

Abstract
Prenatal nicotine exposure has been linked to attention deficit hyperactivity disorder and cognitive impairment, but the sites of action for these effects of nicotine are still under investigation. High-affinity nicotinic acetylcholine receptors (nAChRs) contain the β2 subunit and modulate passive avoidance (PA) learning in mice. Using an inducible, tetracycline-regulated transgenic system, we generated lines of mice with expression of high-affinity nicotinic receptors restored in specific neuronal populations. One line of mice shows functional β2 subunit-containing nAChRs localized exclusively in corticothalamic efferents. Functional, presynaptic nAChRs are present in the thalamus of these mice as detected by nicotine-elicited rubidium efflux assays from synaptosomes. Knock-out mice lacking high-affinity nAChRs show elevated baseline PA learning, whereas normal baseline PA behavior is restored in mice with corticothalamic expression of these nAChRs. In contrast, nicotine can enhance PA learning in adult wild-type animals but not in corticothalamic-expressing transgenic mice. When these transgenic mice are treated with doxycycline in adulthood to switch off nAChR expression, baseline PA is maintained even after transgene expression is abolished. These data suggest that high-affinity nAChRs expressed on corticothalamic neurons during development are critical for baseline PA performance and provide a potential neuroanatomical substrate for changes induced by prenatal nicotine exposure leading to long-term behavioral and cognitive deficits.
Introduction
The comorbidity of prenatal nicotine exposure with psychiatric disorders and cognitive impairment suggests that nicotinic acetylcholine receptors (nAChRs) may play a role in the etiology of these disorders. In utero exposure to nicotine can increase the risk of attention deficit hyperactivity disorder (ADHD), lower IQ, and conduct disorder later in life (Ernst et al., 2001; Wakschlag et al., 2002). Determining the nAChR subtypes and the neuroanatomical loci responsible for these effects is difficult using traditional pharmacological approaches because of the large number of nAChR subtypes, their overlapping pharmacological profiles, and their wide distribution in the brain. The thalamus has been implicated in learning and memory, attention, arousal, and emotionality (Masterman and Cummings, 1997; Kalivas et al., 1999). In addition, the thalamus contains the highest density of high-affinity nAChRs in the brain (composed predominantly of the α4 and β2 nAChR subunits) (Zoli et al., 1998), but virtually nothing is known about the function of nAChRs in this region. There are two main neuronal types in thalamus: GABAergic interneurons and glutamatergic thalamocortical relay neurons (Guillery and Sherman, 2002). Glutamatergic sensory projections, glutamatergic projections from layer VI of the cortex, and cholinergic pedunculopontine projections all synapse onto thalamic neurons (Hallanger et al., 1987). Functional nAChRs are present at several levels of this system on both cell bodies and terminals (Léna and Changeux, 1997; Lu et al., 1998; Corrigall et al., 1999). The enrichment of high-affinity nAChRs in this region make it a potential target for developmental changes caused by alterations in nicotinic receptor function.
Passive avoidance (PA) is a fear-associated learning test sensitive to manipulations of the cholinergic system and a model for emotional learning in humans. Cholinergic deafferentation of thalamus in rodents, as in thiamine-deficient rats with reduced choline-acetyl-transferase levels in thalamus (and other brain areas), is correlated with impairments in PA learning (Nakagawasai et al., 2000). Senescence accelerated mice also have lower levels of acetylcholine in thalamus and hypothalamus and show impaired PA performance that is rectified by acute, pretrial administration of nicotine (Meguro et al., 1994). β2 nAChR subunit knock-out (β2 ko) mice lack all high-affinity binding for nicotine and show abnormally high baseline PA and no improvement in learning as a result of post-training nicotine administration when compared with wild-type siblings (Picciotto et al., 1995). To identify where in the brain nicotinic receptors exert their actions on PA learning, transgenic mice with temporally and spatially restricted expression of high-affinity nAChRs were generated and tested in PA. These mice were used to identify the nAChR subtypes and neuronal populations that appear critical for the long-term behavioral consequences of genetic or pharmacological manipulations of nicotinic neurotransmission.
