

Abstract
Overactivation of NMDA receptors is believed to induce neuronal death by increasing phospholipid hydrolysis and subsequent degradation. We showed previously that NMDA releases choline and inhibits incorporation of [3H]choline into phosphatidylcholine before excitotoxic neuronal death. On the basis of these results, we hypothesized that excitotoxicity results from inhibition of synthesis rather than from increased degradation of phospholipids. We now investigated the effect of NMDA receptor overactivation on synthesis and degradation of major membrane phospholipids in the early stages of the excitotoxic process. Exposure of cortical neurons to neurotoxic concentrations of NMDA increased extracellular choline and activated hydrolysis of phosphatidylcholine and phosphatidylinositol by phospholipase A2 but did not induce significant degradation of phosphatidylcholine, phosphatidylinositol, phosphatidylethanolamine, or phosphatidylserine. In contrast, NMDA strongly reduced the incorporation of [3H]choline and [3H]ethanolamine into their respective phospholipids. Metabolic labeling experiments in whole cells showed that NMDA receptor overactivation does not modify the activity of phosphocholine or phosphoethanolamine cytidylyltransferases but strongly inhibits choline–ethanolamine phosphotransferase activity. This effect was observed well before any significant membrane damage and cell death. Moreover, cholinephosphotransferase activity was lower in microsomes from NMDA-treated cells. These results show that membrane damage by NMDA is preceded by inhibition of phospholipid synthesis and not by phospholipid degradation in the early stages of the excitotoxic process, and that NMDA receptor overactivation decreases phosphatidylcholine and phosphatidylethanolamine synthesis by inhibiting choline–ethanolaminophosphotransferase activity.
- excitotoxicity
- phospholipid synthesis
- NMDA
- choline
- neuronal death
- choline–ethanolamine phosphotransferase activity
Introduction
Membrane phospholipids have a dual role as structural building blocks of cell membranes and as precursors of molecules involved in signal transduction such as the lipid second messengers diacylglycerol (DAG), phosphatidic acid, lysophosphatidic acid, or arachidonic acid (Exton, 1994; Liscovitch and Cantley, 1994; Hodgkin et al., 1998). Enzymatic hydrolysis of membrane phospholipids by phospholipases produces these lipid second messengers, as well as soluble second messengers such as inositol 1,4,5-trisphosphate, and also other molecules such as choline (Cho) that are released to the extracellular medium. It is generally assumed that enzymatic hydrolysis of phospholipids leads to phospholipid degradation and membrane damage. However, the mechanisms by which phospholipases switch from physiological second-messenger production to phospholipid degradation remain unclear.
Excitotoxicity, the process of neuronal death evoked by glutamate receptor overactivation (Rothman and Olney, 1995), has been hypothesized to underlie the neuronal loss observed in both acute and chronic neurodegenerative disorders (Meldrum and Garthwaite, 1990; Coyle and Puttfarcken, 1993). Damaged cellular membranes are the most characteristic feature of excitotoxicity. Excessive Ca2+ entry through glutamate receptors activates a large array of potential neurotoxic mechanisms, which include activation of enzymes such as phospholipases, calpains and other proteases, protein kinases, nitric oxide synthetase, endonucleases, as well as reactive oxygen species (for review, see Sattler and Tymianski, 2000). Increased phospholipid hydrolysis by glutamate receptor overactivation has been proposed to lead to degradation of membrane phospholipids and excitotoxic neuronal death (Farooqui et al., 1997). Moreover, altered brain membrane phospholipid levels, activation or phospholipases, and/or increased phospholipid degradation have been reported in neurodegenerative pathologies (Goto et al., 1988; Nitsch et al., 1992; Svennerholm and Gottfries, 1994; Homayoun et al., 1997). However, the mechanisms by which all of these different neurotoxic processes contribute to the membrane damage associated with excitotoxicity have not been characterized.
Previous results from our laboratory have shown that NMDA receptor overactivation releases Cho before excitotoxic cell death (Zapata et al., 1998; Gasull et al., 2000). Phospholipase inhibitors did not block this release of Cho by NMDA, but it was associated with a strong inhibition of incorporation of [3H]Cho into phosphatidylcholine (PtdCho) (Gasull et al., 2000). On the basis of these findings, we proposed that excitotoxic cell death is produced by inhibition of synthesis and not by increased degradation of membrane phospholipids. We now studied membrane phospholipid catabolism before excitotoxic cell death and investigated which of the steps of phospholipid synthesis is altered by NMDA receptor overactivation.
In neurons, the majority of PtdCho is synthesized through the cytidine diphosphate (CDP)–choline pathway, also named the Kennedy pathway (Kennedy and Weiss, 1956; Kennedy, 1989). This pathway involves three enzymatic steps: phosphorylation of Cho to phosphocholine in the presence of ATP; conversion of phosphocholine to CDP–choline in the presence of cytidine triphosphate (CTP); and transfer of the phosphocholine moiety of CDP–choline to DAG. These three steps are catalyzed by the sequential action of choline kinase, CTP:phosphocholine cytidylyltransferase and cholinephosphotransferase and render, respectively, phosphocholine, CDP-choline, and PtdCho. Likewise, in neurons, the majority of phosphatidylethanolamine (PtdEtn) is synthesized through an analogous CDP–ethanolamine pathway (Araki and Wurtman, 1998).
We now report that exposure of neuronal cortical cells to neurotoxic concentrations of NMDA inhibits the synthesis of the major phospholipid constituents of cell membranes without inducing significant phospholipid degradation. The NMDA-evoked reduction of PtdCho and PtdEtn synthesis is produced by a decrease in choline–ethanolaminephosphotransferase activity, the last step of the Kennedy pathway of phospholipid synthesis.
