

Abstract
Cyclin-dependent kinase 5 (Cdk5) is a proline-directed kinase the activity of which is dependent on association with its neuron-specific activators, p35 and p39. Cdk5 activity is critical for the proper formation of cortical structures and lamination during development. In the adult nervous system, Cdk5 function is implicated in cellular adhesion, dopamine signaling, neurotransmitter release, and synaptic activity. In addition, Cdk5 is also involved in “cross-talk” with other signal transduction pathways. To further examine its involvement in cross-talk with other pathways, we identified proteins that interacted with p35 using the yeast two-hybrid system. We report here that p35 associates with Ras guanine nucleotide releasing factor 2 (RasGRF2) in coimmunoprecipitation and colocalization studies using transfected cell lines as well as primary cortical neurons. Additionally, Cdk5 phosphorylates RasGRF2 both in vitro and in vivo, leading to a decrease in Rac–guanidine exchange factor activity and a subsequent reduction in extracellular signal-regulated kinase 1/2 activity. We show that p35/Cdk5 phosphorylates RasGRF2 on serine737, which leads to an accumulation of RasGRF2 in the neuronal cell bodies coinciding with an accumulation of microtubule-associated protein 1b. The membrane association of p35 and subsequent localization of Cdk5 activity toward RasGRF2 and Rac provide insights into important cellular signaling processes that occur at the membrane, resulting in downstream effects on signal transduction cascades.
Introduction
Cyclin-dependent kinase 5 (Cdk5) is a member of the Cdk family of proteins that are involved in the regulation of the mitotic cell cycle (Pines, 1996). Cdk5 is expressed ubiquitously in a number of tissues, but its expression is most abundant in the nervous system (Hellmich et al., 1992; Tsai et al., 1993). Its activity, however, is restricted to the nervous system because of the tissue-specific expression of its activators p35 and p39 (Tsai et al., 1994; Humbert et al., 2000). p35/Cdk5 activity is essential for neuronal morphogenesis (Nikolic et al., 1996) and survival (Tanaka et al., 2001; Li et al., 2002). Depletion of the Cdk5 gene in knock-out mice results in cortical lamination defects and embryonic lethality (Ohshima et al., 1996). Cdk5 knock-out mice (Cdk5–/–) do not survive to birth and most die in utero by embryonic day (E) 18. p35 knock-out mice (p35–/–), however, survive to birth but exhibit perinatal lethality and onset of seizures (Chae et al., 1997). Brains of Cdk5–/– embryos reveal severe abnormalities with extensively disrupted cerebral cortical layering caused by abnormal neuronal migration, a lack of cerebellar foliation, and degeneration of neurons in the spinal cord and brain stem (Ohshima et al., 1996, 1999; Gilmore et al., 1998; Gilmore and Herrup, 2001).
Over the last decade, >20 functionally diverse proteins have been identified as Cdk5 substrates. Some of these include neurofilaments (NF-H, NF-M) (Guidato et al., 1996; Giasson and Mushynski, 1997; Lee and Johnston, 1997; Pant et al., 1997; Sharma et al., 1998; Bajaj et al., 1999), microtubule-associated proteins MAP1b, MAP2, and tau (Mandelkow et al., 1995; Pigino et al., 1997; Hashiguchi et al., 2002; Otth et al., 2002; Town et al., 2002; Takahashi et al., 2003), β-catenin (Kwon et al., 2000; Kesavapany et al., 2001), presenilin-1 (Lau et al., 2002), dopamine and cAMP regulated phosphoprotein (DARPP)-32 (Bibb et al., 1999), p21-activated kinase 1 (PAK1) (Nikolic et al., 1998), Munc-18, synapsin, and amphiphysin (Matsubara et al., 1996; Shuang et al., 1998; Fletcher et al., 1999; Rosales et al., 2000). Clearly, the function of Cdk5 is related to the phosphorylation of its substrates, and Cdk5 functions include microtubule stability, cell–cell adhesion, dopamine signaling, regulation of actin dynamics through PAK1 signaling, and synaptic transmission. Recent studies have also implicated Cdk5 activity in “cross-talk” with other kinase signal transduction pathways such as the extracellular signal-regulated kinase (ERK) 1/2, c-Jun N-terminal kinase 3 (JNK3) (Li et al., 2002, 2003; Sharma et al., 2002), and neuregulin/Akt pathways.
To further understand this aspect of Cdk5 function, we sought to identify proteins from signal transduction pathways and identify their cross-talk with Cdk5. We used the yeast two-hybrid system to search for novel interacting proteins of p35 to further understand the functions of Cdk5 in the adult nervous system. Ras guanine nucleotide releasing factor 2 (RasGRF2) was identified as a novel interacting protein and substrate of p35/Cdk5. This study shows that p35/Cdk5 phosphorylates RasGRF2 on serine737, thereby modulating the Rac signaling cascade affecting ERK1/2 activity and the subsequent distribution of both RasGRF2 and MAP1b in neurons.
Materials and Methods
All experiments were performed at least three times with identical results.