Materials and Methods
Animals. The cDNA encoding the β2 subunit was generated by reverse transcription (RT)-PCR from mouse brain mRNA (5′, TTT AAG CTT- GCG CGG CTT CAG CAC CAC GGA CAG CGC; 3′, TTT ACT AGT- TCC ACC CAA TAC TAC TGA ACC) and subcloned into pTet-splice (Invitrogen, Gaithersburg, MD). The plasmid was sequenced, and the band containing the Tet-β2 construct and SV40 3′ untranslated region was excised, purified by gel electrophoresis, and microinjected into B6SJL oocytes (Yale University Transgenic Facility). Founder mice carrying the transgene were crossed with Line A NSE-tTA mice on the ICR background (Chen et al., 1998; Kelz et al., 1999) and β2 ko mice on the C57BL/6J background (Picciotto et al., 1995). C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Genotyping was performed by PCR on phenol-extracted tail DNA. Mice were crossed to generate β2 ko or β2 heterozygote (β2 het) offspring with at least one copy of the Tet-β2 and NSE-tTA transgenes. Litters of mice on a mixed genetic background of β2 wild-type (wt), β2 het, and β2 ko genotype with copies of one or both of the transgenes were used for behavioral experiments. β2 ko mice with both transgenes (β2 tr) expressed the β2 subunit inducibly in different brain regions. β2 tr, β2 wt, and het mice with or without the transgenes (wild type), and β2 ko mice with no transgenes or only a single transgene (β2 ko) were compared. Mice were group housed with a maximum of five per cage in a colony room maintained at 22°C on a 12 hr light/dark cycle, with lights on at 7:00 A.M. Food and water were available ad libitum. All animal procedures were in strict accordance withNIH Care and Use of Laboratory Animals Guidelines and were approved by the Yale Animal Care and Use Committee.
PA learning. Testing was performed in a mouse PA chamber (Ugo Basile, Comerio, Italy). On day 1, the mouse was placed in the light chamber and allowed to move freely between the two compartments for 5 min. On day 2, the mouse was placed in the light chamber, and latency to enter the dark chamber was measured. After entry into the dark chamber, the door between compartments closed, and a 2 sec electric shock (0.2 mA) was administered through the grid floor. The animal was removed from the apparatus, given an intraperitoneal injection (10 μl/gm) of 0.9% saline or 10 μg/kg nicotine bitartrate (concentration calculated as free base), and returned to a holding cage. On day 3, mice were again placed in the light chamber, and time to enter the dark chamber was recorded. Mice used in the initial passive avoidance experiment were aged from 2 to 9 months. No differences in PA behavior were seen with age. Another cohort of mice (aged 4–12 months at testing) was divided into two groups (balanced by genotype, sex, and age), and half were given doxycycline (dox) in their drinking water (100 μg/ml) for 5 to 7 weeks before PA testing.
Shock reactivity test. Shock reactivity was measured as described previously (Caldarone et al., 2000). Mice were given a series of 1 sec shocks starting at 0.05 mA and increasing to 1 mA in increments of 0.05 mA, with a 19 sec intershock interval. Mice were scored by two observers for flinch (any observable reaction to the shock), run, jump, or vocalization reactions. For each animal, the experiment was stopped when the mouse had displayed all four reactions. Mean current thresholds to evoke each response were calculated and averaged between observers.
In situ hybridization and equilibrium binding. Mice were decapitated, and brains were removed, frozen on dry ice, and stored at −80°C. Sections (12 μm) were cut at the cryostat, thaw mounted onto chrom-alum-coated slides [0.5% chromium (III) phosphate–0.5% gelatin] for radioligand binding or charged slides (Fisherbrand Superfrost/Plus; Fisher Scientific, Pittsburgh, PA) forin situ hybridization. Sections were dried at room temperature for 20 min and stored at −80°C. The large intracellular loop of the β2 subunit (amino acids 338–456) was subcloned into pBluescript. Antisense and sense cRNA probes were transcribed with T7 or SP6 RNA polymerase in the presence of [35S]UTP (NEN, Boston, MA) and purified through RNA mini-quick-spin columns (Roche, Indianapolis, IN). Brain sections were postfixed in 4% paraformaldehyde in 1× PBS (in mm: 1 KH2PO4, 10 Na2HPO4, 1.37 NaCl, and 2.7 KCl, pH 7.4), acetylated for 15 min in 0.1 mtetraethylammonium and 0.25% acetic anhydride, and dehydrated through an ethanol series. Slides were hybridized overnight at 60°C (106 cpm per slide), washed in 2× SSC (0.3 m sodium acetate and 0.03m sodium citrate), treated with 20 μg/ml RNaseA (Roche), washed in descending concentrations of SSC (to 0.1×) at 55°C, rinsed with water, dried, and exposed to3H-Hyperfilm (Amersham Biosciences, Arlington Heights, IL) for 15 d.
