

Abstract
Periventricular leukomalacia is a form of hypoxic–ischemic cerebral white matter injury seen most commonly in premature infants and is the major antecedent of cerebral palsy. Glutamate receptor-mediated excitotoxicity is a predominant mechanism of hypoxic–ischemic injury to developing cerebral white matter. We have demonstrated previously the protective effect of AMPA–kainate-type glutamate receptor blockade in a rodent model of periventricular leukomalacia. The present study explores the therapeutic potential of glutamate receptor blockade for hypoxic–ischemic white matter injury. We demonstrate that AMPA receptors are expressed on developing human oligodendrocytes that populate fetal white matter at 23–32 weeks gestation, the period of highest risk for periventricular leukomalacia. We show that the clinically available anticonvulsant topiramate, when administered post-insult in vivo, is protective against selective hypoxic–ischemic white matter injury and decreases the subsequent neuromotor deficits. We further demonstrate that topiramate attenuates AMPA–kainate receptor-mediated cell death and calcium influx, as well as kainate-evoked currents in developing oligodendrocytes, similar to the AMPA–kainate receptor antagonist 6-nitro-7-sulfamoylbenzo-(f)quinoxaline-2,3-dione (NBQX). Notably, protective doses of NBQX and topiramate do not affect normal maturation and proliferation of oligodendrocytes either in vivo or in vitro. Taken together, these results suggest that AMPA–kainate receptor blockade may have potential for translation as a therapeutic strategy for periventricular leukomalacia and that the mechanism of protective efficacy of topiramate is caused at least in part by attenuation of excitotoxic injury to premyelinating oligodendrocytes in developing white matter.
- white matter
- glutamate receptor
- periventricular leukomalacia
- topiramate
- oligodendrocyte
- excitotoxicity
Introduction
Premature delivery and improved neonatal intensive care have led to the survival of >55,000 infants born annually weighing under 1500 gm in the United States (Volpe, 2001). Of these survivors, however, 10–15% subsequently exhibit cerebral palsy, and ∼50% have cognitive and behavioral deficits (Hack et al., 2000; Peterson et al., 2000). The predominant form of brain injury underlying this neurological morbidity is selective cerebral white matter injury, termed periventricular leukomalacia (PVL). PVL is a source of significant lifetime morbidity, yet no specific therapy presently exists (Golden et al., 1997).
Diffuse injury to developing oligodendrocytes (OLs) with subsequent hypomyelination is the major neuropathological feature of PVL (Paneth et al., 1990; Iida et al., 1995; Olsen et al., 1997; Skranes et al., 1997; Rorke, 1998; Inder et al., 1999). Perinatal hypoxia–ischemia is thought to be the primary cause of PVL, and the pattern of injury is highly age dependent. In contrast to the term infant in which hypoxia–ischemia results in primarily cortical injury and seizures, hypoxic–ischemic brain injury in the preterm infant appears selective for white matter (Gilles and Averill, 1977; Dambska et al., 1989; Paneth et al., 1990; Rorke, 1998; Volpe, 2001).
The age window of greatest susceptibility to PVL in the human brain is between 24 and 32 weeks gestation, when subcortical white matter is populated predominantly by premyelinating OLs (pre-OLs) (Kinney and Back, 1998; Back et al., 2001), including both OL precursors and immature OLs. In vitro studies have demonstrated that pre-OLs are more vulnerable to injury than mature OLs under conditions of oxidative stress (Back et al., 1998), oxygen–glucose deprivation (OGD) (Fern and Moller, 2000; Yoshioka et al., 2000; Deng et al., 2003), and glutamate receptor (GluR)-mediated excitotoxicity (Itoh et al., 2002; Rosenberg et al., 2003). Glutamate accumulates in brain under hypoxic–ischemic conditions (Benveniste et al., 1984; Andine et al., 1991; Hagberg, 1992), and excessive activation of GluRs on neurons is a major mechanism in hypoxic–ischemic neuronal death (Choi, 1992; Hagberg et al., 1994). Rodent pre-OLs express ionotropic GluRs of the AMPA and kainate but not the NMDA subtypes (Gallo et al., 1994; Patneau et al., 1994; Rosenberg et al., 2003). AMPA–kainate receptor antagonists or OGD induces Ca2+-mediated excitotoxicity in pre-OLs more readily than in mature OLs (Fern and Moller, 2000; Yoshioka et al., 2000; Deng et al., 2003), and this increased vulnerability correlates with transient overexpression of GluRs in pre-OLs compared with mature OLs, both in vitro (Itoh et al., 2002; Rosenberg et al., 2003) and in vivo (Ong et al., 1996; Follett et al., 2000). Furthermore, in a rat model of PVL, white matter injury after unilateral carotid ligation (UCL) and hypoxia can be attenuated by systemic posttreatment with the AMPA–kainate receptor antagonist 6-nitro-7-sulfamoylbenzo-(f)quinoxaline-2,3-dione (NBQX) (Follett et al., 2000).