Materials and Methods
Cell culture. Primary cultures of brain cortical neurons were prepared essentially as described previously (Gasull et al., 2000). Frontolateral cortical lobes were dissected from brains of Sprague Dawley embryonic day 18 fetuses, and cells were chemically dissociated in the presence of trypsin and DNase I. Cortical cells were seeded in poly-l-lysine-coated (10 μg/ml) 24-well plates at a density of 9 × 105 cells/cm2.
Cell death. Cell death was measured by propidium iodide (PI) staining as described previously (Gasull et al., 2001).
Treatments. Treatments were performed in 300 μl of modified Locke-HEPES buffer without Mg2+ (MLH) (in mm: 154 NaCl, 3.6 NaHCO3, 2.3 CaCl2, 5.6 KCl, and 5.6 d-glucose in 5 HEPES buffer, pH 7.4).
Determination of Cho. Extracellular Cho levels were measured in the incubation medium using an HPLC coupled to an enzymatic reactor and an electrochemical detector (BAS, West Lafayette, IN) as described previously (Gasull et al., 2000). Results were expressed in picomoles per milligram of protein. Protein content was measured in each well using the BCA Protein Assay Reagent (Pierce, Rockford, IL).
Extraction of lipids and water-soluble metabolites. After treatment, the incubation buffer was removed, methanol was added, and the methanolic cell suspension was sonicated. Lipid extraction was performed by adding chloroform, incubating 15 min at room temperature, and adding H2O (final proportion of methanol/chlorofom/H2O, 6:6:5, v/v/v) to separate the aqueous and the lipidic phases. Then, tubes were shaken vigorously and centrifuged (10 min at 400 × g). The upper aqueous phases containing radioactive soluble metabolites and the lower organic phases containing radioactive lipids were transferred to new tubes and dried under vacuum centrifugation.
Analysis of lipids by thin-layer chromatography. Lipids were dissolved in 10 μl of chloroform/methanol (4:1, v/v) and spotted onto silica gel plates. When labeled with [3H]Cho, PtdCho was separated in silica gel plates [high-performance thin-layer chromatography (TLC) silica gel-60 plates, 10 × 20 cm, with concentrating zone; Merck, Darmstadt, Germany] using the solvent chloroform/methanol/acetic acid/water (75:45:3:1, v/v/v/v). The phospholipids PtdCho, PtdEtn, phosphatidylinositol (PtdIns), and phosphatidylserine (PtdSer) labeled with [3H]glycerol, [32P]orthophosphoric acid, [3H]myristic acid, or [3H]arachidonic acid were separated by two-dimensional TLC in silica gel plates (TLC silica gel-60 plates, 20 × 20 cm; Merck). The solvent for the first dimension was chloroform/methanol/NH4OH (65:35:10, v/v/v). The solvent for the second dimension was chloroform/methanol/acetone/acetic acid/water (10: 2:4:2:1, v/v/v/v/v).
Analysis of water-soluble metabolites of the CDP–choline and CDP–ethanolamine pathways by TLC. Water-soluble metabolites of the CDP–choline pathway were dissolved in 10 μl of ethanol/H2O (1:1, v/v), spotted onto silica gel plates (TLC silica gel-60 plates, 20 × 20 cm; Merck), and separated using the solvent methanol/0.9%NaCl/ammonia (50:50:5, v/v/v). Water-soluble metabolites of the CDP–ethanolamine pathway were dissolved in water, spotted onto twin silica gel plates (TLC silica gel-60 plates, 10 × 20 cm; Merck), and developed in two different solvents to resolve ethanolamine, phosphoethanolamine, CDP–ethanolamine, and glycerophosphoethanolamine. One of the twin plates was developed using the solvent methanol/0.5% NaCl/ammonia (50:50:5). This solvent does resolve ethanolamine and phosphoethanolamine but does not separate CDP–ethanolamine and glycerophosphoethanolamine. The other twin plate was developed using the solvent ethanol/water/ammonia (62:8:5), which resolves CDP–ethanolamine and glycerophosphoethanolamine. Choline-containing metabolites were visualized with iodine vapors and ethanolamine-containing metabolites by ninhydrin staining. The spots were identified by comparison with known standards. The silica was scraped off the plate, and the radioactivity incorporated was determined.
Analysis of DAG levels. DAG levels were measured as described previously by Preiss et al. (1987). The lipidic content of 50 μl of the organic phase (total volume, 0.9 ml) of each sample or 1,2-dioleoyl-sn-glycerol standards (in a range of 250–8000 pmol) were resuspended by sonication in 20 μl of a mixture of cardiolipin (5 mm), N-octyl glucoside (7.5%), and DETAPAC (1 mm). The lipidic suspension was then incubated at 25°C for 30 min in a final volume of 100 μl of the reaction buffer (in mm: 61 Imidazol-HCl, 50 NaCl, 12.5 MgCl2, 1 EGTA, 0.5 diethylenetriamine pentaacetic acid (DETAPAC), and 2 DTT) in the presence of DAG kinase from Escherichia coli (0.022 U) and 1 μCi of [γ-32P]ATP (1 mm). The reaction was stopped by addition of 0.6 ml of chloroform/methanol/HCl (100:200:1, v/v/v). After 15 min at room temperature, we added 0.25 ml of chloroform and 0.25 ml of water; tubes were shaken vigorously and centrifuged at 2000 × g for 5 min. The upper aqueous phase was removed, and the lower organic phase containing32P-lipids was washed with 0.75 ml of methanol/H2O (1:1, v/v). Aliquots (0.3 ml) of the washed organic phase were centrifuged under vacuum to evaporate the solvent. To separate [32P]phosphatidic acid (PtdOH) from the rest of 32P-lipids, the lipidic pellets were resuspended in 15 μl of chloroform/methanol (4:1, v/v) and spotted onto silica gel plates (TLC silica gel-60 plates, 20 × 20 cm; Merck) that were developed with chloroform/acetone/methanol/acetic acid/H2O (10:4:3:2:1, v/v/v/v/v.). The area corresponding to [32P]PtdOH, identified by comigration with standards, was scraped and counted for radioactivity. Standard values were plotted as disintegrations per minute of [32P]PtdOH versus picomoles of 1,2-dioleoyl-sn-glycerol (250–8000 pmol), and, after linear regression of the data, the amount of DAG in each sample was interpolated and corrected for the total volume of the sample.