For radioligand binding, sections were thawed at room temperature and incubated with 200 pm[125I]A85380 for 30 min in 50 mm Tris-HCl, pH7.4, washed twice in the same buffer, dried, and exposed to 3H-Hyperfilm for 2–7 d.
Tissue preparation and rubidium efflux. Mice were killed, and brain regions were dissected on ice. Samples were homogenized in cold isotonic sucrose (0.32 msucrose with 5 mm HEPES, pH 7.5) in a tissue grinder with 16 strokes by hand. Homogenates were centrifuged at 12,000 × g for 20 min, and pellets were resuspended in different buffers depending on the following assay. Rubidium efflux experiments were performed as described previously (Marks et al., 2002). Briefly, homogenized and centrifuged tissue was resuspended in buffer A (in mm: 140 NaCl, 1.5 KCl, 2 CaCl2, 1 MgSO4, 20 glucose, and 20 HEPES hemisodium, pH 7.5). Aliquots (25 μl) were incubated with 4 μCi of [86Rb+] for 30 min at 22°C in a final volume of 35 μl. Uptake was terminated by filtration of each sample onto a Gelman A/E glass fiber filter under gentle vacuum and washing once with 0.5 ml of buffer A. The washed filters were transferred to a polypropylene platform and superfused with buffer B (135 mm NaCl, 5 mm CsCl, 1.5 mm KCl, 1 mm MgSO4, 2 mm CaCl2, 20 mm glucose, 50 nmtetrodotoxin, 1 μm atropine, 20 mm HEPES hemisodium, and 0.1% bovine serum albumin, pH 7.5). Buffer was applied at a rate of 2.5 ml/min and was actively removed with a second peristaltic pump set for a flow rate of 3.2 ml/min. The buffer was pumped through a 200 μl Cherenkov cell in α-RAM Radioactivity HPLC detector (IN/US Systems, Tampa, FL) to allow continuous monitoring of radioactivity. Samples were stimulated by diverting the application buffer through a 200 μl loop containing the test solution (30 μm nicotine). Stimulation time was 5 sec. Baseline efflux was calculated using points before and after stimulation. Magnitude of [86Rb+] efflux was calculated as stimulation above basal and normalized to basal efflux to allow comparison among regions and genotypes.
Epibatidine binding. Epibatidine binding was performed as described previously (Marks et al., 2002). Samples were incubated in a 96-well polystyrene plate for 2 hr at 22°C. Final incubation was in 30 μl of binding buffer. Binding reactions (200 pm [125I] epibatidine) were terminated by filtration onto Gelman A/E glass fiber paper that had been treated with 0.5% polyethylenimine. Samples were washed six times with cold load buffer. Bound ligand was quantified using a Packard Cobra Gamma Counter. Counting efficiency was 85%. Blanks were determined with 100 μm nicotine, and all results presented are specific binding.
GABA release. [3H]GABA release from synaptosomes was performed as described previously (Lu et al., 1998). Homogenates from cortex, thalamus, hippocampus, and striatum were centrifuged at 12,000 × g for 20 min. The resulting crude synaptosomal pellet was resuspended in uptake buffer (in mm: 128 NaCl, 2.4 KCl, 1.2 KH2PO4, 1.2 MgSO4, 3.2 CaCl2, 25 HEPES, and 10 glucose, pH 7.5) and incubated with 1 mmaminooxyacetic acid at 37°C for 10 min. [3H]GABA (0.1 μm) and unlabeled GABA (0.25 μm) were added and incubated 10 min at 37°C. Aliquots were collected onto glass fiber filters and washed with 0.5 ml of perfusion buffer (119 mm NaCl, 3.6 mm KCl, 1.2 mmMgSO4, 10 mm CsCl, 3.2 mm CaCl2, 25 mm HEPES, 10 mm glucose, and 0.1% BSA, pH 7.5). Perfusion buffer was pumped over synaptosomes at 1.8 ml/min (RT). Release was stimulated by a 12 sec exposure to 30 μml-nicotine. Data (as counts per minute released per fraction) were corrected for baseline release from fractions collected before and after the agonist stimulation. Data were then normalized to baseline, and fractions in which release exceeded baseline by 10% or more were summed. Units of release are as fraction of baseline.