The protective efficacy of AMPA–kainate receptor antagonists against receptor-mediated Ca2+ influx and excitotoxic injury to pre-OLs in vivo and in vitro suggests that these agents may have clinical potential in the treatment of PVL. Existing AMPA–kainate blockers have not been proven suitable for clinical use. Thus, the discovery of clinically relevant compounds is of tremendous importance. In this study, we examined the potential for translating this therapeutic strategy to clinical use. First, we investigated whether GluRs, the therapeutic target of this strategy, are actually expressed on pre-OLs in immature human white matter during the period of risk for PVL. Second, we examined the therapeutic efficacy and safety of the FDA-approved drug topiramate [2,3:4,5-bis-O-(1-methyl ethylidene)-β-d-fructopyranose sulfamate] in our in vivo model of PVL. Although we have previously reported protective in vivo efficacy of the AMPA–kainate receptor antagonist NBQX in this model, this agent is not clinically available because of severe renal toxicity and other adverse effects. Topiramate is currently in use as an anti-epileptic drug for adults and children over the age of 3 years. Third, we investigated the specific mechanism of protection by topiramate in complementary culture and animal models of GluR-mediated pre-OL injury.
Materials and Methods
In vivo white matter injury model. All procedures were approved and in accordance with guidelines set by the institutional animal care and use committee. Hypoxic–ischemic injury was generated in postnatal day (P) 7 rats by UCL–hypoxia (6% for 1 hr) as described previously (Follett et al., 2000). For treatment trials, rat pups received four intraperitoneal injections of 0.1 ml of topiramate (10, 30, or 50 mg/kg) or saline over 48 hr, commencing immediately after completion of hypoxia.
Human tissue. The brain tissue was determined to be “normal,” without significant brain pathology, on the basis of the following combined criteria (Back et al., 2001): nonneurological cause of death by clinical criteria and no abnormality by neuropathological analysis. Fetal tissue between 17 and 21 gestational weeks was obtained after spontaneous abortion. The older premature infants (23–32 gestational weeks) were stillborn or died of lung immaturity in the neonatal intensive care unit. All patients died within days after birth, and none presented with dysmorphic features suggestive for a genetic disease. Postmortem intervals were <24 hr, except two cases, one from each age group, with 51 and 53 hr postmortem intervals. No brain malformations or lesions were detected at the macroscopic or microscopic neuropathological examination.
Immunocytochemistry and histological analysis. OL maturation was evaluated by immunocytochemistry (ICC) with antibodies to the stage-specific OL markers O4, O1, and myelin basic protein (MBP) in rats killed at P9 or P11. Histological and ICC analysis on rodent tissue were performed on serial 20–30 μm coronal sections, as detailed previously (Follett et al., 2000). Sections were blocked and incubated overnight with anti-MBP (1:1000; SMI-99, Sternberger Monoclonals, Baltimore, MD), O4, or O1 mouse monoclonal IgM antibody (Ab), rinsed, and then incubated with the appropriate secondary Ab (Molecular Probes, Eugene, OR) for 1 hr at room temperature. Lesion size was assessed by a blinded observer, with reproducibility demonstrated by a second blinded observer, using a semiquantitative scale described previously in detail (Follett et al., 2000). MBP was used as a marker for white matter injury, rated on a scale of 0 (no injury) to 3 (severe injury) as compared with the contralateral side, in three stereotactically similar sections per animal. Scores were normalized to a percentage of the mean injury in control littermates and analyzed for significance by Student's t test (p < 0.05).
ICC of human tissue was performed using similar protocols. Fetal parietal brain sections fixed with 4% paraformaldehyde were cryoprotected, cut at 50 μm thickness, blocked, and incubated overnight with O4 monoclonal surface Ab followed by incubation with Alexa 568 anti-mouse IgM (1:1000; Molecular Probes). For serial double-labeling, sections were rinsed, blocked, permeabilized, incubated overnight with anti-GluR4 (1:200; Chemicon, Temecula, CA), rinsed, and incubated with biotin-conjugated anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA) followed by avidin-conjugated fluorescein (1:10,000; Vector Laboratories). Primary blank controls were used for each condition, and Ab specificity of GluR4 was further demonstrated with peptide blocking. Quantification was performed by counting O4+ cells (>250 cells for each age) on five random 60 μm2 white matter fields per section, evaluating the OLs for the presence of GluR4 expression, and determining the percentage of O4+ OLs expressing GluR4. Data are presented as mean ± SD and evaluated for significance (p < 0.05) with Student's t test.
Neurobehavioral assessment. Additional litters were used for assessment of correlative motor deficits (Crawley, 2000). After UCL–hypoxia, animals were raised with their dams for an additional 2 weeks to P21. Motor strength and function were evaluated by determining the normal climbing behavior of P21 animals at different incline angles and comparing the behavior of untreated hypoxic–ischemic animals (n = 8) and littermates treated with topiramate at 30 mg/kg (n = 7). Animals were scored by points as follows: 1 for hanging on, 2 for turning and going down, 3 for walking up the incline without sliding, and 4 for running up. Four trials were averaged for each animal, and significance was determined by one-way ANOVA with post hoc all pair-wise multiple comparison by Bonferroni t test. A gait analysis was performed at the maximum angle that an animal could climb to evaluate symmetry.
In situ ICC and Co2+ uptake. Coronal brain slices (300 μm) from decapitated P7 Long–Evans rat pups were collected in oxygen-rich rat artificial CSF (ACSF) at 4°C, stabilized, and then maintained at 22°C. Slices were incubated with O4 surface Ab (1:200) in rat ACSF for 40 min, rinsed, and then incubated with anti-mouse IgM-conjugated Alexa 568 (1:500; Molecular Probes) for 40 min. Slices were then transferred to a modified ACSF solution containing low Ca2+ and 5 mm CoCl2 and stimulated with kainate (100 μm) in the presence of the NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (APV; 100 μm) alone or with NBQX (100 μm) or topiramate (10–100 μm) for 20 min at room temperature (Sanchez et al., 2001). After washes with EDTA (2 mm), the Co2+ was precipitated with ammonium sulfide and visually enhanced with silver nitrate. Whole sections were fixed, and the number of Co2+-containing OLs was determined in a 60 μm2 field centered on the pericallosal white matter, using optical dissection microscopy at a fixed depth to control for penetration variability.