Antibodies and reagents
Polyclonal and monoclonal anti-FLAG antibodies (Sigma, St. Louis, MO) were used at 1:1000 for immunocytochemistry in transfected cells, 1:1000 for Western blotting, and 1:100 for immunoprecipitations (IPs). Anti-c-myc monoclonal 9E10 antibody (Ab) was obtained from Sigma. Polyclonal anti-c-myc antibodies were purchased from Sigma. Anti-p35 (C19) and Cdk5 (J3 and C8) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were used at 1:1000 dilution to detect transfected proteins and at 1:200 to detect endogenous protein. Monoclonal anti-Xpress epitope tag Ab (Invitrogen, Carlsbad, CA) was used at 1:500 dilution for immunoprecipitation studies. Anti-RasGRF2 monoclonal antibodies and goat polyclonal antibodies were obtained from Transduction Laboratories (Lexington, KY) and Santa Cruz Biotechnology, respectively. Both were used at 1:500 for Western blotting, 1:100 for immunocytochemistry in neuronal cells, and 1:1000 for immunocytochemistry in transfected cells. Monoclonal anti-phosphoserine–threonine-proline Ab was obtained from Cell Signaling Technology (Beverly, MA). Total ERK1/2, phospho-ERK1/2, and phospho-MEK1/2 antibodies were obtained from Cell Signaling Technology. Secondary horseradish peroxidase-conjugated antibodies were obtained from Amersham Biosciences (Piscataway, NJ) and used at 1:2000 dilutions. Secondary fluorescence-conjugated Alexa-Fluor 633, Oregon Green, and Texas Red antibodies (Molecular Probes, Eugene OR) were used at 1:400 dilutions. Monoclonal anti-p35 Ab was obtained from Upstate Biotechnology (Lake Placid, NY), and 4 μg was used per immunoprecipitation. PD98059 (50 μm) was obtained from Calbiochem (La Jolla, CA), and treatment of cortical neurons was performed for 16 hr.
Plasmids
p25, p35, wild-type, and dominant-negative Cdk5 cDNAs in pcDNA3.1C vector were gifts from Dr. L. H. Tsai (Harvard Medical School, Cambridge, MA). RasGRF22–463 fragment isolated from the yeast two-hybrid screen was excised from pACT2 library vector and inserted into pCMVmyc vector (Clontech, Cambridge, UK). Full-length FLAG epitope-tagged mouse RasGRF2 cDNA in pcDNA3 was a gift from Dr. Mike Moran (MDS Proteomics, Toronto, Ontario).
Mammalian cell culture and transfection
Primary rat cortical neurons, Chinese hamster ovary (CHO), and COS-7 cells were grown as described previously (Kesavapany et al., 2001, 2003). For transfection of cortical neurons, neurons were plated onto glass coverslips that had been coated with poly-l-lysine and were transfected after 3 d in culture (3-DIC) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. CHO and COS-7 cells were grown in F12/HAM and Advanced-DMEM (Invitrogen), respectively, both containing 10% (v/v) fetal bovine serum supplemented with 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. CHO and COS-7 cells were transfected using Lipofectamine (Invitrogen) essentially according to the manufacturer's instructions. Total cell lysates were prepared by scraping the cells into ice-cold 1× PBS, washing them twice, and resuspending them in 100 μl. An equal volume of 2× SDS sample buffer was added, and the samples were heated for 95°C for 5 min. Soluble cell lysates were obtained by scraping the cells into ice-cold IP lysis buffer (see below), kept on ice for 20 min, and centrifuged for 30 min at 4°C. Lysates were then mixed with an equal volume of 2× SDS sample buffer–DTT and heated for 5 min at 95°C before separation by SDS-PAGE.
Yeast two-hybrid system
Yeast two-hybrid experiments were performed using the yeast two-hybrid System 3 (Clontech) essentially according to the manufacturer's instructions. p10 (residues 1–99), p25 (residues 99–307), and p35 “baits” were generated by PCR with EcoRI linkers and subcloned into the yeast GAL4 binding-domain vector pGBKT7. These constructs were sequenced to confirm 100% fidelity. Library screens were performed by comating yeast Y190 with p25 bait and a “prey” pretransformed adult human brain cDNA library in GAL4 DNA activation domain vector pACT2 (EcoRI and XhoI) according to the manufacturer's instructions (Clontech). After Trp–, Leu–, His–/25 mm 3-aminotriazole (3-AT) selection, vigorously growing clones were picked after 7 d and subjected to β-galactosidase assays. Briefly, yeast colonies were freeze-fractured by immersion in liquid nitrogen and placed on a presoaked filter paper containing phosphate buffer, pH 7.0, and 33 mg/ml X-galactopyranoside. Color generation was observed for 8 hr at 30°C. pACT2 plasmids were rescued and isolated using the RPM yeast plasmid isolation kit (BIO101; Q-Biogene, Carlsbad, CA), and the inserts were sequenced. To precisely map the domain(s) of RasGRF2 that interacted with the bait, cDNA constructs corresponding to the pleckstrin homology (PH) domains, a Dbl homology (DH) domain, a coil-coiled (CC) motif, and calcium–calmodulin binding (IQ) motif domains of RasGRF22–463 were generated using PCR and inserted as EcoRI/XhoI fragments into pGADT7. Bait (p25 and p35) domain constructs were comated and colonies were tested for interaction strength using the β-galactosidase assays as described above.