Results
Inducible, region-specific expression of β2 subunit-containing nAChRs
Transgenic mice were generated that express the β2 subunit of the nAChR under the control of a tetracycline-regulated promoter (TetOp-β2). Founder mice were crossed with β2 ko mice carrying a transgene encoding the neuron-specific enolase promoter driving expression of the tetracycline transactivator (NSE-tTA). In β2 tr mice, all β2 subunit-containing nAChRs in the brain were expressed from the tetracyline-regulatable transgene. Equilibrium binding using the nicotinic agonist [125I]epibatidine in brain slices identified three TetOp-β2 lines of mice with increased densities of high-affinity binding sites (data not shown). Region-specific patterns of binding were similar in all three lines, and one line was picked for further characterization. This line was crossed to three different NSE-tTA founder lines (Chen et al., 1998;Kelz et al., 1999). Equilibrium binding studies using [125I]A85380, a nicotinic ligand specific to β2-containing nAChRs (Mukhin et al., 2000), showed distinct patterns of expression in each of the NSE-tTA lines (Fig.1). High-affinity nicotinic binding in β2 tr(CT) mice was restricted to cortex and thalamus. β2 tr(VN) mice expressed high-affinity nAChRs in the optic tract, visual nuclei of the thalamus, and superior colliculus. β2 tr(VTA) mice showed high-affinity nicotinic binding predominantly in nucleus accumbens, substantia nigra, ventral tegmental area, and striatum. In contrast, β2 het siblings of the β2 tr mice showed A85380 binding throughout the brain, and β2 ko mice had no detectable binding. As a result of the enrichment of nicotine binding in thalamus and cortex in the β2 tr(CT) transgenic mice, this line was selected for further characterization in neurochemical and behavioral paradigms.

Transgenic mice with region-specific expression of β2 subunit-containing nAChRs. Mice from three transgenic lines carrying the NSE promoter driving expression of the tetracycline transactivator (NSE-tTA) (Chen et al., 1998; Kelz et al., 1999) were crossed with mice carrying the TetOp promoter driving the β2 subunit (TetOp-β2) on a β2 ko background. [125I]A85380binding is shown in β2 het, β2 ko, and three lines of β2 tr mice (CT, VN, and VTA). Bregma coordinates are shown for the four levels (Paxinos and Franklin, 1997).
Inducible expression in β2 tr(CT) mice is restricted to corticothalamic projection neurons
Nicotinic ligand binding was performed on membranes isolated from several brain regions to provide a quantitative measure of high-affinity nAChRs expressed in β2 tr(CT) mice (Fig.2A). There was a clear effect of β2 subunit gene dosage on epibatidine binding in most brain areas across β2 wt, het, and ko mice, as has been reported previously (Marks et al., 1999); however, β2 tr(CT) mice showed significant epibatidine binding in the cortex and thalamus only. In situhybridization was performed using the intracellular loop region of the β2 subunit as a probe in β2 tr(CT) mice. β2 subunit mRNA was detected specifically in layer VI of the cerebral cortex, CA1, and dentate gyrus of the hippocampal formation and throughout the dorsal striatum, but no specific hybridization was seen in the thalamus (Fig.2B). This pattern is similar to that seen for other genes of interest driven by the same NSE-tTA line (Chen et al., 1998;Kelz et al., 1999). Ectopic expression of the β2 subunit transgene in cells not normally expressing the heteromeric nAChRs does not result in the production of assembled nAChRs or high-affinity nicotinic radioligand binding because the β2 subunit can only form a functional channel in the presence of its partner subunits (Luetje and Patrick, 1991). The equilibrium binding data coupled with the in situhybridization studies suggest that β2 tr(CT) mice express functional high-affinity nAChRs only in corticothalamic projection neurons.