OL cultures. Highly enriched primary rat pre-OLs were isolated from mixed glial cultures, characterized by ICC detection of cell-specific markers, and maintained in a chemically defined medium, as described previously (Rosenberg et al., 2003). For OGD, cultures were transferred to deoxygenated, glucose-free medium and incubated in an anaerobic chamber at 37°C for 2 hr (Fern and Moller, 2000). Topiramate (RWJ Pharmaceutical Research Institute, Raritan, NJ), NBQX, and all other pharmacological agents (Sigma, St. Louis, MO) were applied 10 min before exposure of the cells to kainate or OGD. Cell viability was assessed at 24 hr as described previously (Deng et al., 2003). Ca2+ influx was assessed by incubation with 45CaCl2 (8 μCi/ml) at room temperature for 10 min. Radioactivity of the whole-cell lysate was counted by liquid scintillation. Electrophysiological recordings on pre-OLs were performed using conventional patch-clamp techniques in whole-cell mode as published previously (Dai et al., 2001; Rosenberg et al., 2003). Kainate (100 or 300 μm) and topiramate (50 or 100 μm; 20 min preincubation) were applied through a gravity perfusion system. Additional experiments were performed with application of AMPA (100 μm) and cyclothiazide (CTZ; 50 μm), after a 1 min pretreatment with CTZ (50 μm) alone.
Analysis of pre-OL proliferation and maturation in vivo and in vitro. Incorporation of the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) into DNA was used to measure the proliferative behavior of pre-OLs in vitro and in vivo. Pre-OLs in culture were exposed to BrdU (10 mm) during the last 2 hr of the experiment, labeled with A2B5 Ab, and fixed with ice-cold methanol. A2B5 was used as a marker in this culture system because most A2B5+ cells are also O4+. Additionally, because the A2B5 marker decreases with further maturation, this marker is less likely than O4 to include the postproliferative OL population (Deng et al., 2003). In cell cultures or tissue sections, DNA was denatured with 2N HCl, returned to neutral pH with buffer solution, and incubated with anti-BrdU Ab (Chemicon) followed by the appropriate fluorescent-conjugated secondary Ab. All culture experiments were repeated in triplicate and are reported as mean ± SEM. Statistical comparisons on the data of all experiments were performed using one-way ANOVA, followed by Tukey post hoc analysis, and significance was accepted for p < 0.05.
To assess the effects of protective doses of NBQX (20 mg/kg) or topiramate (30 mg/kg) on normal white matter development, rats were treated as above on P7–8 except without hypoxic–ischemic insult and killed at P9 or P11. For analysis of proliferation, animals received concurrent injections of BrdU. Rat pups were given BrdU (120 mg/kg every 12 hr) during the treatment protocol at P7–8 and killed at P9, and 30 μm coronal sections were labeled with O4 Ab, fixed, and processed for BrdU labeling (Magavi et al., 2000). The sections were all triple labeled with a nuclear marker, BrdU, and O4. Localization was verified by confocal microscopy. BrdU+ cells (n = 40 sections; >450 cells) with all three labels present were counted in the corpus callosum and external capsule of sections stereotactically similar to the region evaluated for injury.
Results
GluRs are present on pre-OLs in human premature white matter
GluR expression was evaluated by ICC on postmortem fetal human parietal white matter from brains determined to be normal by neuropathological examination. Cases were collected from the autopsy services of the Departments of Pathology of Children's Hospital and Brigham and Women's Hospital, Boston, with permission according to approved hospital research protocols. OLs in white matter were labeled with the pre-OL surface marker O4 (Gard and Pfeiffer, 1990; Back et al., 2002) and double-labeled for GluR4 (Wenthold et al., 1992), which is the AMPA receptor subunit that is expressed consistently by pre-OLs in rodents (Gallo et al., 1994; Patneau et al., 1994; Itoh et al., 2002; Liu et al., 2002). Previous studies demonstrate that the O4 Ab labels the pre-OLs that are prevalent in white matter during gestational weeks 17–32 in the human (Kinney and Back, 1998; Back et al., 2001). Analysis of very young brains (17–21 weeks gestation; n = 3 cases) showed many pre-OLs (Fig. 1A) throughout the white matter but with little to no colocalization with GluR4 (13.3 ± 14.9% of O4+ cells expressed GluR4) (Fig. 1B,C). In contrast, analysis of parietal white matter later in gestation (23–32 weeks; n = 4 cases) demonstrated a significant increase in O4+ cells coexpressing GluR4 (88.5 ± 8.3% of O4+ cells expressed GluR4; p < 0.004) (Fig. 1D–F). Hence, these results indicate increased expression of AMPA receptors on pre-OLs in human white matter during the age window of increased susceptibility for PVL relative to earlier ages.

Pre-OLs in human parietal white matter express GluRs during the window of increased vulnerability to PVL. ICC analysis of very young brains (17–21 weeks gestation; n = 3) shows many O4+ OLs (red) throughout the white matter (A) but little colocalization with GluR4 (green) (B), as demonstrated in C by the overlay (yellow). Analysis of parietal white matter later in gestation (23–32 weeks; n = 4) demonstrates increased density of O4+ cells (D), with significantly greater (p < 0.004) colocalization of GluR4 (E), as demonstrated in F by the overlay (yellow). Scale bar, 10 μm.