Analysis of cholinephosphotransferase activity in microsomes. Cholinephosphotransferase activity was assayed by measuring the incorporation of the radiolabeled substrate CDP-[methyl-14C]choline [specific activity (S.A.), 54 mCi/mmol] into PtdCho as described previously (Voziyan et al., 1993). After treatment, cortical cells were homogenized in a Dounce homogenizer in 10 mm Tris-HCl buffer, pH 7.4, containing 150 mm NaCl and 0.2 mm PMSF. Homogenates were centrifuged (10 min at 1000 × g and 4°C), the supernatant obtained was centrifuged (60 min at 100,000 × g and 4°C), and the pellet obtained from this second centrifugation was used to determine cholinephosphotransferase activity in the microsomes fraction. Cholinephosphotransferase activity was assayed in a mixture containing 175 mm Tris-HCl, pH 8.5, 8 mm MgCl2, 0.5 mm EGTA, 1 mg/ml BSA, 2 mm 1,2, dioleoyl-rac-glycerol in 0.1% taurocholate (sonicated before addition to assay mixture), 0.4 mm CDP-[methyl- 14C]choline, and 25 μg of microsomal protein. The mixture was incubated at 37°C for 15 min in a shaking water bath. The reaction was stopped by addition of methanol/chloroform (2:1, v/v). Lipid extraction was performed by adding chloroform, and, after 15 at room temperature, H2O was added to separate two phases (final proportion of methanol/chloroform/H2O, 7.5:6.2:4, v/v/v), tubes were shaken and centrifuged at 700 × g for 5 min, and the lower lipidic fraction was kept. This lipidic fraction was further purified by adding 4 vol of a mixture of methanol/H2O/chloroform (48:47:3, v/v/v) and separating phases by centrifugation (700 × g for 5 min). The radioactivity incorporated into the lipidic fraction was measured. Protein content was measured in each well using the BCA Protein Assay Reagent (Pierce).
Statistical analysis. All data are presented as mean ± SEM. Data from experiments investigating concentration dependence were fitted to a sigmoid function using nonlinear regression in the GraphPad PRISM computer program (GraphPad, San Diego, CA). Statistical significance of differences was determined using Student's t test or two-way ANOVA followed by Student's t test.
Results
Cho released by NMDA receptor overactivation does not originate from phospholipid degradation
Exposure of cortical neurons to neurotoxic concentrations of NMDA increased extracellular Cho (Fig. 1A). To determine whether this release of Cho originates from degradation of membrane phospholipids, we studied the effect of NMDA on the levels of radioactivity present in prelabeled PtdIns, PtdSer, PtdCho, and PtdEtn, the major phospholipid constituents of membranes. To do so, cells were labeled with [3H]Cho, [3H]glycerol, [32P]orthophosphoric acid, [3H]myristic acid, or [3H]arachidonic acid for 30 min, the radioactive marker was removed, and cells were incubated with either vehicle or NMDA (300 μM) for 20 min. We used these five different radioactive markers to ensure the labeling of a wide range of phospholipids. [3H]Cho incorporates into the head group of PtdCho, [3H]glycerol incorporates into the phospholipid backbone of phospholipids, [32P]orthophosphoric acid incorporates into the phosphate moiety of phospholipids, and [3H]myristic acid incorporates into the sn-1 position of phospholipids. Finally, [3H]arachidonic acid labels a small, rapid turnover pool of PtdCho at the sn-2 position that is more accessible to phospholipase A2 (PLA2). We found that NMDA receptor overactivation reduced the radioactivity present in PtdCho and PtdIns prelabeled with [3H]arachidonic acid by 40 and 60%, respectively (Fig. 1F). However, NMDA receptor overactivation, despite increasing extracellular Cho (Fig. 1A), did not change the amount of radioactivity present in PtdCho prelabeled with [3H]Cho (Fig. 1B) or in PtdIns, PtdSer, PtdCho, or PtdEtn prelabeled with [3H]glycerol (Fig. 1C), [32P]orthophosphoric acid (Fig. 1D), or [3H]myristic acid (Fig. 1E). These results indicate that NMDA receptor overactivation may increase phospholipid hydrolysis and produce second messengers and choline release without inducing phospholipid degradation.

A, NMDA receptor overactivation increases extracellular Cho levels. Neuronal cortical cell cultures were incubated in 300 μl of MLH containing either vehicle (open bars) or NMDA (300 μm) (filled bars) for 20 min. Incubation buffer was removed after treatment to determine extracellular Cho levels. Values are expressed in picomoles per milligram of protein and are mean ± SEM of three experiments. *p < 0.05, significantly different from control cells. B–F, Effect of treatment with NMDA on prelabeled membrane phospholipids. Cells were incubated for 30 min in 300 μl of MLH containing the radioactive markers [methyl-3H]Cho (1 μCi/well; S.A., 75 Ci/mmol) (B), [3H]glycerol (5 μCi/well; S.A., 2.6 Ci/mmol) (C), [32P]orthophosphoric acid (2 μCi/well) (D), [3H]myristic acid (10 μCi/well; S.A., 40 Ci/mmol) (E), or [3H]arachidonic acid (10 μCi/well; 217 Ci/mmol) (F). The radioactive markers were washed, and cells were incubated for 20 min in MLH containing either vehicle (open bars) or NMDA (300 μm) (filled bars). The lipidic fraction was obtained, phospholipids were separated by two-dimensional TLC, and the radioactivity in the spots identified as PtdCho, PtdEtn, PtdSer, and PtdIns was measured. Data are expressed in disintegrations per minute per 1000 and are mean ± SEM of three experiments. *p < 0.05, significantly different from control cells.