Coimmunoprecipitation of p25 and p35 with RasGRF22–463 and full-length RasGRF2
CHO cells were transfected using Lipofectamine (Invitrogen) according to manufacturer's instructions with empty pcDNA3.1 only, RasGRF22–463/pCMVmyc alone, or with p25 and p35 separately. Cells were harvested 24 hr after transfection and lysed in ice-cold IP lysis buffer containing 50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1 mm EGTA, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 5 μg/ml pepstatin, and 1 mm PMSF. Immunoprecipitations were performed as described previously (Kesavapany et al., 2003). RasGRF22–463 was immunoprecipitated using the monoclonal anti-c-myc Ab. Ig was captured using Protein G Sepharose beads (Sigma) overnight at 4°C. p25 and p35 were immunodetected using C19 Ab. To detect full-length RasGRF2, p25 and p35 were cotransfected into CHO cells, and cell lysates were prepared as described above. p25 and p35 were immunoprecipitated using the monoclonal anti-Xpress epitope tag Ab. Full-length RasGRF2 was immunodetected using the polyclonal anti-RasGRF2 Ab. For rat brain coimmunoprecipitation studies, 10% rat brain homogenates were prepared using ice-cold IP lysis buffer (described above). Briefly, rat brain was homogenized with 40 strokes of a Dounce homogenizer on ice and centrifuged for 30 min at 4°C at 14,000 rpm. Supernatants were pre-cleared using Protein G Sepharose beads, and 250 μg of total protein was used in immunoprecipitation experiments using the monoclonal anti-p35 Ab. Samples were separated on 4–20% gradient acrylamide gels (Novex) and transferred to nitrocellulose membranes (Amersham Biosciences) using the Novex XCell II blotting module for 2 hr at 100 mA. Immunodetection of full-length RasGRF2 was performed using polyclonal anti-RasGRF2 Ab. p25 and p35 were detected using C19 (anti-p35 Ab). Secondary incubation of appropriate species peroxidase-conjugated secondary antibodies (Amersham Biosciences) were performed for 1 hr at room temperature. Signals were visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL). Membranes were stripped using stripping buffer (100 mm 2-mercaptoethanol, 2% SDS, 62.5 mm Tris-Cl, pH 6.8) for 30 min at 50°C with agitation and reprobed as required.
Immunocytochemical analyses
Cortical neurons and transfected COS-7 cells grown on glass coverslips were fixed with 4% (w/v) paraformaldehyde in PBS for 20 min, permeabilized in 0.1% (v/v) Triton X-100 in PBS for 20 min, blocked with 5% (v/v) fetal bovine serum–PBS for 30 min, and then probed with primary antibodies, diluted in blocking solution at room temperature for 1 hr. To examine endogenous RasGRF2 staining, 7-DIC cortical neurons were used. Transfections of cortical neurons were performed on 3-DIC neurons. Primary antibodies were then visualized using donkey anti-mouse or donkey anti-rabbit Igs coupled to Alexa-Fluor 633, Oregon Green, or Texas Red (Molecular Probes) diluted in blocking solution for 1 hr at room temperature. Nuclei were counterstained using Hoescht 33342 (Sigma). Coverslips were washed in PBS between incubations and mounted in Gel/Mount (Biomeda, Hayward, CA). Fluorescence images were acquired using a 63× oil-immersion objective on a Zeiss LSM510 confocal microscope. Images were combined using Zeiss LSM510 image software and managed in Adobe Photoshop.
Phosphorylation of RasGRF2 by p35/Cdk5
In vitro phosphorylation of RasGRF2 by p35/Cdk5. CHO cells transfected with FLAG-RasGRF2 were immunoprecipitated using monoclonal anti-FLAG Ab. Active Cdk5 was immunoprecipitated from a 10% adult rat brain homogenate using C8 Ab. Immunoprecipitates were washed three times with lysis buffer and then once with 1× kinase buffer containing 20 mm Tris-Cl, pH 7.4, 1 mm EDTA, 1 mm EGTA, 10 mm MgCl2, 10 μm sodium fluoride, and 1 mm sodium orthovanadate. Kinase assays were performed as described previously (Veeranna et al., 1998) with and without 5–10 μm roscovitine (ROS) (Calbiochem). Samples were separated by SDS-PAGE, and gels were stained with Coomassie and then destained, dried, and exposed to autoradiography. Parallel Coomassie-stained bands were excised before drying for protein identification and confirmation by electrospray mass spectroscopy to confirm that the band was indeed RasGRF2. Recombinant glutathione S-transferase (GST)-Cdk5 and GST-p35 fusion proteins were expressed and in vitro phosphorylation reactions were performed as described previously (Amin et al., 2002). To identify the site of Cdk5 phosphorylation, phosphopeptide analysis of phosphorylated RasGRF2 protein was performed as described previously (Sihag et al., 1999). Briefly, phosphorylated RasGRF2 was resolved by SDS-PAGE, Coomassie stained, and then excised from the gel. The protein was subjected to in-gel tryptic digestion and then subjected to two-dimensional thin-layer chromatography (TLC) on 100 mm cellulose-coated TLC plates. TLC plates were exposed to autoradiography, and “hot” spots were scraped off and phosphopeptides were analyzed by matrix-assisted laser desorption/ionization (MALDI) mass spectroscopy. Spectra obtained were compared with fragmentation patterns predicted for tryptic digests of mouse FLAG-RasGRF2 protein.
Site-directed mutagenesis. Once the phospho-site was identified, site-directed mutagenesis was performed using the XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions using wild-type FLAG-RasGRF2 as template, mutating serine717 and serine737 to alanine.