Post-insult treatment of topiramate attenuates white matter injury in a rodent model of PVL
The effect of topiramate treatment was evaluated in an immature rat model of PVL. We have previously adapted a well described model of UCL–hypoxia (6% O2, 1 hr) in the P7 rat to produce selective subcortical white matter injury (Follett et al., 2000). Major features of this resulting lesion include ipsilateral white matter cell death by in situ end labeling (ISEL), loss of immature OLs and MBP expression, and white matter gliosis with relative axonal and cortical neuronal sparing. Impairment of skilled limb use correlates with the contralateral white matter injury in the developing rodent (Tomimatsu et al., 2002), and white matter injury appears to persist into adulthood (Yager and Asselin, 1999).
After hypoxia, rat pups were treated intraperitoneally with topiramate at 10 (n = 15), 30 (n = 9), or 50 (n = 10) mg/kg or saline vehicle with repeated doses given every 12 hr for 48 hr, consistent with the half-life of topiramate (Schneiderman, 1998). The injury was assessed by a blinded observer for degree of MBP loss in ipsilateral cerebral white matter (Fig. 2A–E) and with hematoxylin and eosin histology on serial sections. Similar to our results with NBQX (Follett et al., 2000), there was a significant attenuation of lesion severity in rats treated post-insult with 30 mg/kg topiramate, as compared with vehicle-treated controls (Fig. 2B) (p < 0.01), but no significant effect at lower doses. In addition, topiramate (30 mg/kg) significantly attenuated cell death evaluated by ISEL at 48 hr after hypoxia–ischemia (data not shown). In addition, there was no significant difference in ISEL staining in these topiramate-treated rats compared with normoxic littermate control, indicating that therapeutic doses of topiramate do not contribute to constitutive cell death present in the developing brain. The higher dose of 50 mg/kg of topiramate also had no significant protective effect (Fig. 2B), similar to our experience with higher doses of NBQX (data not shown). Notably, these higher doses of AMPA–kainate receptor antagonists result in sedation in rats, and higher doses of topiramate inhibit carbonic anhydrase, which may result in metabolic acidosis and subsequent respiratory depression. There was no significant difference in mortality or weight gain among any of the treatment groups. These results revealed that topiramate (30 mg/kg) was the optimal dose for protection as a posttreatment, similar to our findings with a 20 mg/kg dose of NBQX (Follett et al., 2000).

Topiramate protects against hypoxic–ischemic white matter injury in vivo. A, Hematoxylin and eosin staining of a coronal brain section from a pup killed at P11 after UCL–hypoxia at P7, demonstrating ipsilateral white matter injury with relative sparing of the overlying cortex. Inset demonstrates regions shown in C–F. B, Dose–response to topiramate in vivo, demonstrating attenuation of MBP injury at P11 with systemic topiramate treatment after hypoxia–ischemia at P7. C, D, Loss of MBP is seen in the P11 pup ipsilateral to a carotid ligation (C), after UCL–hypoxia at P7, and compared with the contralateral side (D). Systemic treatment post-insult with topiramate (30 mg/kg) attenuates this injury in a littermate pup, ipsilateral (E) and contralateral (F). Scale bar, 50 μm.
Topiramate attenuates motor deficits after hypoxia–ischemia in a rodent model of PVL
White matter injury in the premature human infant presents as clinical spastic diplegia in the growing child. Because topiramate at 30 mg/kg attenuated the histological white matter injury in the immature rat, we evaluated whether these cellular changes might be correlated with improvement in observable motor deficits. Muscle strength and coordination were evaluated by the ability of the rats to climb a variably inclined plane. We found that a significant contralateral limb dysfunction exists in the rat at P21 after UCL–hypoxia at P7, consistent with the results of others (Tomimatsu et al., 2002), as well as a significant decrease in motor deficit in rats treated with topiramate for 48 hr after hypoxia (Fig. 3). All normal P21 rats can scamper up a 30° incline (score of 4) and climb up a 45° incline without sliding (score of 3). After UCL–hypoxia at P7, animals tested at P21 performed significantly worse than controls (mean score, 2.75 ± 0.01 at 30° and 1.88 ± 0.12 at 45°) and had significantly improved scores with topiramate treatment (mean score, 3.64 ± 0.11 at 30° and 2.68 ± 0.12 at 45°; p < 0.001 for all comparisons; one-way ANOVA with post hoc pair-wise analysis). Topiramate treatment at P7–9 (30 mg/kg every 12 hr for 2 d; n = 5) in normal littermates did not alter behavior at P21. These data demonstrate that topiramate can improve functional outcome after hypoxia–ischemia.

Topiramate attenuates motor deficits in a rodent model of hypoxic–ischemic white matter injury. Normal P21 rats can scamper up a 30 ° incline and climb up a 45 ° incline without sliding. Animals tested at P21 after UCL–hypoxia at P7 demonstrate significant loss of function on inclined planes as compared with controls (68.8 ± 2.5 and 46.9 ± 3.1% of normal P21 score at 30 ° and 45 °, respectively). Littermates treated with topiramate had significantly improved scores (91.1 ± 2.9 and 67.0 ± 3.2% of normal pups at 30 ° and 45 °; p < 0.001 for all pair-wise comparisons).
