of hippocampal neurons with glutamate induced a significant increase in the amount of CaMKIIα that coimmunoprecipitated with p35 (Fig. 5A). Surprisingly, no increase in the coimmunoprecipitation of α-actinin-1 and p35 was detected under the same treatment conditions (Fig. 5A,B). Glutamate treatment also did not affect CaMKIIα coimmunoprecipitation with α-actinin-1 significantly (Fig. 5B). In fact, a slight decrease in association between CaMKIIα and α-actinin-1 was seen in some experiments, but this effect did not prove to be significant in repeated experiments (Fig. 5B).

Glutamate treatment of dissociated hippocampal neurons enhances the association of CaMKIIα and p35.A, Dissociated hippocampal neurons in culture for 2 weeks were treated (Glu/Gly) with buffer (−) or 100 μmglutamate/10 μm glycine (+) for 5 min. Crude synaptosome fractions prepared from these neurons were used for the immunoprecipitation of p35. Then 10% of the proteins used for the immunoprecipitations (10% Input) and the p35-associated proteins (p35 IP) were resolved by SDS-PAGE. The amounts of CaMKIIα (top), α-actinin-1 (middle), and Cdk5 (bottom) were analyzed by immunoblotting. Quantitative analysis from three independent experiments is presented in the histogram. The amount of CaMKIIα, α-actinin-1, and Cdk5 immunoprecipitated with p35 is presented as a percentage of the values that were measured in control treatments and is the mean ± SEM of three independent experiments. *Statistically different (p < 0.05) from control values by Student's t test. B, The same lysates described in A were used for immunoprecipitation with α-actinin-1 antibody. Then 10% of the proteins used for the immunoprecipitations (10% Input) and the α-actinin-1-associated proteins (α-actinin-1 IP) were resolved by SDS-PAGE; the amounts of p35 (top) and CaMKIIα (bottom) were analyzed by immunoblotting. Quantitative analysis from three independent experiments is presented in the histogram. The amount of CaMKIIα and p35 immunoprecipitated with α-actinin-1 is presented as a percentage of the values that were measured in control treatments and is the mean ± SEM of three independent experiments.C, Cdk5 kinase activity is unaltered by glutamate treatment of hippocampal neurons. Dissociated hippocampal neurons were treated with either buffer (Control) or 100 μm glutamate/10 μm glycine (Glu/Gly), as described in A. The Cdk5/p35 complex was immunoprecipitated with p35 antibody, and Cdk5 kinase activity was measured in an in vitro kinase assay toward Histone H1. The Cdk5 kinase activity is presented as a percentage of the values that were measured from control-treated neurons and is the mean ± SEM from three independent experiments.
We also wanted to determine whether glutamate treatment could result in altered Cdk5 kinase activity. Toward this end we used the p35 antibody to immunoprecipitate the p35/Cdk5 complex from lysates of control and glutamate-stimulated hippocampal neurons. We detected no change in Cdk5 kinase activity after comparing the Cdk5 kinase activity of these precipitates in an in vitro kinase assay by using Histone H1 as a substrate (Fig. 5C).
CaMKII activation is required for the glutamate-dependent increase in the association of p35 with CaMKIIα
Our data suggest that the stimulation of glutamate receptors in neurons can enhance the association of CaMKIIα with the Cdk5 activators. To determine whether Ca2+influx was required for this effect, we treated hippocampal slices with 100 μm glutamate/10 μm glycine for 5 min in the presence or absence of 5 mm EDTA. As in the dissociated hippocampal neurons, the treatment of hippocampal slices with glutamate enhanced the coimmunoprecipitation of CaMKIIα with p35 (Fig.6A). Pretreatment of the slices with 5 mm EDTA to reduce extracellular free calcium blocked the glutamate effect, suggesting that calcium influx is critical (Fig. 6A). The treatment of hippocampal slices with glutamate also resulted in an increase in the coimmunoprecipitation of CaMKIIα with p39, which could be abolished by pretreatment with EGTA (Fig. 6B).