In vivo phosphorylation of RasGRF2. Mouse brain homogenates (10%) of age-matched wild-type and p35 knock-out mice were made as described previously. RasGRF2 was immunoprecipitated using the anti-RasGRF2 goat polyclonal antibody overnight at 4°C. Samples were washed three times with lysis buffer and prepared for SDS-PAGE by addition of 2× SDS-sample buffer, heating at 95°C for 5 min, and separation on 4–20% polyacrylamide gels followed by transfer onto nitrocellulose membranes. Phosphorylated species were detected using the anti-phosphoserine–threonine–proline Ab (Cell Signaling Technology), after which membranes were stripped and the presence of RasGRF2 was confirmed using the polyclonal RasGRF2 Ab.
Active Rac and Ras assays. Active Rac and Ras levels mediated by RasGRF2 were assayed using the EZ-Detect Rac1 and Ras Activation Kits (Pierce Biotechnology) according to the manufacturer's instructions. For the Rac assays, empty vector (EV), RasGRF2, RasGRF2+p35+Cdk5, and RasGRF2+Cdk5 were cotransfected into CHO cells, and cells were harvested in the provided lysis–binding–wash buffer. For the Ras assays, wild-type Ras was cotransfected with the above transfections. An equal amount of EV lysate was incubated with GTP as a positive control. Lysates were incubated with GST-Pak1-PBD, which specifically interacts with active Rac1, and GST-Raf1, which interacts with active Ras. The bound complexes were then incubated with glutathione-Sepharose beads provided for 1 hr at 4°C. GST-bound active Rac1 and Raf were washed three times in buffer and eluted from the GST beads using 2×SDS sample buffer containing 5% β-mercaptoethanol (Sigma) and heated for 5 min. Samples were separated using 4–20% SDS-PAGE and subjected to Western blotting using the monoclonal anti-Rac1 and Ras Ab supplied in the kit. Samples of cell lysates were also run to detect the total amount of Ras and Rac present.
Results
p35 interacts with RasGRF2 in a yeast two-hybrid screen
To identify possible binding partners and substrates of p35/Cdk5, we used p25 as bait in a yeast two-hybrid screen of a human brain cDNA library. This bait was initially selected because it contains a common protein association region described previously (Wang et al., 2000). Several vigorously growing yeast colonies were obtained after selection with Trp–, Leu–, His–/25 mm 3-AT. After rescue of the prey plasmids from yeast, they were transformed into Escherichia coli DH5α to obtain single plasmids. One clone identified in the screen contained a cDNA insert encoding amino acid residues 2–463 of RasGRF2 (Fig. 1A). To further map the domains of interactions between the bait and prey plasmid, p10, p25, and p35/pGBKT7 (GAL-4 DNA binding domain-BD) (Fig. 1B) were cotransformed with RasGRF22–463/PGADT7 (GAL-4 DNA activation domain-AD) into yeast strain AH109 and tested for β-galactosidase activity (Fig. 1C). p10 did not interact with RasGRF22–463; however, both p25 and p35 interacted with RasGRF22–463 with equal strength using β-galactosidase activity assays, thus confirming our initial screening result. RasGRF2 is a widely expressed guanine nucleotide exchange factor (GEF) that contains a number of protein motifs such as two PH domains, a DH domain, a CC motif, an IQ motif, a Ras exchanger motif (REM), a cyclin-destruction box (CDB), and a Cdc25 motif (Fig. 1A). Because RasGRF22–463 contains some of these domains, precise mapping of the domain(s) of interaction with p25 and p35 were performed. GAL4-AD constructs of the DH, IQ, and CC domains were produced and cotransformed with p10, p25, and p35 to map the domains of interaction. Cotransformants were subjected to β-galactosidase assays to examine interaction strength, and the results are summarized in Figure 1C. p25 had the highest affinity to the IQ domain, whereas it had the lowest affinity with the DH domain. It had a slightly stronger interaction with the PH and CC domains compared with the DH domain. Both p25 and p35 interacted weakly with the DH domain. Similar to p25, p35 interacted strongest with the IQ domain of RasGRF2, weakly with the CC and DH domains, and slightly stronger with the PH domain. This suggests that calcium levels may play a role in RasGRF2 interaction with p35 and p25, because the IQ domain is the calcium sensor.

p35 interacts with RasGRF2 in a yeast two-hybrid screen. A, Schematic illustration of RasGRF2. RasGRF2 is a protein of 1262 amino acids containing a number of distinct domains. It has two PH domains, one at its N terminal. This is followed by CC, IQ, and DH domains. A REM is followed by a CDB, and at its C terminus it contains a Cdc25 motif. Fragment (RasGRF22–463) isolated from the screen is shown as a dark box above the full-length protein. B, Schematic illustration of the Cdk5 activator p35. p35 consists of two distinct domains. p10 (aa residues 1–99) and p25 (aa residues 100–307). cDNA fragments corresponding to p10, p25, and p35 were constructed and inserted in-frame into the pGBKT7 yeast DNA binding-domain vector. C, Mapping of p35 domain interaction with RasGRF2. Yeast cotransformed with different domains of p35 and RasGRF2 were grown on media lacking tryptophan, leucine, and histidine and containing 25 mm 3-aminotriazole. β-galactosidase assays were performed, and interactions were judged compared with the color generation of the positive control. p10, p25, and p35 were cotransformed with RasGRF22–463. Interaction strength is indicated as follows: ++++, very strong interaction (color generation was visible after 1 hr); +++, strong interaction (color visible after 2 hr); ++, medium strength interaction (color generation was visible after 4 hr); +, weak interaction (color weakly visible between 4 and 6 hr). Minus sign refers to an absence of interaction (no color seen).