NMDA receptor overactivation inhibits phosphatidylcholine synthesis but not incorporation of [3H]Cho into phosphocholine or CDP–choline
We next investigated the effect of treatment with NMDA on the incorporation of [3H]Cho into the metabolites of the CDP–choline pathway. PtdCho can be synthesized through the CDP–choline pathway or by the conversion of PtdEtn to PtdCho via repeated methylation of PtdEtn by the enzyme phosphatidylethanolamine-N-methyltransferase. Activity of this latter enzyme has been detected in brain (Blusztajn and Wurtman, 1981), but it is well established that, in nucleated mammalian cells, except hepatocytes, PtdCho is synthesized almost exclusively through the CDP–choline Kennedy pathway (Kennedy, 1989; Vance, 1991; Kent, 1995; Walkey et al., 1998). However, to rule out the possibility that, in neurons, under our experimental conditions, PtdCho might be synthesized through the repeated methylation of PtdEtn, we labeled our primary neuronal cultures with [3H]Etn and measured the presence of radioactivity in PtdCho as well as in other membrane phospholipids. Nevertheless, radioactivity was exclusively detected in membrane PtdEtn (results not shown).
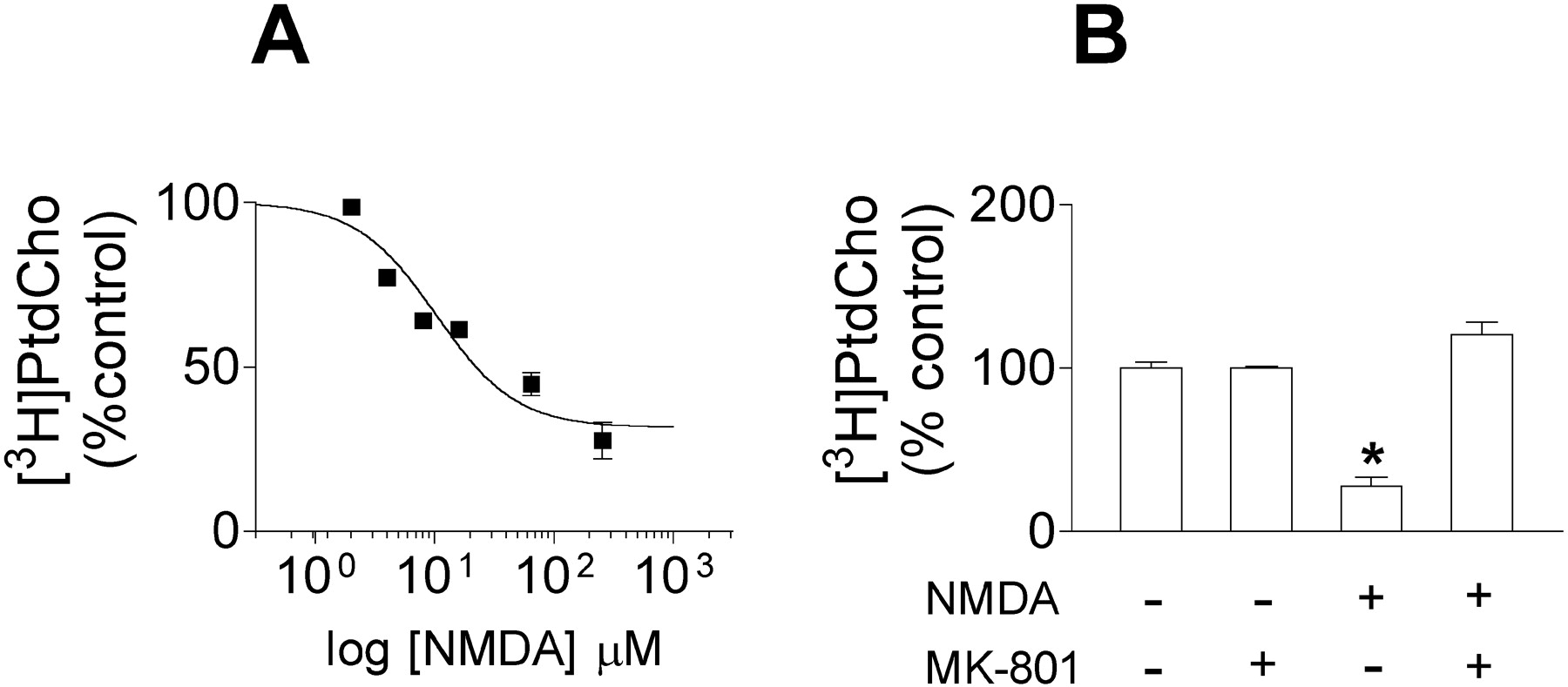
Treatment of cortical neuronal cells with NMDA produced a concentration-dependent reduction of the incorporation of [3H]Cho into membrane PtdCho with an EC50 with 95% confidence intervals of 10 (5–20) μM (Fig. 2A). The maximal effect of NMDA was a 68% reduction of the incorporation of [3H]Cho into membrane PtdCho, and (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine hydrogen maleate (MK-801) (10 μM), a noncompetitive NMDA receptor antagonist, completely blocked the effect of NMDA (Fig. 2B). These results suggest that NMDA reduces PtdCho synthesis by an action at NMDA receptors. However, one alternative interpretation could be that the inhibition of [3H]Cho incorporation into PtdCho might result from dilution of extracellular [3H]Cho with the unlabeled Cho released to the medium by NMDA receptor overactivation. To rule out this possibility, we performed experiments in the presence of 10 μM extracellular [3H]Cho (1.33 Ci/mmol), a concentration that prevents any significant dilution of [3H]Cho by unlabeled Cho released from the cell by NMDA. In these experiments, we measured the incorporation of [3H]Cho into the metabolites of the Kennedy pathway at different time points in the presence of either vehicle or NMDA (300 μM). Under these conditions, NMDA receptor overactivation, again, inhibited the incorporation of [3H]Cho into PtdCho (Fig. 3C). In contrast, NMDA did not change the rate of incorporation of [3H]Cho into phosphocholine (Fig. 3A) or CDP–choline (Fig. 3B) when compared with controls. These results indicate that treatment with NMDA does not significantly modify choline kinase and CTP: phosphocholine cytidylyltransferase activities. These enzymes, which catalyze the sequential conversion of Cho into phosphocholine and CDP–choline, require ATP and CTP, respectively, as cosubstrates. Therefore, treatment with NMDA does not change the concentration of these cosubstrates to significantly alter the rate of activity of these two enzymes.