Ca2+ influx and CaMKII activity are required for the glutamate-mediated enhanced association of the Cdk5 activators and CaMKIIα. *Statistically different (p < 0.05) from control treatments; **statistically different (p < 0.05) from Glu/Gly treatment by one-way ANOVA. A, Hippocampal slices were treated with either buffer or 100 μm glutamate/10 μm glycine (Glu/Gly) in the absence or presence of 5 mmEDTA for 5 min. Crude synaptosome fractions prepared from these neurons were used for the immunoprecipitation of p35. The amounts of CaMKIIα (top) and Cdk5 (bottom) associated with p35 were analyzed by immunoblotting. Quantitative analysis from three independent experiments is presented in the histogram. The amount of CaMKIIα immunoprecipitated with p35 is presented as a percentage of the values that were measured in control treatments and is the mean ± SEM of three independent experiments. B, Hippocampal slices were treated with 100 μm glutamate/10 μm glycine (Glu/Gly) in the absence or presence of 5 mm EGTA for 5 min. The lysates were used for the immunoprecipitation of p39, and the amounts of CaMKIIα (top) and Cdk5 (bottom) associated with p39 were analyzed by immunoblotting. Quantitative analysis from three independent experiments is presented in the histogram.C, Hippocampal slices were treated with either buffer or 100 μm glutamate/10 μm glycine (Glu/Gly) in the presence or absence of 10 μm each KN62, KN93, and KN92 or 0.5 μm H-89 for 5 min. Crude synaptosome fractions prepared from these neurons were used for the immunoprecipitation of p35. The amounts of CaMKIIα (top) and Cdk5 (bottom) associated with p35 were analyzed by immunoblotting. Quantitative analysis from three independent experiments is presented in the middlehistogram. The amount of CaMKIIα immunoprecipitated with p35 is presented as a percentage of the values that were measured in control treatments and is the mean ± SEM of three independent experiments. In the bottom histogram crude synaptosome fractions prepared from the treated slices described above were assayed for total and Ca2+/CaM-independent CaMKII kinase activity (see Materials and Methods). Autonomous CaMKII activity is defined as the Ca2+/CaM-independent activity expressed as a percentage of the total CaMKII activity. The autonomous CaMKII activity in slices is expressed as a percentage of the autonomous CaMKII activity in control slices and is the mean ± SEM from three independent experiments.
A relationship between CaMKII activation and its association with the Cdk5 activators was investigated by the treatment of hippocampal slices with kinase inhibitors. Pretreatment with the CaMKII inhibitors KN62 and KN93 reduced the glutamate-mediated increase in the association of p35 with CaMKII (Fig. 6C). The structurally similar but inactive analog of KN93 (KN92) and the PKA inhibitor H-89 had no effects. None of these treatments altered the association between p35 and Cdk5, also measured by coimmunoprecipitation. Consistent with previous reports (Fukunaga et al., 1992; Tan and Chen, 1997), glutamate treatment of the slices stimulated CaMKII autonomous activity, which was abolished by the pretreatment of slices with KN62 and KN93, but not by KN92 or H-89 (Fig. 6C). This suggests that the glutamate-mediated activation of CaMKII may regulate the formation or maintenance of the p35/CaMKIIα complex. Both KN62 and KN93, although commonly used as CaMKII inhibitors, have been reported to inhibit other members of the Ca2+/CaM-dependent family, and we cannot rule out that the inhibition of other family members in our assay may contribute to the regulation of the CaMKIIα/p35 complex.
Activation of NMDA receptor enhances the association of p35 and CaMKIIα
To identify the specific neurotransmitter receptors involved in the glutamate-mediated stimulation of p35/CaMKII coimmunoprecipitation, we treated hippocampal slices with glutamate in the presence of glutamate receptor antagonists (Fig. 7). Ionotropic glutamate receptor antagonists were effective in reducing the glutamate-stimulated increase in p35/CaMKII association. A combination of antagonists, used to block all glutamate receptors, completely abolished the glutamate effect, but the NMDA receptor antagonist MK801 alone reduced the association virtually to basal levels (Fig. 7). The non-NMDA receptor antagonist CNQX also caused a smaller but statistically significant reduction, whereas treatment with the mGluR antagonist AIDA had no effect (Fig. 7). These results suggest that the CaMKIIα/p35 association triggered by glutamate treatment occurs predominantly via NMDA receptor signaling.