RasGRF22–463 and full-length RasGRF2 interact with p25 and p35 in mammalian cells and brain
A disadvantage of the yeast two-hybrid system is its propensity to exhibit false positives or an interaction that is not reproduced in mammalian cells. To confirm the mammalian relevance of the interaction found in yeast, we used coimmunoprecipitations to confirm that p25 and p35 can interact with the fragment of RasGRF2 identified from the yeast two-hybrid screen in another system. The fragment of RasGRF2 isolated in the yeast two-hybrid screen (RasGRF22–463) was subcloned into the pCMV-Myc epitope-tagged vector and cotransfected with p25 and p35 into CHO cells. The monoclonal anti-c-myc Ab (9E10) was used to immunoprecipitate myc-RasGRF22–463, and p25 and p35 were immunodetected using the polyclonal C19 Ab. Both p25 and p35 coimmunoprecipitated with RasGRF22–463 (Fig. 2A). To further confirm that the interaction was not a fragment artifact of the yeast two-hybrid system, p25 and p35 were cotransfected with full-length FLAG-RasGRF2, and p25 and p35 were immunoprecipitated using monoclonal anti-Xpress Ab (Xpress is an epitope engineered into pcDNA3.1C vectors). Full-length RasGRF2 was then immunodetected using the monoclonal anti-RasGRF2 Ab. p25 and p35 were immunodetected using C19 (anti-p35 Ab). In this case, however, p35 interacted with RasGRF2 but p25 did not (Fig. 2B). A possible reason that the fragment of RasGRF2 interacted with both p25 and p35 but the full-length RasGRF2 only interacted with p35 may be attributed to stearic hindrances present in the full-length RasGRF2 that are absent in the fragment. When p25 and p35 were cotransfected with RasGRF22–463, both p25 and p35 were able to interact with this fragment. When full-length RasGRF2 is cotransfected with p25 and p35, however, the folding of full-length RasGRF2 may mask the epitope that p25 is able to bind to. p35, with its N-terminal extension and because it is a larger protein, may interact with the full-length RasGRF2 at a region that p25 cannot bind to. To investigate whether this interaction also occurs in brain tissue, we immunoprecipitated p35 from brain and immunodetected RasGRF2. Indeed, we found that p35 and RasGRF2 coimmunoprecipitates in rat brain (Fig. 2C), thus confirming our yeast two-hybrid data.

p25 and p35 coimmunoprecipitate with RasGRF2 in transfected cells and adult rat brain. A, CHO cells were cotransfected with EV, p25/RasGRF22–463 (GRF2 FRAG), p35/RasGRF22–463, and RasGRF22–463 alone. RasGRF22–463 was immunoprecipitated using the monoclonal anti-c-myc 9E10 Ab. Presence of RasGRF22–463 was confirmed using the polyclonal anti-c-myc Ab (top panel). p25 and p35 were detected using polyclonal C19 Ab (bottom panel). Plus and minus signs refer to the presence and absence of immunoprecipitating anti-c-myc Ab, respectively. Samples of cellular lysates before immunoprecipitation were resolved: 1, p25/RasGRF22–463; 2, p35/RasGRF22–463; 3, empty vector. Single asterisk refers to the position of RasGRF22–463; double asterisk refers to the position of p35; triple asterisk refers to the position of p25. The polyclonal anti-myc Ab cross-reacts with the light chain of the monoclonal 9E10 Ab, explaining the band above 24 kDa seen in the top panel. B, CHO cells were cotransfected with EV, p35/FLAG-GRF2, p25/FLAG-GRF2, and FLAG-GRF2. p25 and p35 were immunoprecipitated using the mouse monoclonal anti-Xpress Ab, and the presence of p25 and p35 was confirmed using C19 Ab (top panel). The presence of full-length GRF2 was detected using the monoclonal anti-RasGRF2 Ab (bottom panel). Full-length RasGRF2 coimmunoprecipitates with p35 but not with p25. Plus and minus signs refer to the presence and absence, respectively, of immunoprecipitating anti-Xpress epitope tag Ab. Transfected cell lysates were also resolved: 1, p25/RasGRF2; 2, p35/RasGRF2; 3, EV. Single asterisk refers to the presence of p35; double asterisk refers to the presence of p25; triple asterisk refers to the presence of full-length RasGRF2. C, p35 was immunoprecipitated from rat brain lysate using the monoclonal anti-p35 Ab, and the presence of p35 was confirmed using C19 Ab (top panel) RasGRF2 was immunodetected using the polyclonal anti-RasGRF2 Ab (bottom panel). Plus and minus signs refer to the presence and absence, respectively, of immunoprecipitating Ab. Samples of CHO cells transfected with p35 and GRF2 (CHO) and rat brain lysate (RB) were resolved as markers.