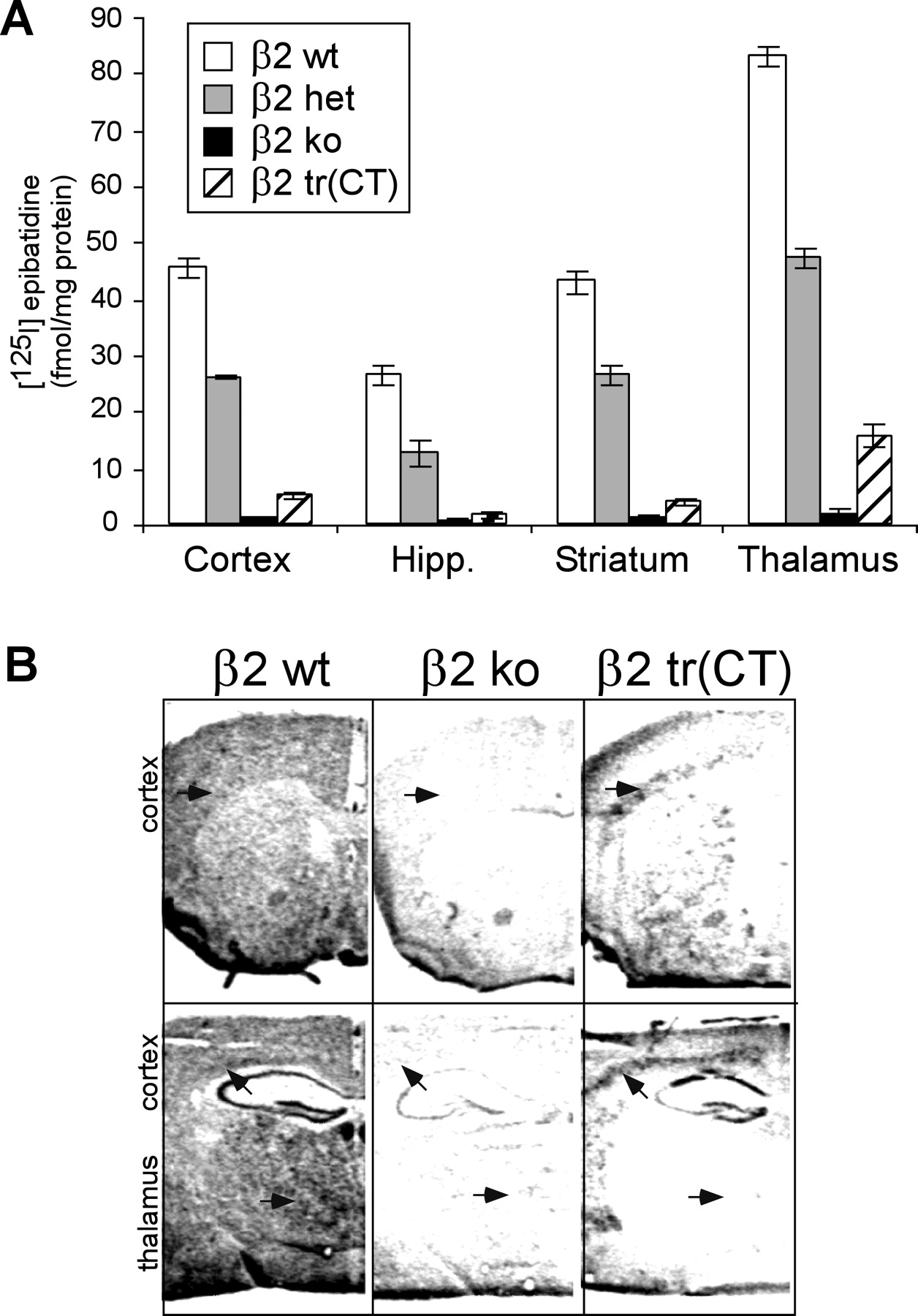
β2 subunit-containing nAChRs are expressed in the cortex and thalamus of β2 tr(CT) mice. A, Quantitiative epibatidine binding in microdissected brain regions of β2 wt, het, ko, and β2 tr(CT) transgenic mice (n = 3 for each genotype). ANOVA showed a significant effect of genotype on binding (p< 0.0001). Post hoc Newman–Keuls tests showed the β2 wt and β2 het mice to be different from each other and all other genotypes for all brain areas (p < 0.05). The β2 tr(CT) mice had greater binding than β2 ko mice in the cortex and thalamus (p < 0.05). Binding in the hippocampus (Hipp.) and striatum did not differ between β2 tr(CT) and β2 ko mice (p > 0.05).B, In situ hybridization with an antisense riboprobe against the large intracellular loop region of the β2 subunit at the level of the thalamus (bregma +1.10 mm) and prefrontal cortex (bregma −1.70 mm). Arrows point to the thalamus and layer VI of the cortex.
Functional characterization of β2 nAChRs in corticothalamic terminals
To confirm that nicotinic binding in the thalamus represents functional, terminal nAChRs and to identify the neurotransmitter released from these neurons, synaptosomal fractions were collected from several brain areas and were assayed for rubidium [86Rb] efflux (Fig.3A) and [3H]GABA release (Fig. 3B). The [86Rb+] efflux assay measures the opening of functional nicotinic receptor channels in synaptosomes in response to nicotine stimulation (Marks et al., 1995). The expected gene dosage response in [86Rb+] efflux stimulated by nicotine was seen across β2 wt, het, and ko mice (Fig. 3A) (Marks et al., 1999). A significant increase in [86Rb+] efflux was seen in the thalamus of β2 tr(CT) compared with β2 ko mice. Efflux was similar to that seen in β2 het mice, suggesting that β2 tr(CT) mice express physiologically relevant levels of high-affinity nAChR (Fig. 3A). There was no increase in [86Rb+] efflux in cortical synaptosomes from β2 tr(CT) mice above levels seen in β2 ko mice, demonstrating that nicotinic binding in this brain region is on cell bodies rather than neuronal terminals. Furthermore, no release was seen in striatum or hippocampus of β2 tr(CT) mice, as expected from the lack of nicotinic binding in these brain areas.