Activation of the NMDA receptor enhances the association of CaMKIIα and p35. Hippocampal slices were treated with either buffer or 100 μm glutamate/10 μmglycine in the absence (Glu) or presence of 10 μm MK801 (Glu/MK801), 100 μmCNQX (Glu/CNQX), 100 μm AIDA (Glu/AIDA), and 10 μm MK801 plus 100 μm CNQX plus 100 μm AIDA (Glu/MK801+CNQX+AIDA) for 5 min. Hippocampal slices also were treated with either 100 μm NMDA in the absence (NMDA) or presence of 10 μm MK801 (NMDA/MK801) or 10 μm KN62 (NMDA/KN62) or 100 μm AMPA in the absence (AMPA) or presence of 100 μm CNQX (AMPA/CNQX) or 10 μm KN62 (AMPA/KN62) for 5 min. Crude synaptosome fractions prepared from these neurons were used for the immunoprecipitation of p35. The amounts of CaMKIIα (top) and Cdk5 (bottom) associated with p35 were analyzed by immunoblotting. Quantitative analysis from three independent experiments is presented in the histogram. The amount of CaMKIIα immunoprecipitated with p35 is presented as a percentage of the values that were measured in control treatments and is the mean ± SEM of three independent experiments. aStatistically different (p < 0.05) from control treatment;bstatistically different (p < 0.05) from Glu treatment; cstatistically different (p < 0.05) from NMDA treatment;dstatistically different (p < 0.05) from AMPA treatment by one-way ANOVA.
In further support, the treatment of hippocampal slices with NMDA dramatically increased the association of CaMKII with p35, mimicking the effect of glutamate (Fig. 7). The NMDA-mediated association could be abolished by pretreatment with the receptor antagonist MK801 or with the CaMKII inhibitor KN62. Stimulation with AMPA also enhanced the association between p35 and CaMKIIα, but not as efficiently as with NMDA. Together, our data suggest that the activation of CaMKII by NMDA receptor favors the association of CaMKIIα with p35.
DISCUSSION
Several presynaptic and postsynaptic proteins have been identified as Cdk5 substrates, and a role for Cdk5 in synaptogenesis and synaptic transmission is beginning to emerge (Dhavan and Tsai, 2001). In this study we report that CaMKIIα and α-actinin-1 interact with Cdk5 via its activators p35 and p39. CaMKIIα and α-actinin-1 bind distinct domains of the Cdk5 activators and can also interact with each other. Triple colocalization of CaMKII and α-actinin-1 with Cdk5 and its activators is detected in neurons, suggesting that these proteins can exist together in a complex. Ca2+ augments the association of CaMKIIα and α-actinin-1 with p35 and p39 and also enhances their interaction with each other. Stimulation of glutamate receptors also could increase the association of the Cdk5 activators with CaMKIIα, and this effect is mediated primarily by the NMDA receptors. Together, these results suggest that Cdk5, via its activators, forms a complex with CaMKIIα and α-actinin-1 that may be modulated by synaptic activity.
Interaction of Cdk5 and CaMKIIα with α-actinin-1
The α-actinin family members are actin cross-linking proteins that bind and tether cell surface receptors, ion channels, and adhesion complexes to the actin cytoskeleton (Blanchard et al., 1989; Otey et al., 1990; Knudsen et al., 1995; Wyszynski et al., 1997; Cukovic et al., 2001). Both α-actinin-1 and α-actinin-2 are present in the PSD and in dendritic spines (Wyszynski et al., 1997, 1998; Walikonis et al., 2000). α-Actinin-2 binds directly to both the NR1 and NR2B NMDA receptor subunits (Wyszynski et al., 1997) and is thought to modulate receptor function by anchoring the receptor to the actin cytoskeleton. This is supported by studies showing that depolarization of the actin cytoskeleton is associated with the depression of NMDA receptor-mediated transmission (Rosenmund and Westbrook, 1993; Paoletti and Ascher, 1994). Disruption of the actin cytoskeleton also results in a complete loss of CaMKII from the PSD and α-actinin from spines (Allison et al., 1998, 2000). The association of CaMKII and Cdk5 with α-actinin-1 may serve to anchor the kinases to the subsynaptic cytoskeleton and perhaps modulate the association of the two kinases to the NMDA receptor and other substrates at the PSD.