Full-length RasGRF2 colocalizes with p35 in transfected COS-7 cells and in primary cortical neurons
Previous studies using transfected p35 showed that a proportion of p35 localizes to the cell periphery and cell body (Nikolic et al., 1996; Kesavapany et al., 2001, 2003). Because the interaction of p25 and RasGRF2 is absent in CHO cells, we investigated the p35–RasGRF2 interaction. Full-length RasGRF2 and p35 were cotransfected with p35 into COS-7 cells to confirm that these two proteins exist in similar cellular compartments. Immunocytochemistry was used to determine whether the proteins could be found in similar cellular compartments and whether they colocalized with each other. RasGRF2 had a cytoplasmic staining pattern (Fig. 3a,d) and was absent in the nucleus (Fig. 3b). Immunocytochemical analyses of cotransfected cells revealed that p35 colocalized with RasGRF2 in the cell body and along the membrane (Fig. 3d–f). p35 was found mainly in the cell perikarya and along the cell membrane, with slight staining in the nucleus (Fig. 3e). p35 has been shown to have a perikaryal distribution in neurons, localizing to the cell membrane, along the axon, and in the neurite tips of cultured cortical neurons (Nikolic et al., 1996; Kesavapany et al., 2001, 2003). The localization of RasGRF2, however, has not been studied extensively. We used immunocytochemistry to determine whether endogenous p35 colocalizes with endogenous RasGRF2 as well as whether the transfected proteins colocalize in a similar manner in neurons as they do in transfected COS-7 cells. Cortical neurons cultured for 7-DIC were used for the endogenous detection because expression of RasGRF2 is regulated developmentally and detected weakly at 5-DIC (data not shown) but more strongly at 7-DIC. In 7-DIC neurons, RasGRF2 staining was detected strongly in the cell periphery as well as along the axon (Fig. 3g). p35 was also strongly detected in the cell body, along the axonal processes, and some was found in the nucleus (Fig. 3h). The overlay shows colocalization of the endogenous proteins in yellow (Fig. 3i). Endogenous and transfected RasGRF2 was expressed in the neuronal periphery and along the axons in the transfected cells but not in the nucleus (Fig. 3g,j), and it clearly colocalized with endogenous p35 in the cell perikarya and along the length of the neurite (Fig. 3l). To ensure that we were not observing a p35 staining artifact, both RasGRF2 and p35 were cotransfected in 3-DIC cortical neurons so that both primary antibodies could be used at lower concentrations to ensure specificity. Immunocytochemical analyses showed that RasGRF2 and p35 colocalized in these double-transfected neurons. Both were found to be present in the cell periphery and along the neurite (Fig. 3m–o).
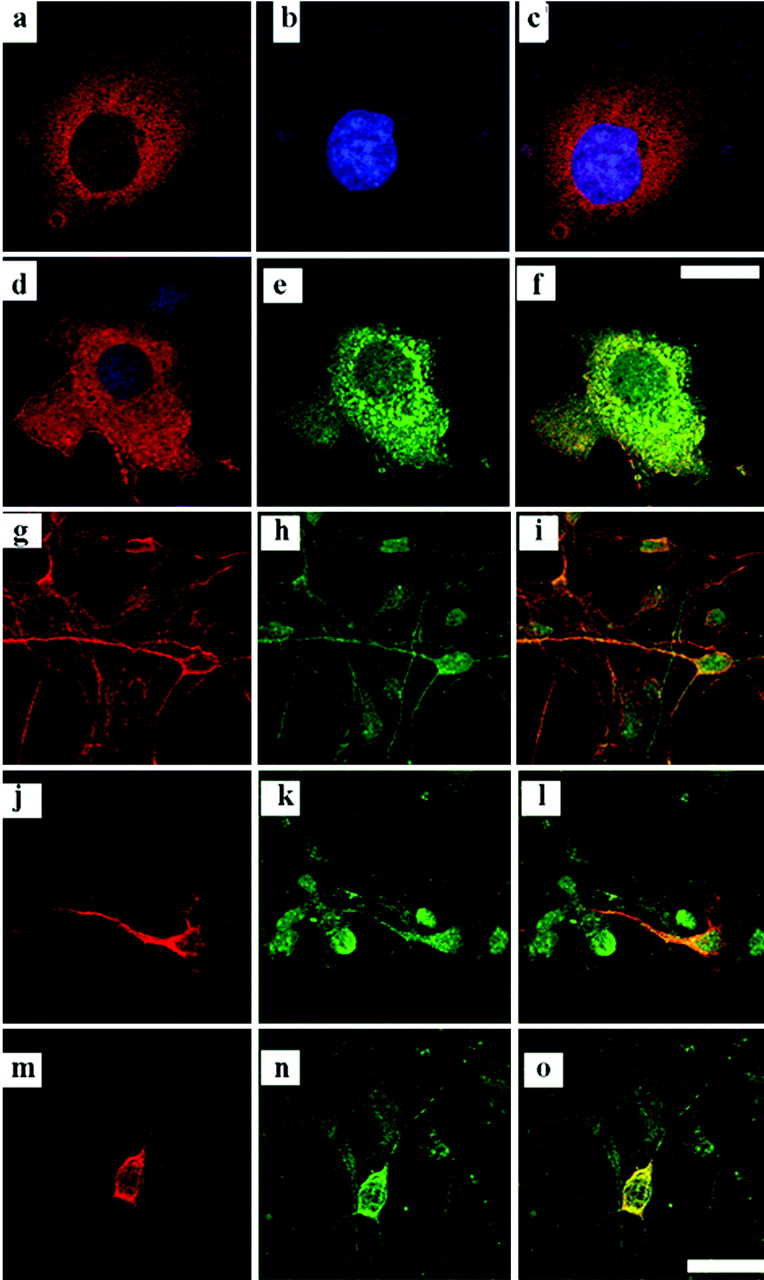
p35 colocalizes with RasGRF2 in transfected COS-7 cells and in cortical neurons. a–c, Full-length RasGRF2 was transfected into COS-7 cells and immunostained using the monoclonal anti-RasGRF2 Ab, and this was visualized using the anti-Texas Red-conjugated secondary Ab (a). Counterstaining of the nucleus was performed using Hoescht 33342 (b). Overlay of the two stains is shown in c. d–f, To examine colocalization between RasGRF2 and p35, COS-7 cells were cotransfected with p35 and RasGRF2. RasGRF2 was immunostained with Texas Red (d), and p35 was immunostained with Oregon Green (e). The overlay showing colocalization is seen in yellow (f). Scale bar, 20 μm. g–i, 7-DIC cortical neurons were immunostained for endogenous RasGRF2 and p35. RasGRF2 was immunostained using the monoclonal anti-RasGRF2 Ab and visualized with Texas Red (g). p35 was immunostained using C19 and visualized using Oregon Green (h). The overlay shows colocalization of the two proteins in yellow (i). Colocalization is visible in the cell body and along processes of the neurons. j–l, 3-DIC cortical neurons were transfected with RasGRF2. RasGRF2 was detected using the monoclonal anti-RasGRF2 Ab and visualized using Texas Red (j), endogenous p35 was detected using C19 and visualized using Oregon Green (k), and the overlay shows the colocalization of the two proteins in yellow (l), with a distribution pattern similar to the endogenous staining described above. m–o, 3-DIC cortical neurons were cotransfected with RasGRF2 and p35. RasGRF2 was detected using the monoclonal anti-RasGRF2 Ab and visualized using Texas Red (m), p35 was detected with C19 and visualized using Oregon Green (n), and the overlay shows colocalization in yellow (o). Scale bar, 20 μm.