β2 tr(CT) mice have functional expression of β2 subunit-containing nAChRs on thalamic terminals of cortical projection neurons. A, Effect of genotype on nicotine-evoked rubidium efflux (30 μm nicotine) from synaptosomal preparations of microdissected brain regions (n = 4 for each genotype). ANOVA showed a significant effect of genotype on [86Rb] efflux in all brain areas (p < 0.0001). Post hoc Newman–Keuls tests showed the β2 wt mice to be different from all other genotypes in all brain areas (p < 0.05). The β2 tr(CT) mice differed from β2 ko mice only in the thalamus (p < 0.05). β2 tr(CT) did not differ from β2 het mice in [86Rb]efflux from thalamic synaptosomes (p > 0.05). B, Effect of genotype on nicotine-evoked GABA release. [3H]GABA release was measured in response to a 12 sec exposure to 30 μm nicotine in four brain regions (n= 4 for each genotype). ANOVAs of each brain area showed a significant genotype effect on nicotine evoked [3H]GABA release in all brain areas (p < 0.0001).Post hoc Newman–Keuls tests showed the β2 wt and β2 het mice to be different from all other genotypes in all brain areas (p < 0.05). There was no increase in [3H]GABA release in the β2 tr(CT) compared with β2 ko mice in any brain area (p > 0.05). No significant differences were seen for baseline release for any genotype in any brain area. Hipp., Hippocampus.
Both GABAergic and glutamatergic terminals in the thalamus express high-affinity nAChRs in wild-type mice. We therefore measured [3H]GABA release from synaptosomes in response to treatment with 30 μm nicotine to identify the neuronal subtypes expressing functional high-affinity nAChRs in β2 tr(CT) mice. As expected, β2 wt and het mice showed increased [3H]GABA release in response to nicotine treatment compared with β2 ko mice (Lu et al., 1998). In contrast, β2 tr(CT) mice show no increased GABA release over the level seen in β2 ko mice in any of the brain areas tested (Fig. 3B). This implies that these transgenic mice do not express high-affinity nAChRs in GABAergic neurons. Thus, nicotinic binding, in situ hybridization, and release data together suggest that β2 tr(CT) mice express high-affinity nAChRs in glutamatergic, layer VI cortical neurons projecting to the thalamus that are functional and can regulate neurotransmitter release from synaptic terminals (Turner and Salt, 1998).
Corticothalamic β2 nAChR expression restores baseline PA behavior
We tested PA behavior in β2 tr(CT) mice and compared these mice with sibling controls of different genotypes (Fig.4). In contrast to β2 ko mice, which showed hypersensitive PA learning compared with control siblings (Picciotto et al., 1995), β2 tr(CT) mice had PA performance similar to wild-type mice and had a significantly shorter latency to enter the dark chamber than β2 ko mice (Fig. 4). Thus, restricted expression of β2 subunit-containing receptors to corticothalamic efferents is sufficient for normal PA behavior.

Expression of β2 subunit-containing receptors in corticothalamic terminals is sufficient for normal baseline PA performance. PA performance was evaluated in wild-type (β2 het and β2 wt), β2 ko, and β2 tr(CT) mice [wild type,n = 17; β2 ko, n = 21; β2 tr(CT), n = 29]. A repeated-measures ANOVA of the baseline PA performance with day (testing vs training) as the within-subject variable and genotype [wild type, β2 ko, and β2 tr(CT)] as the between-subject variable revealed an interaction of day and genotype (F(2,64) = 4.311;p < 0.05). Post hoc Newman–Keuls tests showed the β2 ko mice to have a longer latency to enter the dark compartment on testing than the wild-type and β2 tr(CT) mice (p < 0.05).