Ca2+ was found to enhance the association of CaMKIIα and α-actinin-1 with the Cdk5 activators. We observed a significant increase in association of p35 and p39 with CaMKIIα and α-actinin-1 after the addition of 10 μm calcium (Fig.4). Measurements of NMDA receptor-dependent calcium influxes have demonstrated increases in calcium concentration in distal CA1 spines to as much as 20–40 μm in organotypic slice culture and acute slices, similar to the system we used (Petrozzino et al., 1995;Sabatini et al., 2001). The threefold to fourfold increase in association between the Cdk5 activators and CaMKIIα after the addition of 10–50 μm calcium is very comparable with the increase in association observed after the stimulation of NMDA receptors in hippocampal slices or neuronal cultures. However, the physiological relevance of the 20- to 30-fold increase in the association of these proteins at calcium concentrations higher than 300 μm is unclear.
α-Actinin-1 contains a functional EF-hand motif that can bind Ca2+, and the association with Ca2+ inhibits its ability to bind and cross-link actin. Ca2+ also regulates the association of α-actinin with the NMDA receptor (Wyszynski et al., 1997; Zhang et al., 1998; Krupp et al., 1999). A Ca2+-dependent association with α-actinin might serve to regulate Cdk5 and CaMKII interactions with the actin cytoskeleton as well as the NMDA receptor subunits. Although increasing the concentration of Ca2+ in brain lysate enhanced the association of α-actinin-1 with CaMKIIα and Cdk5, the glutamate treatment of hippocampal neurons did not produce a similar increase in complex formation. Further investigation is required to determine more closely whether physiological or pathological changes in intracellular Ca2+levels can regulate the association of α-actinin with the Cdk5 activators or CaMKIIα.
The association of α-actinin-1 with the Cdk5 activators may be regulated by neurotoxicity. Exposure to neurotoxic insults induces a calpain-mediated cleavage of p35 and p39 to generate p25 and p29, respectively (Lee et al., 2000; Patzke and Tsai, 2002). Cleavage of p35 and p39 results in a longer half-life than the full-length activator and a loss of the N-terminal myristoylation signal (Patrick et al., 1999; Patzke and Tsai, 2002). Generation of p25 or p29, therefore, results in prolonged activation and mislocalization of Cdk5, causing cytoskeletal disruptions and cell death. Indeed, the accumulation of p25 has been implicated in the pathogenesis of neurodegenerative diseases such as Alzheimer's disease and amyotrophic lateral sclerosis (Dhavan and Tsai, 2001). We have demonstrated that α-actinin-1 cannot interact with p25 or p29. Exposure to neurotoxic insults therefore may disrupt the association of the Cdk5 activators with α-actinin-1, contributing to the deregulation of Cdk5 activity and subcellular distribution.
Our interaction data and triple colocalization of CaMKIIα and α-actinin-1 with Cdk5 and its activators suggest that these proteins form a complex together. This is formally possible because CaMKIIα and α-actinin-1 bind distinct sites on p35 and p39. Further, whereas CaMKIIα and p35/p39 bind to the C terminus of α-actinin-1, p35 and p39 also can bind independently on at least one additional site in the spectrin-repeat region of α-actinin-1. It is therefore also possible that the Cdk5 activators and CaMKII can bind to α-actinin-1 simultaneously. α-Actinin-4 recently was found to associate with CaMKII in a ternary complex that also contained the PSD protein Densin-180 (Walikonis et al., 2001). The Cdk5 activators may interact with Densin-180 via their association with either α-actinin-1 and/or CaMKIIα.
Interaction of Cdk5 and CaMKIIα
CaMKII interacts with the C-terminal Cdk5 binding region of p35 and p39. Our coimmunoprecipitation data suggest that CaMKII associates with Cdk5 in vivo, presumably via its interaction with the Cdk5 activators. Several p35 binding proteins have been identified, including β-catenin, neurofilament subunits, and tau, which can bind to the C terminus of p35 simultaneously with Cdk5 (Patrick et al., 1999; Kesavapany et al., 2001). Further, CaMKII colocalizes with Cdk5 and its activators in mature hippocampal neurons, supporting our interaction data that CaMKII can form a complex with p35/Cdk5 and p39/Cdk5.
Enhanced association between p35 and CaMKII was observed after an increase in the calcium concentration in brain lysates and with the glutamate treatment of hippocampal cultures and slices. We have reported previously that glutamate-mediated neurotoxic stress and calcium influx induce the calpain-dependent cleavage of p35 and p39 (Lee et al., 2000; Patzke and Tsai, 2002). The experiments in this study were, however, not performed under comparable conditions to activate calpain, and we have not observed a significant increase in the cleavage of p35 or p39 in our studies.