NBQX and topiramate attenuate excitotoxicity, kainate-evoked currents, and Ca2+ influx in pre-OLs in vitro
Excitotoxicity induced by OGD or kainate in pre-OLs in vitro has been shown to be Ca2+-mediated and secondary to AMPA–kainate receptor activation (Fern and Moller, 2000; Yoshioka et al., 2000; Deng et al., 2003). Thus, we evaluated the effects of NBQX and topiramate on kainate or OGD-induced excitotoxicity and Ca2+ influx in pre-OL cultures. Pre-OLs were incubated with NBQX or topiramate, or both, each at concentrations ranging from 0 to 100 μm, and exposed to either kainate (300 μm) or OGD (2 hr), with cell survival assessed at 24 hr. Topiramate or NBQX each attenuated kainate-induced pre-OL death in a dose-dependent manner, with EC50 values of 28.9 ± 3.0 and 12.3 ± 1.8 μm, respectively (Fig. 4A). OGD-induced pre-OL death was significantly reduced by topiramate (EC50 = 12.4 ± 2.4 μm; p < 0.05) or NBQX (EC50 = 9.26 ± 1.6 μm; p < 0.05) (Fig. 4B). Moreover, the effects of topiramate and NBQX were additive when the two agents were applied together at the concentrations that were not saturated for each compound. Thus in these paradigms, topiramate appears to be acting in a manner similar to the AMPA–kainate receptor antagonist NBQX; however, because topiramate is known to have multiple mechanisms of action, we next determined what other mechanisms might play a role in the protection. Using the OGD model on pre-OLs in vitro, we evaluated the individual and combined effects of compounds that mimic each of the additional known mechanisms for topiramate: GABAA receptor agonist activity, blockade of Na+ and voltage-gated Ca2+ channels, and carbonic anhydrase inhibition (White, 1997; Schneiderman, 1998; Shank et al., 2000; Skradski and White, 2000). No protection was observed with nontoxic dose ranges of the Ca2+ channel blockers nimodipine (2.5–100 μm) and nifedipine (10–100 μm), for GABA (0.1–2mm), for the Na+ channel blocker tetrodotoxin (TTX; 0.1–0.5 mm), or for the carbonic anhydrase inhibitor acetazolamide (0.1–1mm) when each was applied alone. In addition, there was no protection with combined administration of TTX (200 μm), nimodipine (10 μm), GABA (200 μm), and acetazolamide (200 μm). Taken together, these data strongly support the possibility that AMPA–kainate blockade is the predominant mechanism of OL protection in this model.

NBQX and topiramate protect against excitotoxic cell death and Ca2+ influx in pre-OLs in vitro. Dose–response curves demonstrating protection of topiramate (closed circles), NBQX (open circles), and the combination of the two compounds (triangle) against kainate (300 μm) excitotoxicity (A) and OGD-induced cell death (B). *p < 0.001 with ANOVA–Tukey analysis, indicating the lowest concentration with significant protection. C, Topiramate and NBQX prevent kainate (300 μm, for 10 min)- or OGD-induced 45Ca2+ uptake. D, Topiramate (30 μm) blocks kainate (300 μm, for 10 min)-induced Co2+ uptake.
To determine whether protection against excitotoxicity in vitro was associated with a reduction in Ca2+ influx, pre-OLs were incubated with NBQX (100 μm) or topiramate (30 μm) before adding kainate (300 μm) or after exposure to OGD (2 hr), and 45Ca2+ uptake was measured after 10 min for each condition. NBQX (10 μm), topiramate (30 μm), and, to a greater degree, topiramate (30 μm) plus NBQX (10 μm), attenuated kainate-evoked 45Ca2+ uptake (p < 0.01, 0.01, 0.001, respectively), whereas topiramate (30 μm) or NBQX (100 μm) fully blocked OGD-induced 45Ca2+ uptake (p < 0.001) (Fig. 4C). Additional experiments using Co2+ uptake as an alternate indicator of Ca2+ influx demonstrated that topiramate (30 μm) also blocks kainate (300 μm, for 10 min)-induced Co2+ uptake (Fig. 4D). A selective effect of topiramate on AMPA–kainate receptor-mediated Ca2+ influx has also been reported in cultured cerebellar granule cells (Skradski and White, 2000), and our results indicate that topiramate is as effective as NBQX in preventing OGD-induced pre-OL death.
NBQX has been shown previously to block kainate-induced currents in pre-OLs (Rosenberg et al., 2003). To further characterize the mechanism of action of topiramate in the models, we directly examined AMPA–kainate-evoked currents using whole-cell patch-clamp recordings in cultured pre-OLs. Topiramate significantly attenuated kainate (100 μm)-evoked currents in pre-OLs (Fig. 5), with a reduction of 62.4 ± 2.4% (50 μm topiramate; n = 8; p < 0.001) and 11.1 ± 1.9% (100 μm topiramate; n = 7; p < 0.0001) of control responses (n = 8), consistent with its action on hippocampal and cortical neurons in vitro (Gibbs et al., 2000; Skradski and White, 2000). Topiramate (100 μm) also significantly inhibited responses to kainate (300 μm; n = 6; p < 0.0001). Because AMPA may be a more selective agonist than kainate for AMPA receptors, we also tested the modulation effect of topiramate on responses to AMPA in the presence of CTZ (50 μm). We found that the responses to AMPA (100 μm) were also significantly reduced to 54.6 ± 5.4% by 50 μm topiramate (n = 7; p < 0.0001) and to 18.5 ± 1.9% by 100 μm topiramate (n = 7; p < 0.0001), respectively. In addition, the rise time and the decay time constants of the AMPA receptor-mediated response were slowed by topiramate (data not shown), further supporting negative modulation of AMPA receptors as a mechanism of action of topiramate. The demonstration that topiramate can directly attenuate AMPA–kainate-evoked receptor currents and Ca2+ influx, together with the lack of effect of agents mimicking other known actions of topiramate, strongly supports the possibility that the primary mechanism of efficacy of topiramate here is AMPA–kainate receptor blockade.