A, NMDA induces a concentration-dependent decrease of the incorporation of [3H]Cho into PtdCho. Neuronal cortical cultures were incubated in MLH in the presence of 1 μCi/well, [3H]Cho (S.A., 75 Ci/mmol) and varying concentrations of NMDA. Lipids were extracted after 1 hr of treatment to determine the radioactivity incorporated into PtdCho. Data are mean ± SEM of three experiments. B, NMDA-evoked decrease in [3H]Cho incorporation into PtdCho is blocked by MK-801. Cells were incubated in 300 μl of MLH containing 1 μCi of [3H]Cho (S.A., 75 Ci/mmol) and vehicle or NMDA (250 μm) in the presence of vehicle or MK-801 (10 μm). Lipids were extracted after 1 hr of treatment to determine the radioactivity incorporated into PtdCho. Data are mean ± SEM of three experiments. *p < 0.05, significantly different from control cells.

Time course analysis of the effect of NMDA on incorporation of [3H]Cho into metabolites of the CDP–choline pathway in the presence of 10μm extracellular Cho. The incorporation of [3H]Cho into phosphocholine (A), CDP–choline(B), and PtdCho (C) was measured in neuronal cortical cultures at different times after addition of 4 μCi/well [3H]Cho in the presence of 10 μm unlabeled Cho (final S.A., 1.3 Ci/mmol) and either vehicle (○) or NMDA (300 μm) (•). Results are expressed in disintegrations per minute per 1000 and are mean ± SEM of three experiments. *p < 0.05, significantly different from control cells.
NMDA receptor overactivation reduces the incorporation of [3H]Cho into membrane PtdCho by decreasing cholinephosphotransferase activity
To establish which reaction of the CDP–choline pathway is altered after NMDA receptor overactivation, we performed pulse-chase experiments. To do so, cells were prelabeled for 10 or 30 min with 10 μM [3H]Cho (0.33 Ci/mmol) (pulse) and then incubated with either vehicle or NMDA (300 μM) for 10, 20, or 30 min in the presence of 250 μM unlabeled Cho (chase). In these experiments, inhibition of a given enzymatic step of the pathway should induce accumulation of its radioactive substrate and decrease the incorporation of radiolabel in its products.
Radioactive phosphocholine decreased during the chase, in both 10 min (results not shown) and 30 min pulse experiments. This effect was not significantly different in control and NMDA-treated cells (Fig. 4A). In contrast, NMDA receptor overactivation induced accumulation of radioactive CDP–choline (Fig. 4B) and decreased the incorporation of label into PtdCho when compared with controls (Fig. 4C). These results show that activation of the NMDA subtype of glutamate receptors inhibits PtdCho synthesis by selectively decreasing cholinephosphotransferase activity, the last step of the CDP–choline pathway. This was confirmed further with experiments using [3H]cytidine instead of [3H]Cho as radiolabel. In these experiments, cells were prelabeled for 30 min with [3H]cytidine (21.5 Ci/mmol), the radiolabel marker was removed, and the cells were incubated with either vehicle or NMDA (300 μM) for 20 min. We found that the levels of [3H]CDP–choline labeled with [3H]cytidine were significantly higher in NMDA-treated cells compared with controls (NMDA, 8199 ± 572 dpm; control, 4267 ± 301 dpm; mean ± SEM of three experiments; p < 0.05), showing again that the rate of use of CDP–choline for PtdCho synthesis is decreased by NMDA in cortical neurons.

Pulse-chase studies on phosphatidylcholine biosynthesis pathway using [3H]Cho as radiolabel. Neuronal cortical cultures were incubated for 30 min in 300 μl of MLH containing 10 μm [3H]Cho (0.33Ci/mmol) (pulse), washed, and incubated in MLH containing 250 μm unlabeled Cho in the presence of either vehicle (○) or NMDA (300 μm) (•) (chase). The radioactivity present in phosphocholine (A), CDP–choline (B), and PtdCho (C) was measured at different times after the addition of NMDA. Phosphocholine, CDP–choline, and PtdCho were separated by TLC, and the radioactivity present in each anabolite was measured. Results are expressed in disintegrations per minute and are mean ± SEM of three experiments. *p < 0.05, significantly different from control cells.
Microsomal cholinephosphotransferase activity is lower in neuronal cortical cells treated with NMDA
The enzymes with cholinephosphotransferase activity are integral membrane-bound proteins, which localize in the endoplasmic reticulum, Golgi, and nuclear membranes (McMaster and Bell, 1997; Henneberry et al., 2002). To investigate the effect of NMDA receptor activation on cholinephosphotransferase activity, we measured this activity in microsomes obtained from neuronal cortical cells previously incubated for 45 min with either vehicle or NMDA (300 μm). The assay was performed in the presence of Mg2+, the cation activator of cholinephosphotransferase, a fixed concentration of CDP–choline (400 μM), and varying concentrations of DAG. We found that cholinephosphotransferase activity was lower in microsomes of NMDA-treated cells than in microsomes of control cells, at least for a range of DAG concentrations between 125 and 1000 μM (Fig. 5).