We showed previously that nicotine is ineffective in improving PA performance in β2 ko mice (Picciotto et al., 1995). In the current study, although nicotine was able to enhance PA performance in wild-type mice [saline, 23.7 ± 5.2 sec (n = 17); nicotine, 56.4 ± 16.8 sec (n = 9); Newman–Keuls test; p < 0.05], it had no effect on β2 tr(CT) mice [saline, 31.8 ± 6.6 sec (n = 29); nicotine, 26.7 ± 1.6 sec (n = 11)]. Thus, expression of high-affinity nAChRs on corticothalamic neurons rescues baseline performance, although it is not sufficient to restore nicotine-mediated enhancement of PA. These data suggest that nAChRs in other brain regions, such as the amygdala, striatum, or hippocampus, may be critical for the nicotine-mediated enhancement of PA learning.
The restored baseline PA learning in β2 tr(CT) mice was attributable to β2 subunit expression rather than transgene insertion, because no difference in baseline PA behavior was seen in mice carrying either the NSE-tTA transgene or the TetOp-β2 transgene alone (Fig.5A). The ability of transgenic expression from a different locus to restore a phenotype altered in the β2 ko mice also confirms that the original phenotype was not attributable to effects of flanking genes surrounding the mutant allele (Banbury Conference on Genetic Background in Mice, 1997; Bolivar et al., 2000). Furthermore, the difference in PA performance in β2 tr(CT) mice cannot be explained by altered pain sensitivity because no differences were seen in shock reactivity at intensities similar to those used in PA training (Fig. 5B).

The PA phenotype is not attributable to transgene insertion or alteration of shock sensitivity. A, The insertion of the individual transgenes on the β2 het or β2 ko backgrounds does not affect PA performance. β2 het and β2 ko mice with either the TetOp-β2 or NSE-tTA transgene alone were tested for PA behavior (β2 het/TetOp-β2, n = 8; β2 het/NSE-tTA, n = 9; β2 ko/TetOp-β2,n = 15; β2 ko/NSE-tTA, n = 12). Repeated-measures ANOVA with day (testing vs training) as the within-subject variable and genotype (β2 het vs β2 ko) and transgene (TetOp-β2 vs NSE-tTA) as between-subject variables showed an interaction of day and genotype (F(1,40) = 9.015; p< 0.01) but no main effect or interaction with single transgenes.B, The altered PA phenotype is not a result of differential shock reactivity. Mice of different genotypes [β2 wt and β2 het, β2 ko and β2 tr(CT)] were tested for response to increasing levels of shock. (β2 wt and β2 het were combined as “wild types” for subsequent analysis). Mean lowest shock that evoked the responses flinch, run, and vocalize for each of the three genotypes (n = 7 per genotype) is plotted. ANOVA showed no effect of genotype (p > 0.05 in all cases).
Developmental expression of β2 nAChRs is critical for baseline PA
One possible role for high-affinity nAChRs in thalamus could be to modulate sensory processing through thalamus to cortex by altering synaptic strength in glutamatergic neurons during development. To determine whether high-affinity nAChRs act acutely to regulate PA behavior in the adult or whether the receptors are critical earlier in development, adult mice were administered 100 μg/ml dox in their drinking water for 30 d to abolish expression of high-affinity nAChRs in β2 tr(CT) mice (Fig.6A). dox administration had no effect on PA performance of mice of any genotype (Fig.6B). These data suggest that expression of β2 subunit-containing nAChRs in corticothalamic efferents during development results in a long-lasting alteration of this pathway that facilitates PA learning in the adult.

Expression of the β2 subunit during development is necessary for normal PA learning. A, β2 het, β2 ko, and β2 tr(CT) mice were treated with dox (100 μg/ml) in their drinking water for 4 weeks. No change was seen in [125I]A85380 binding in β2 wt (data not shown) or β2 het mice after dox treatment, but no binding was detectable in β2 tr(CT) mice administered dox. B, Four weeks of dox treatment in adult mice does not alter PA performance in β2 tr(CT) mice [β2 het, no dox, n = 4; β2 het with dox, n = 6; β2 ko, no dox,n = 4, β2 ko with dox, n = 9; β2 tr(CT), no dox, n = 10; β2 tr(CT) with dox,n = 11]. A repeated-measures ANOVA with day (training vs testing) as the within-subject variable and dox treatment (on or off) and genotype [β2 het, β2 ko, and β2 tr(CT)] as the between-subject variables showed an interaction of day and genotype (F(2,39) = 13.382;p < 0.01) but no main effect or interaction with dox.