Topiramate attenuates AMPA–kainate receptor currents in pre-OLs in vitro. Representative traces show that AMPA (100 μm; n = 8) with CTZ (A) or kainate (100 μm; n = 8) (B) evoked a large sustained inward current response significantly inhibited by topiramate in patch-clamp recordings from cultured pre-OLs. C, Bar graph of pooled data demonstrates significant attenuation by topiramate of AMPA-induced responses [topiramate at 50 μm (n = 7) and 100 μm (n = 7); p < 0.0001] and kainate-induced responses [topiramate at 50 μm (n = 8; p < 0.001) and 100 μm (n = 7; p < 0.0001)].
GluR-mediated Ca2+ uptake is blocked by NBQX and topiramate in pre-OLs in situ
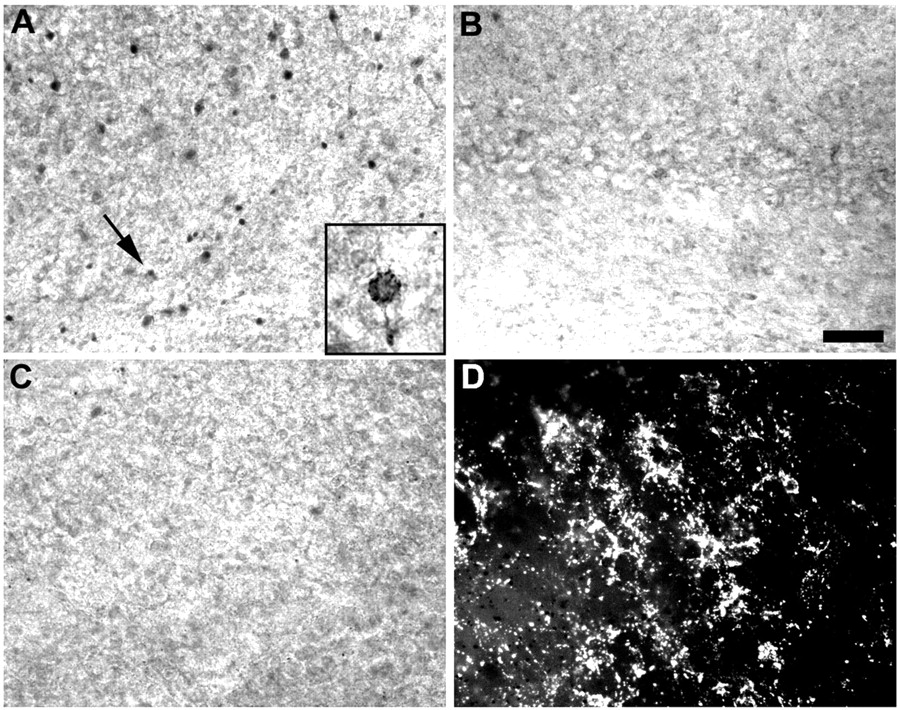
Given the protective efficacy of NBQX and topiramate on AMPA–kainate receptor-mediated Ca2+ influx in vitro, we examined their effects on pre-OLs in situ. Kainate-induced Co2+ uptake in pre-OLs was examined in the pericallosal white matter of P7 coronal brain slices maintained in vitro. The divalent cation Co2+ can be used as an indicator of Ca2+, because it substitutes for Ca2+ via GluR channels but not via voltage-gated Ca2+ channels (Pruss et al., 1991; Sanchez et al., 2001). Pre-OLs identified in slices by vital immunostaining with O4 Ab showed Co2+ uptake in 20 ± 6.4 OLs per 0.01 mm2 in pericallosal white matter after 20 min incubation with kainate (100 μm) (Fig. 6A). In all experimental conditions, slices were coincubated with APV (100 μm) to block secondary Co2+ entry via NMDA receptor activation. Kainate-induced Co2+ uptake was reduced by either NBQX (100 μm) (Fig. 6B) or topiramate (50 μm) (Fig. 6C,D), significantly decreasing the number of Co2+-positive cells to 3.4 ± 4.4 OLs per 0.01 mm2 and 5.4 ± 4.1 OLs per 0.01 mm2, respectively (p < 0.001; ANOVA). These data indicate that topiramate, like NBQX, blocks AMPA–kainate receptor-mediated Ca2+ influx in pre-OLs in situ within intact white matter, further suggesting that this is a mechanism of the protection observed in vivo.

Co2+ uptake in OL precursors in situ is blocked by NBQX and topiramate. A–D, NBQX and topiramate block Co2+ uptake via AMPA–kainate receptors in immature white matter. A, Kainate evokes Co2+ uptake in white matter of P7 rats in cells identified by ICC as O4+ OLs. Inset shows high-power image of O4 cell (arrow) filled with Co2+. Scale bar, 10 μm. NBQX (100 μm) (B) and topiramate (50 μm) (C) significantly block Co2+ uptake in the white matter of adjacent slices (p < 0.001; ANOVA). D, ICC for O4+ pre-OLs in the same section as C demonstrates that topiramate blocks Co2+ uptake in OL precursors. Scale bar, 50 μm.