NMDA decreases CPT activity in microsomes. Microsomal CPT activity was determined by measuring the incorporation of radioactivity into PtdCho by the transfer of the phosphocholine moiety of CDP-[methyl-14C]choline to DAG. Cells were incubated for 45 min in MLH containing either vehicle (open bars) or NMDA (300 μm) (filled bars). After treatment, cells were homogenized and processed to obtain the microsomal fraction. The activity CPT in microsomes (25 μg of microsomal protein) obtained from control or NMDA-treated cells was assayed in a mixture containing 400 μm CDP-[methyl-14C]choline (2Ci/mol), 8 mm Mg2+, and 125, 250, 500, or 1000 μm DAG. Data are mean ± SEM of three experiments. *p < 0.05, significantly different from control cells.
NMDA does not modify the availability of DAG
Reduced substrate availability is one of the factors that might explain NMDA-evoked inhibition of cholinephosphotransferase activity. The NMDA-evoked accumulation of radioactive CDP–choline shown in Figure 4 indicates that NMDA receptor overactivation either prevents the use of this substrate by cholinephosphotransferase or reduces the availability of the cosubstrate. Thus, we investigated the effect of NMDA on the mass of DAG, the other substrate of cholinephosphotransferase. To do so, neuronal cultures were treated with either vehicle or NMDA (300 μM) for 10, 20, and 30 min. We found that NMDA did not induce any change in DAG mass when compared with control cells (Table 1). Therefore, a reduction of substrate availability does not account for NMDA-evoked inhibition of cholinephosphotransferase activity.
Levels of DAG
NMDA receptor overactivation inhibits ethanolaminephosphotransferase activity
Cholinephosphotransferase activity may be catalyzed by two different enzymes (Henneberry and McMaster, 1999; Henneberry et al., 2000, 2002). One of these enzymes exhibits both cholinephosphotransferase and ethanolaminephosphotransferase activity. Moreover, in neurons, PtdEtn can be synthesized through the CDP–ethanolamine pathway or by decarboxylation of PtdSer. However, the contribution of the last pathway to the synthesis of PtdEtn in brain is very small (Araki and Wurtman, 1998). Indeed, when we labeled our primary neuronal cultures for 30 min with [3H]Ser and measured the presence of radioactivity in PtdEtn and PtdSer, only a 7% of radioactivity was detected in PtdEtn and a 93% of radioactivity was detected in PtdSer. Identical ratio was observed in cells treated with NMDA. Therefore, to investigate the effect of NMDA receptor overactivation on the synthesis of PtdEtn, we focused on the different enzymes of the CDP–ethanolamine pathway and performed metabolic labeling experiments with [3H]Etn (0.49 Ci/mmol). In these experiments, 10–30 min exposure to NMDA (300 μM) decreased to one-third the incorporation of [3H]Etn to phosphoethanolamine, indicating that NMDA receptor overactivation inhibits the activity of ethanolamine kinase. This effect of NMDA is not followed by a decrease in radioactive CDP–ethanolamine. On the contrary, Figure 6C shows a small increase of labeled CDP–ethanolamine by NMDA. Moreover, pulse-chase experiments using radiolabeled ethanolamine showed that NMDA receptor overactivation induced accumulation of radioactive CDP–ethanolamine and decreased radioactive PtdEtn (Fig. 7), indicating that NMDA inhibits ethanolaminephosphotransferase activity.

Time course analysis of the effect of NMDA on the incorporation of [3H]Etn into PtdEtn and metabolites of the CDP–ethanolamine pathway. Neuronal cortical cultures were incubated for up to 30 min in 300 μl of MLH containing 1.5 μCi of [3H] Etn (0.49Ci/mmol) in the presence of either vehicle (○) or NMDA (•) (300 μm). Lipids were extracted to determine the radioactivity present inethanolamine (A), phosphoethanolamine (B), CDP–ethanolamine (C), and PtdEtn (D) at different times after the addition of NMDA (300 μm). Results are expressed in disintegrations per minute per 1000 and are mean ± SEM of three experiments. * indicates significantly different from control cells; p < 0.05.

Pulse-chase experiments show that NMDA receptor overactivation inhibits the incorporation of [3H]Etn into membrane PtdEtn by decreasing ethanolaminephosphotransferase activity. Neuronal cortical cell cultures were incubated for 30 min in 300 μl of MLH containing 1 μCi of [3H]Etn (27 Ci/mmol) (pulse), washed, and incubated in MLH containing 250 μm unlabelled Etn in the presence of either vehicle (○) or NMDA (•) (300 μm) (chase). Phosphoethanolamine (A), CDP–ethanolamine (B), and PtdEtn (C) were separated by TLC, and the radioactivity was measured. Results are expressed in disintegrations per minute per 1000 and are mean ± SEM of three experiments. * indicates significantly different from controls; p < 0.05.
Accumulation of CDP–choline and CDP–ethanolamine precede NMDA-evoked membrane damage and cell death
PI is excluded by the plasma membrane of viable cells but passes through the plasma membrane of damaged cells. In these damaged cells, PI reaches the nucleus, in which it fluoresces when interacting with DNA. We studied the time course of cell death induced by NMDA receptor overactivation as measured by PI fluorescence. We found that continuous exposure to neurotoxic concentrations of NMDA (250 μM) does not induce significant cell death within the first hour of treatment. However, exposure to NMDA for 10 min already produces a marked accumulation of CDP–choline and CDP–ethanolamine (Fig. 8). Thus, inhibition of choline–ethanolaminephosphotransferase by NMDA receptor overactivation precedes by at least 1 hr membrane damage associated with excitotoxicity.