Discussion
We generated transgenic mice with inducible, region-specific expression of β2 subunit-containing nAChRs in corticothalamic projection neurons. Using these mice, we showed that alterations in neurotransmission through nAChRs during development can affect PA performance in the adult. Nicotine treatment during development is known to have neurodevelopmental and behavioral consequences. In humans, maternal smoking has been associated with babies of lower birth weight, increased risk of sudden infant death syndrome, and increased risk of psychiatric disorders in childhood and adolescence (Ernst et al., 2001), including ADHD (Milberger et al., 1996; Milberger et al., 1998). Cognitive impairments and hyperactivity have also been reported in animals exposed to nicotine during development (Genedani et al., 1983; Sorenson et al., 1991; Pennington et al., 1994; Shacka et al., 1997). A blunting of both cholinergic and catecholaminergic function is also seen in rats prenatally exposed to nicotine (Navarro et al., 1988,1989; Oliff and Gallardo, 1999). Thus, expression of nAChRs during prenatal and postnatal development is likely to be critical for normal cognitive behavior in the adult.
The first 2 weeks of life are a critical period for maturation of glutamatergic synapses on corticothalamic neurons, with targeting of glutamate receptor subunits such as mGluR1a to the distal dendrites of these synapses during the second postnatal week of rat development (Liu et al., 1998). Chronic nicotine treatment at this time has been shown to enhance glutamatergic (NMDA receptor-mediated) transmission in the neocortex of rats (Aramakis and Metherate, 1998), resulting in a subsequent increase in NMDA receptor subunit NR2A mRNA expression in layer VI of the auditory cortex that lasts for 2 weeks (Hsieh et al., 2002). These data suggest that expression of high-affinity nAChRs during this period of development can affect maturation of glutamatergic synapses. In rat, the NSE promoter drives expression early in the development of layer VI neurons and remains on throughout development, with a dip on postnatal day 10, before rising again to adult levels by day 20 (Hamre et al., 1989). Our data suggest that the presence of β2 subunit-containing nAChRs during these critical periods of synaptogenesis is essential for the long-term modulation of PA performance.
The nicotine-induced enhancement of PA learning in adult mice is not mediated by nAChRs on corticothalamic efferents, however. We hypothesize that nAChRs in other brain regions are critical for the effect of nicotine on PA learning in adulthood, and potential brain regions that could be explored include the amygdala (Riekkinen et al., 1993), striatum (Sandberg et al., 1984; Giordano et al., 1998), or hippocampus (Pope et al., 1985; Bailey et al., 1986), all of which have been identified as critical sites for plastic changes associated with consolidation of PA learning (McGaugh et al., 1996).
High-affinity nAChRs are normally expressed throughout the brain and are likely to affect the development and function of synaptic activity in many brain areas. Mutations in the α4 and β2 subunits of the nAChR in several human families result in autosomal dominant nocturnal frontal lobe epilepsies (Sutor and Zolles, 2001), which have been associated with an increased probability of learning and developmental disorders (Gunduz et al., 1999; Verrotti et al., 2000). It should be noted, however, that it is unclear whether the cognitive impairment in these patients results directly from altered nAChR activity during development or indirectly from repeated seizures, which are known to cause cognitive impairment. We showed that mutation of the β2 subunit in mice results in abnormal sensitivity to PA learning and have identified corticothalamic efferents as the anatomical locus for this effect. Abnormalities in nAChRs in human subjects may also result in developmental changes in corticothalamic signaling that could affect emotional learning. Prenatal exposure to nicotine in humans may desensitize and functionally downregulate high-affinity nAChRs, as is seen with chronic nicotine exposure in mice (Marks et al., 1993;Zachariou et al., 2001). Thus, chronic exposure to nicotine during development could alter nicotinic neurotransmission in corticothalamic circuits and lead to inappropriate emotional learning during adulthood.
Footnotes
This work was supported by National Institute on Drug Abuse Grants DA00436, DA14241, DA10455, and DA84733 (M.R.P.) and DA03194 and DA00197 (A.C.C). We thank Dr. Eric Nestler for the generous gift of NSE-tTA mice and valuable input on many aspects of this study. We thank Drs. Ronald Duman and Angus Nairn for reading this manuscript and for helpful conversations about this work. We thank Drs. Max Kelz and Jingshan Chen for providing information and primers for characterizing NSE-tTA transgenic mice.
Correspondence should be addressed to Marina R. Picciotto, Department of Psychiatry, Yale University School of Medicine, 34 Park Street, Third Floor Research, New Haven, CT 06508. E-mail:marina.picciotto{at}yale.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}