Proliferation and maturation of pre-OLs are not affected by treatment protocols
AMPA–kainate receptor blockade may affect OL proliferation and differentiation under certain conditions (Gallo et al., 1996; Yuan et al., 1998) and thus could confound the observed protective effect of topiramate or NBQX in the cell survival analysis. To examine the possible effects on OL proliferation, the percentage of BrdU+ cells in the A2B5+ populations was counted in cultures exposed to protective concentrations of topiramate (30 μm), NBQX (100 μm), or vehicle, in conditions with and without platelet-derived growth factor (PDGF) and basic fibroblast growth factor (bFGF) (10 ng/ml each). To determine the effects of topiramate or NBQX on OL differentiation, we measured cell morphology and the sequential emergence of a panel of OL stage-specific markers (A2B5, O4, O1, and MBP), after 0, 2, 6, and 10 d of maturation. Neither topiramate nor NBQX at protective concentrations altered OL proliferative capability (Fig. 7A) or differentiation pattern (data not shown).

Maturation and proliferation of OLs is not affected by protective doses of NBQX or topiramate. A, Control cultures exposed to either NBQX (100 μm) or topiramate (30 μm) for 24 hr show no significant difference in BrdU incorporation or cell number. Sister cultures show significant proliferation when exposed to growth factors PDGF and bFGF, but there is no additional effect of NBQX or topiramate. ICC assessment with OL stage-specific markers for O4, O1, and MBP confirmed no maturational difference between treated and untreated sister cultures. B, Double-labeling with BrdU and O4 demonstrates no significant difference in number of proliferating O4 cells in subcortical white matter of the corpus callosum and external capsule in pups treated with protective dose regimens of topiramate (30 mg/kg) or NBQX (20 mg/kg) from untreated littermates. C, Overlay image of BrdU+ nucleus (red) in pre-OLs labeled with O4 (green). Scale bar, 10 μm. D, ICC analysis with O4 (top), O1 (middle), and MBP (bottom) stage-specific OL markers in untreated (left panels) and topiramate-treated (right panels) littermates show no qualitative difference in maturation. Scale bar, 50 μm.
We next examined the effect of topiramate and NBQX on pre-OL proliferation and maturation in vivo, in the treatment protocols shown to be protective after hypoxia–ischemia. BrdU-injected P7 rat pups were treated intraperitoneally with topiramate (30 mg/kg; n = 7), NBQX (20 mg/kg; n = 7), or vehicle. There was no significant difference in the number of O4+ pre-OLs incorporating BrdU in subcortical white matter of stereotactically similar coronal sections after rats were killed at the conclusion of the treatment window at P9 (n = 20 per group) (Fig. 7 B, C). Consistently, analysis with stage-specific markers (O4, O1, and MBP) in serial sections demonstrated no qualitative difference in maturation at either P9 (Fig. 7D) or P11 (data not shown). Therefore, protective dosing regimens of topiramate or NBQX did not alter pre-OL proliferation or maturation, making it unlikely that those effects contributed to the observed protection against hypoxic–ischemic injury.
Discussion
The findings presented here support the hypothesis that Ca2+-mediated excitotoxicity to pre-OLs is a contributing factor in age-dependent, selective white matter injury, and they suggest a potential role for AMPA–kainate receptor antagonists as an age-specific therapeutic strategy for PVL. AMPA receptor expression in developing human brain appears to increase during white matter development, in a pattern similar to the previously demonstrated increase in AMPA receptor expression on pre-OLs in the rat. The onset of GluR expression in pre-OLs in human brain occurs coincident with the ages of increased risk for PVL, similar to the observation in rodents that increased vulnerability of white matter to injury during development correlates with increased expression of AMPA–kainate receptors on OLs (Follett et al., 2000; Rosenberg et al., 2003). Because AMPA receptor blockade is protective in the rodent model, the presence of GluRs in developing human white matter justifies consideration of GluR blockade as a potential therapeutic strategy for hypoxic–ischemic injury in the premature infant.
We demonstrate the increased presence of AMPA receptors on developing oligodendrocytes in human fetal parietal white matter between 23 and 32 weeks of gestation, coincident with the time period of highest vulnerability for PVL. GluRs are expressed during development on other glial cells (Burnashev et al., 1992; Iino et al., 2001), and neuronal–glial interactions mediated by glutamate have been postulated to be important for neuronal migration and cortical development (Gallo and Ghiani, 2000). Furthermore, glutamate and GluR antagonists modulate OL proliferation and differentiation in vitro (Gallo et al., 1996; Yuan et al., 1998). Hence, although the physiological significance of the developmental regulation of GluR expression in human pre-OLs in immature white matter remains unclear, these receptors may play a role in normal white matter development and myelination.
A major finding in this study is that post-insult treatment with topiramate prevents the hypomyelinating injury caused by hypoxia–ischemia at P7, similar to the effects of the AMPA receptor antagonist NBQX (Follett et al., 2000). Importantly, there is an associated improvement in neuromotor function. Many studies of injury protection have only demonstrated an effect of pretreatment, a more clinically limited paradigm. Because premature infants are typically maintained in a continuously monitored, intensive-care setting, treatment commencing even within a few minutes after an insult is feasible.