RasGRF2 is phosphorylated by p35/Cdk5
p35/Cdk5 is an active, neuronal-specific kinase complex. To determine whether p35/Cdk5 phosphorylates RasGRF2, in vitro kinase assays were performed. In Figure 4A, in vitro kinase assays show that active Cdk5, immunoprecipitated from rat brain, phosphorylates RasGRF2. To show that this was a specific effect of Cdk5, the specific Cdk5 inhibitor roscovitine (10 μm) was added, and this reduced the levels of phosphorylation (GRF2 + c8 + ROS). Positive controls using histone H1, with and without roscovitine, were also performed to confirm that, first, there was Cdk5 kinase activity in the immunoprecipitates, and second, that this activity was inhibited by the use of roscovitine (H1 + c8 +/– ROS). In addition, to further confirm that Cdk5 was responsible for RasGRF2 phosphorylation and to rule out the involvement of other kinases, we also performed the in vitro phosphorylation reactions using recombinant and purified GST-Cdk5 and GST-p35.

RasGRF2 is phosphorylated by p35/Cdk5. A, Active Cdk5 was immunoprecipitated using C8 Ab from adult rat brain lysates. RasGRF2 substrate was obtained by transfecting FLAG-RasGRF2 into CHO cells and immunoprecipitating the protein using the monoclonal anti-FLAG Ab. Kinase assays were resolved by SDS-PAGE and Coomassie stained and destained, and gels were dried and subjected to autoradiography. The top panel is the Coomassie stained gel; the bottom panel is the corresponding autoradiograph. GRF2 refers to the presence of RasGRF2 immunoprecipitates, whereas –GRF2 refers to the absence of RasGRF2 immunoprecipitating Ab; –C8 refers to an absence of immunoprecipitating C8 Ab. Assays were performed in the presence of 10 μm roscovitine to specifically inhibit Cdk5 (+ROS). As a positive control for Cdk5 activity, kinase assays were performed using histone H1 as a Cdk5 substrate, with and without ROS. Similar results were obtained in the presence of 5 μm roscovitine. B, To identify the Cdk5 phosphorylation site of RasGRF2, in vitro kinase assays of RasGRF2 were performed. RasGRF2 was isolated by SDS-PAGE, excised from the gel, and subjected to in-gel tryptic digestion. The peptides were then resolved by thin-layer chromatography, and the phosphorylated species were visualized by autoradiography of the cellulose plate. The phosphopeptide identified from the autoradiograph of the cellulose TLC plate (B1) was scraped off the plate and subjected to MALDI mass spectroscopy. The peptide identified was LAVSRTSS*PVRARKLS (indicated beneath autoradiograph) (B), where serine737 was found to be the phospho-site. The open circle indicates where the tryptic digest of phosphor-RasGRF2 was applied to the plate for electrophoresis. C, To eliminate the possibility that another kinase immunoprecipitated with Cdk5 to phosphorylate RasGRF2, we used recombinant Cdk5 and p35 in in vitro kinase assays. RasGRF2 wild-type (WT RasGRF2) and two serine-to-alanine mutants corresponding to serine717 (S717A GRF2) and serine737 (S737A GRF2) were transfected and immunoprecipitated from CHO cells, and assays were performed with GST and GST-Cdk5+glutathione S-transferase-p35 (Cdk5 p35). ROS (5 μm) and H1 were used as controls. Top panel corresponds to the Coomassie-stained gel, and the bottom panel is the corresponding autoradiograph. D, RasGRF2 was immunoprecipitated from age-matched wild-type (WT) and p35 knock-out (KO) mouse brain lysates using the anti-RasGRF2 Ab. Samples were then resolved by SDS-PAGE, transferred onto nitrocellulose, and probed for the presence of phosphoserine–threonine species as well as RasGRF2 itself. The top panel shows equal amounts of RasGRF2 immunoprecipitated. Samples of lysates are shown to the right. The middle panel shows the presence of phosphorylated serine–threonine–proline RasGRF2 species in wild-type mouse brain that is absent in the p35 knock-out mouse brain. The third panel shows the presence of RasGRF2, and the bottom panel shows the samples probed with anti-phosphotyrosine 4G10 Ab. +/– refers to the presence and absence of RasGRF2 immunoprecipitating Ab. A sample of wild-type mouse brain lysate was also probed with 4G10 to confirm that the Ab was working.