A, NMDA induces a rapid accumulation of both CDP–choline and CDP–ethanolamine. Neuronal cortical cultures were preincubated for 30 min in 300 μl of MLH containing [3H]Cho (0.33 Ci/mmol) or [3H]Eth (27 Ci/mmol) (pulse), washed, and incubated for 10 min in MLH containing 250 μm unlabelled Cho or Etn, respectively, in the presence of either vehicle (open bars) or NMDA (filled bars) (300 μm) (chase). Data are mean ± SEM of three experiments. * indicates significantly different from control cells; p < 0.05. B, Time course of neuronal death by NMDA. Cell death was measured with propidium iodide staining in cortical cell cultures at several times after continuous treatment with vehicle (○) or 250 μm NMDA (•). Data are mean ± SEM of three experiments. *p < 0.05, significantly different from control cells.
Discussion
The present results show that there is no phospholipid degradation in the early stages of the excitotoxic process. Thus, exposure to neurotoxic concentrations of NMDA did not reduce the radioactivity present in the major phospholipid classes PtdCho, PtdEtn, PtdIns, and PtdSer in neurons prelabeled with [3H]Cho, [3H]glycerol, [32P]orthophosphoric acid, or [3H]myristic acid (Fig. 1B–E). Nonetheless, NMDA reduced the amount of radioactivity present in PtdCho and PtdIns prelabeled with [3H]arachidonic acid (Fig. 1F). This effect might be related to the preferential labeling by [3H]arachidonic acid of a small, rapid turnover pool of PtdCho that is more accessible to phospholipase A2. However, this result is consistent with several studies showing that NMDA receptor activation enhances PLA2 activity and releases [3H]arachidonic acid from the sn-2 position of PtdCho and PtdIns (Sanfeliu et al., 1990; Farooqui et al., 1997). Thus, the reduction of [3H]arachidonic acid labeling in PtdCho and PtdIns by NMDA is most likely attributable to activation of PLA2 but not to NMDA-evoked phospholipid degradation, because this effect of NMDA on PtdCho and PtdIns prelabeled with [3H]arachidonic acid is not observed when PtdCho and PtdIns are prelabeled with any of the other radioactive markers used (Fig. 1B–E). This indicates that a subsequent addition of an unlabeled fatty acid into the sn-2 position transforms the lysophospholipids produced by NMDA-evoked activation of PLA2 into the original phospholipids. Thus, our results show that the enhanced hydrolysis of PtdIns and PtdCho and subsequent release of arachidonic acid evoked by NMDA does not lead to phospholipid degradation or membrane damage, at least in the early stages of the excitotoxic process. These findings are consistent with previous results showing that NMDA receptor-evoked release of Cho is independent of phospholipase activity and is associated with a decrease in PtdCho synthesis (Zapata et al., 1998; Gasull et al., 2000). Accordingly, we studied the activity of the different enzymes of the CDP–choline pathway to identify the mechanism by which NMDA receptor overactivation inhibits PtdCho synthesis.
Metabolic labeling experiments showed that NMDA receptor overactivation induced a significant decrease of the incorporation of radioactivity into membrane PtdCho as early as 10 min after the beginning of treatment and at least 1 hr before significant membrane damage and cell death (Figs. 3, 8). This effect was concentration dependent (Fig. 2A) and was produced by an action at NMDA receptors because it was completely blocked by MK-801(10 μM) (Fig. 2B), a noncompetitive antagonist of the NMDA subtype of glutamate receptors (Wong et al., 1986). The rapid time course of the effect of NMDA on PtdCho synthesis is consistent with our previous results showing that NMDA receptor overactivation releases Cho almost immediately after treatment (Zapata et al., 1998; Gasull et al., 2000).
In contrast to the marked effect of NMDA on incorporation of [3H]Cho into PtdCho, NMDA receptor overactivation did not modify the rate of incorporation of radioactivity into phosphocholine or CDP–choline. Choline kinase and CTP:phosphocholine cytidylyltransferase, the enzymes that catalyze the sequential conversion of Cho into phosphocholine and CDP–choline require ATP and CTP, respectively. Indeed, treatments that deplete cell energy resources, e.g., glucose deprivation, elicit a marked decrease in choline kinase activity (Gasull et al., 2002). In cortical cells, neurotoxic concentrations of NMDA produce a time-dependent reduction of cell energy resources that may reach a 20% decrease of cell energy charge 30 min after treatment (Ankarcrona et al., 1995). However, the lack of effect of NMDA receptor overactivation on the activity of the energy-dependent enzymes of the CDP–choline pathway observed in our experiments indicates that reduction of cell energy resources by NMDA receptor overactivation is either not sufficient or it follows a slower time course than the inhibition of PtdCho synthesis evoked by NMDA.
Pulse-chase experiments using [3H]Cho showed that NMDA receptor overactivation does not modify the rate of use of phosphocholine (Fig. 4A). Conversely, treatment with NMDA decreased the rate of use of CDP–choline (Fig. 4B), showing that NMDA receptor overactivation inhibits PtdCho synthesis by selectively reducing cholinephosphotransferase activity. Experiments using [3H]cytidine as a radioactive marker provide strong and additional support for this interpretation, because NMDA receptor overactivation induced also accumulation of [3H]CDP–choline in [3H]cytidine-prelabeled neurons. These results show that NMDA receptor overactivation inhibits PtdCho synthesis by specifically reducing cholinephosphotransferase activity but leaves choline kinase and CTP:phosphocholine cytidylyltransferase activities unaltered.