Although topiramate has multiple mechanisms of action, our results demonstrate that its known pharmacological effects other than GluR blockade, either alone or in combination, do not appear to contribute significantly to the observed protection. Consistent with the results of others (Fern and Moller, 2000; Tekkok and Goldberg, 2001), we found that the sodium channel blocker TTX is not protective against OGD-induced OL death in vitro, suggesting that any such action of topiramate would similarly not be a relevant factor in OGD-induced OL injury. In addition, although topiramate can block voltage-gated Ca2+ channels and inhibit carbonic anhydrase, we have seen no protective efficacy of either Ca2+ channel blockers or carbonic anhydrase inhibition in our in vitro model, making this mechanism unlikely. Similarly, although topiramate enhances GABA receptor-mediated currents, there is no attenuation of OGD-induced OL death in these pre-OL cultures in the presence of GABA.
In this study we show that topiramate diminishes both kainate and AMPA-induced current amplitude and rise and decay time constants on pre-OLs in a dose-dependent manner, consistent with previous reports regarding its effect on neuronal AMPA–kainate currents (White, 1997; Schneiderman, 1998; Shank et al., 2000; Skradski and White, 2000). Additionally, the effects of topiramate and NBQX are not additive at saturating concentrations of either agent, further suggesting that topiramate is not protective via an alternative mechanism. The specific mechanisms of action of topiramate on the AMPA–kainate receptor are not yet clearly elucidated. Topiramate may interact directly with AMPA–kainate receptors or may modulate receptor activity either by binding to receptor phosphorylation sites or by modulating the activity of the protein kinases or protein phosphatase active at these sites (Shank et al., 2000; Gryder and Rogawski, 2003).
Multiple mechanisms are likely to contribute to hypoxic–ischemic injury to pre-OLs in developing white matter. Data in several paradigms suggest that pre-OLs are highly vulnerable to excitotoxicity mediated via Ca2+-permeable AMPA–kainate receptors (Fern and Moller, 2000; Follett et al., 2000; Itoh et al., 2002; Deng et al., 2003; Rosenberg et al., 2003). Here, we have further demonstrated Ca2+ influx in pre-OLs, induced by kainate or AMPA in parallel models of excitotoxic injury, including in situ brain slices from rats at an age of peak vulnerability to hypoxia–ischemia-induced excitotoxicity. This response is effectively blocked by both NBQX and topiramate, supporting a role for AMPA–kainate receptor-mediated Ca2+ influx in this model of immature white matter injury.
The dose of topiramate most effective in our in vivo model (30 mg/kg) results in a steady-state rodent brain concentration of 20–25 μm, consistent with the efficacious doses in most of the in vivo models in which topiramate is active and correlating with the steady-state CSF concentrations between 3 and 57 μm demonstrated in epilepsy patients effectively treated with topiramate (Shank et al., 2000; Christensen et al., 2001; May et al., 2002). Similarly, our in vitro data reveal an EC50 for topiramate against OGD toxicity of 12.8 μm, falling in this clinically relevant range. It is interesting that the protective efficacy of topiramate decreases with higher doses of topiramate. This decreased efficacy at higher doses is consistent with a similar response of decreased anti-epileptic activity seen at higher doses of topiramate (White et al., 1997; Koh and Jensen, 2001). Reasons for this decrease in efficacy are unclear but may include increased inhibition of carbonic anhydrase and other systemic actions apparent at high serum levels (White et al., 1997). Furthermore, a dose of 50 mg/kg in rodents results in a serum concentration of ∼100 μm, well above levels used clinically in humans (Christensen et al., 2001; May et al., 2002).
In considering the appropriateness of translation of this experimental therapeutic strategy to human premature newborns, it is notable that limited protective doses of NBQX or topiramate do not appear to interfere with normal OL development, because GluR antagonists have also been reported to affect pre-OL proliferation and maturation in vitro (Gallo et al., 1996; Yuan et al., 1998). The in vivo evaluation of the effects of systemic administration of protective doses of AMPA–kainate receptor antagonists in normal rats demonstrates no quantitative or qualitative changes in OL proliferation or constitutive death in developing white matter, as well as no discernable changes in the neurobehavioral performance attributable to treatment alone.
In summary, these results demonstrate protective efficacy of experimental AMPA–kainate receptor antagonists in both in vitro and in vivo models of hypoxic–ischemic injury to pre-OLs. We have demonstrated the presence of a transient overexpression of AMPA receptors on oligodendrocytes during development that coincides with their vulnerability to excitotoxicity in cell culture, in the rat, and in developing human parietal cortex. Our data suggest that hypoxic–ischemic injury to pre-OLs during this stage of development occurs via activation of Ca2+-permeable AMPA–kainate receptors. In addition, the clinically available drug topiramate, known to attenuate neuronal AMPA receptor currents, has therapeutic efficacy in each of these models of PVL. The results of the present study justify consideration of an age-specific therapeutic strategy involving AMPA–kainate receptor antagonists for the treatment of premature infants at risk for PVL.
Footnotes
This work was supported by National Institutes of Health (NIH)–National Institute of Child Health and Human Development (NICHD) Grant HD01359 (P.L.F.), NIH–National Institute of Neurological Disorders and Stroke Grants NS31718 (F.E.J.) and NS38475 (J.J.V., F.E.J.), the Charles H. Hood Foundation (P.L.F.), the United Cerebral Palsy Foundation, and R. W. Johnson Pharmaceutical Research Institute and Mental Retardation Research Center Grant P30 NICHD HD18655. We thank Drs. Hannah Kinney and Rebecca Folkerth for their advice with the human tissue studies.
Correspondence should be addressed to Dr. Frances E. Jensen, Enders 348, Department of Neurology, Children's Hospital, 300 Longwood Avenue, Boston, MA 02115. E-mail: frances.jensen{at}childrens.harvard.edu.
DOI:10.1523/JNEUROSCI.0477-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244412-09$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}