The effects of glutamate on neurons are mediated by several different types of receptors, all of which can lead to increases in cytosolic calcium (Nakanishi et al., 1998). Our pharmacologic evidence, however, indicates that the increased association between p35 and CaMKII is dependent mainly on NMDA receptor activity, because NMDA treatment mimicked the effect of glutamate and MK801 reduced the increases that were stimulated by glutamate or NMDA virtually to basal levels. Furthermore, because the reduction in free calcium ions or the inhibition of CaMKII also effectively blocks the glutamate and NMDA effects, our data suggest that calcium entry via NMDA receptors activates CaMKII and triggers its association with the Cdk5 activators in synaptic sites.
In contrast, the inhibition of metabotropic glutamate receptors did not change the glutamate-mediated increases in p35/CaMKII complex association, although these receptors can trigger calcium release from internal stores (Hermans and Challiss, 2001), activate Cdk5 (Liu et al., 2001), and interact directly with calmodulin (Minakami et al., 1997). A small component of the glutamate effect appeared to be mediated by AMPA receptor activity, and, although a direct AMPA receptor mechanism cannot be ruled out, it is possible that the modest changes observed with the AMPA agonists and antagonists are attributable to secondary effects of depolarization on NMDA receptor activation. Thus our results, together with other reports that NMDA receptor and CaMKII activities are coupled closely (Bayer and Schulman, 2001), indicate that the calcium-dependent association of p35 and CaMKII stimulated by glutamate is mediated predominantly by NMDA receptor activity.
Glutamate stimulation of hippocampal neurons, although resulting in a CaMKII activity-dependent increase in the association of p35 with CaMKII, did not result in changes in Cdk5 kinase activity. It is possible that the Cdk5 association with CaMKII, rather than regulating Cdk5 activity, modulates its synaptic localization and association with synaptic proteins. CaMKII is reported to translocate from dendritic shafts into the PSD after NMDA receptor stimulation (Strack et al., 1997; Shen and Meyer, 1999). The autophosphorylation-dependent and -independent association of CaMKII with the NMDA receptor subunits is thought to play a role in its translocation as well as in anchoring CaMKII at the PSD (Bayer et al., 2001). At the PSD, CaMKII can phosphorylate the NR2A and NR2B receptor subunits (Omkumar et al., 1996; Strack et al., 2000). Phosphorylation of the GluR1 subunit of the AMPA receptor by CaMKII enhances channel conductance, which might contribute to long-term potentiation (Derkach et al., 1999). Cdk5 also has been shown to phosphorylate the NR2A subunit of the NMDA receptor, and the inhibition of Cdk5 activity reduces channel conductance (Li et al., 2001). The increased association of p35 with CaMKII after neuronal activity might facilitate the localization and association of Cdk5 in the vicinity of the NMDA receptor and its other putative substrates at the PSD.
The multiple mechanisms that modulate synaptic strength have been shown to involve cross talk among complex signal transduction pathways of the Src family tyrosine kinases, protein kinase A, protein kinase C, mitogen-activated protein kinase, and CaMKII (Soderling and Derkach, 2000; Grant and O'Dell, 2001; Soderling et al., 2001). Functional consequences of the association of CaMKII and Cdk5, as well as their shared regulatory and target interactions with proteins such as α-actinin-1 and the NMDA receptor, may elucidate novel mechanisms that are important in synaptic transmission and plasticity.
Footnotes
↵* R.D. and P.L.G. contributed equally to this work.
This work was supported by National Institutes of Health grants to L.-H.T. We thank Dr. H. Schulman for providing CaMKII constructs; Dr. Anne Young for the use of her animal procedure facility; Dr. Lily Moy for assistance with statistical analysis; and Dr. Janet Volker, Dr. Lily Moy, and Benjamin Samuels for critical reading of this manuscript.
Correspondence should be addressed to Dr. Li-Huei Tsai, Department of Pathology, Harvard Medical School, Armenise Building, Room 342, 200 Longwood Avenue, Boston, MA 02115. E-mail:li-huei_tsai{at}hms.harvard.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}