It is well established that Ca2+ influx through the NMDA subtype of glutamate receptors, but not a general increase in the amount of cytosolic Ca2+, plays a central role for triggering the cascade of pathophysiological events leading to excitotoxic cell death (Sattler and Tymianski, 2000). The inhibition of cholinephosphotransferase activity and PtdCho synthesis after treatment with NMDA observed in our studies is most likely produced by Ca2+entry through NMDA receptors. Indeed, we showed previously that Cho release by NMDA is blocked in the absence of extracellular Ca2+ (Gasull et al., 2000). On the other hand, Ca2+ has been reported to decrease cholinephosphotransferase activity in vitro (Taniguchi et al., 1986). However, whether the reduction of cholinephosphotransferase activity in vivo by NMDA receptor activation is produced by a direct or an indirect effect of Ca2+ on the activity of the enzyme is at present unknown. There is little knowledge on the factors that regulate cholinephosphotransferase activity in neuronal cells, probably because most studies focused on the regulation of the activity of CTP:phosphocholine cytidylyltransferase, the enzyme that is rate-limiting in PtdCho synthesis (Kent, 1997). However, it has been reported that the supply of DAG, one of the substrates of cholinephosphotransferases, is also rate limiting for PtdCho synthesis (Kolesnick and Hemer, 1990; Jamil et al., 1992) and that treatment with nerve growth factor, which increases DAG levels, enhances, in turn, cholinephosphotransferase activity in PC12 cells (Araki and Wurtman, 1997). Moreover, cholinephosphotransferase activity has been reported to be dependent on the concentration of cation activators such as Mg2+ (McMaster et al., 1996). Altogether, the reduction of cholinephosphotransferase activity by NMDA receptor overactivation could result from several possible mechanisms: first, a direct modification of the enzyme or alteration of its membrane localization by a Ca2+ dependent cytosolic factor; second, a change in the availability or localization of its substrates; and third, reduced availability of the cation activator. Our results support the first mechanism, because we found that NMDA receptor overactivation neither reduced CDP–choline (Fig. 4B) nor decreased the mass of DAG (Table 1). Nonetheless, we observed that, when substrate and Mg2+ concentrations are controlled in microsomes from control and NMDA-treated cells, cholinephosphotransferase activity is still inhibited (Fig. 5).
In addition, NMDA receptor overactivation reduced the incorporation of [3H]Etn into PtdEtn. Treatment with NMDA decreased the incorporation of [3H]Etn into PtdEtn and also induced accumulation of labeled CDP–ethanolamine. These results indicate that NMDA receptor overactivation, in addition to decreasing cholinephosphotransferase activity, reduces ethanolaminephosphotransferase activity (Figs. 6, 7). However, in this latter case, the effect of NMDA is not as selective as the one observed on the CPD–choline pathway, because analysis of PtdEtn metabolites showed that NMDA also inhibits ethanolamine kinase activity, the enzyme that catalyzes the first step of the PtdEtn synthesis pathway. Despite this, NMDA did not reduce the amount of radioactive CDP–ethanolamine, indicating that inhibition of ethanolamine kinase activity does not really contribute to the effect of NMDA on PtdEtn synthesis. The cytidylyltransferase reaction is a rate-limiting step for PtdEtn biosynthesis, and the high molar ratio of phosphoethanolamine to CDP–ethanolamine allows for a significant reduction of phosphoethanolamine without altering CDP–ethanolamine levels and PtdEtn synthesis. Subsequent pulse-chase experiments provided strong support for this interpretation because NMDA induced accumulation of labeled CDP–ethanolamine. In summary, these experiments clearly show that NMDA receptor overactivation inhibits PtdEtn synthesis by inhibiting ethanolaminephosphotransferase activity.
There are two different genes encoding enzymes with cholinephosphotransferase activity in mammals (Henneberry and Mc-Master, 1999; Henneberry et al., 2000, 2002). The cpt1 gene codes for a CDP–choline-specific cholinephosphotransferase for the exclusive synthesis of PtdCho, whereas cept1 codes for a dual-specificity choline–ethanolaminephosphotransferase that can use both CDP–choline and CDP–ethanolamine as substrates to synthesize both PtdCho and PtdEtn. Expression of cpt1 varies 100-fold in the main body organs and is very low in brain (Henneberry et al., 2000), whereas cept1 is equally present in the main body organs including brain (Henneberry and McMaster, 1999). Therefore, in neurons, the same enzyme most likely catalyzes the last step of the Kennedy pathway for the synthesis of PtdCho and PtdEn. The observation that NMDA receptor overactivation inhibits both cholinephosphotransferase and ethanolaminephosphotransferase activities provides support for this interpretation and suggests that NMDA inhibits phospholipid synthesis by decreasing the activity of CEPT1.
In summary, our results show that, in the early stages of the excitotoxic process, NMDA receptor overactivation alters membrane phospholipid composition by reducing de novo synthesis of PtdCho and PtdEtn without altering phospholipid degradation. This effect of NMDA is produced by a reduction of choline–ethanolamine phosphotransferase activity. Because PtdCho and PtdEtn are the major structural building blocks of cellular membranes, a decrease in its synthesis is likely to be responsible for the alterations of membrane structure and function associated with excitotoxic necrosis. Therefore, the present findings provide additional support to the hypothesis that cytoplasmic membrane damage during excitotoxicity, instead of being a passive process of cell destruction, is produced by alteration of biochemical processes involved in membrane building.
Footnotes
This work was supported by Grants BFI2001–1035 from Dirección General de Investigación, Ministerio de Ciencia y Tecnologia (MCyT) of Spain, and PI020555 from Fondo de Investigación Sanitaria, Ministerio de Sanidady Consumo of Spain (R.T.). T.G. was supported by a contract from “Programa Ramon y Cajal” of MCyT of Spain. We thank Juana Maria Hurtan for technical assistance.
Correspondence should be addressed to Ramon Trullas, Neurobiology Unit, Institut d'Investigacions Biomèdiques de Barcelona, Consejo Superior de Investigaciones Científicas, Institut d'Investigacions Biomèdiques August Pi i Sunyer, Rosselló 161, 08036 Barcelona, Spain. E-mail: rtonbi{at}iibb.csic.es.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234100-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}