p35/Cdk5 phosphorylates RasGRF2 on serine737
We used the RasGRF2 phosphorylated by GST-p35/Cdk5 and performed phosphopeptide mapping and MALDI mass spectroscopy to identify the site phosphorylated by Cdk5. In Figure 4B, we show the autoradiograph of the tryptic digest. The spot identified was scraped off the TLC plate and subjected to MALDI analysis, and it was identified as the peptide LAVSS*PVRA, which corresponded to serine737 of RasGRF2. Serine737 is included in an excellent consensus phosphorylation sequence for Cdk5 (S/T P X K/R), where X is any amino acid (Veeranna et al., 1995). Accordingly, we produced a serine737 to alanine737 mutant (S737A) and a serine717 to alanine717 mutant (S717A), in which this sequence was KS*PRLC, as a control. Using recombinant GST-Cdk5 and GST-p35 in in vitro kinase assays, we confirmed that RasGRF2 is phosphorylated by p35/Cdk5 and, additionally, that serine737 was indeed the site targeted (Fig. 4C). The S717A RasGRF2 mutant produced similar phosphorylation as wild-type RasGRF2 (Fig. 4C). In Figure 4D, we show Cdk5 phosphorylation of RasGRF2 in vivo. RasGRF2 immunoprecipitations from wild-type mice showed phosphorylation of serine–threonine residues in RasGRF2 with the anti-phosphothreonine–serine–proline Ab. This signal was absent from immunoprecipitates of RasGRF2 from p35 knock-out mice, showing that p35/Cdk5 is responsible for RasGRF2 phosphorylation in brain. The involvement of a tyrosine kinase associated with RasGRF2 was discounted when the immunoprecipitates were probed for phosphorylated tyrosine residues using 4G10 Ab. No signal was detected in mouse brain immunoprecipitates, although total lysates showed a number of phosphotyrosine bands, confirming that the Ab was working (Fig. 4D). The membranes were stripped and reprobed with anti-RasGRF2 Ab to confirm that the immunoprecipitations had pulled down RasGRF2 (Fig. 4D, bottom panel). Additionally, FLAG-RasGRF2 immunoprecipitations were phosphorylated using nonradioactive ATP, and the immunoprecipitates were probed using 4G10. We could not detect RasGRF2 tyrosine phosphorylation (data not shown).
p35/Cdk5 downregulates RasGRF2-mediated Rac activity but does not affect RasGRF2-mediated Ras activity
Previous reports have shown that Ras and Rac are membrane-associated GTPases that are able to activate mitogen-activated protein kinase (MAPK) cascades in the cell in response to extracellular signals received at the cell surface. The activation of Ras and Rac have been shown to be controlled directly by RasGRF2 affecting both Ras and Rac pathways, thereby affecting the activation of the MAPK and stress-activated protein kinase pathways (Fan et al., 1998). To determine whether p35/Cdk5 was involved in cellular Rac activity, we cotransfected EV, RasGRF2, RasGRF2+p35+Cdk5, and RasGRF2+Cdk5 into CHO cells and performed Rac activity assays using the harvested lysates (Fig. 5A). EV lysate showed a basal level of Rac activity (Fig. 5A, lane 1) that increased when GTP was added (Fig. 5A, lane 5). In the presence of RasGRF2, Rac activity increased compared with basal levels (Fig. 5A, lane 2). The cotransfection of p35/Cdk5 with RasGRF2 reduced levels of active Rac back to basal levels (Fig. 5A, lane 3). This observation was confirmed when inactive Cdk5 (without p35) was cotransfected with RasGRF2, in which active Rac levels were similar to levels of active Rac seen when only RasGRF2 was transfected (Fig. 5A, lane 4). To investigate whether p35/Cdk5 affects RasGRF2-mediated Ras activity, we performed the same transfections as those done for the Rac activity. In Figure 5B we show that p35/Cdk5 does not significantly affect Ras activity (Fig. 5B, compare lanes 2, 3, and 4).

p35/Cdk5 and RasGRF2 mediate cellular Rac activity and ERK1/2 activity. A, CHO cells were transfected with EV-GTP (lane 1), RasGRF2 (lane 2), RasGRF2+p35+Cdk5 (lane 3), RasGRF2+Cdk5 (lane4), and EV+GTP (lane5). CHO cell lysates were incubated with GST-PAK1 and GST beads for 1 hr. Beads with bound active Rac were washed three times in lysis–binding–wash buffer and eluted by addition of 2× SDS-sample buffer/β-mercaptoethanol and boiling for 5 min. Eluted, active Rac samples were resolved by SDS-PAGE, transferred onto nitrocellulose, and immunodetected using the monoclonal anti-Rac1 Ab provided in the kit at 1:1000 dilution. p35/Cdk5 reduced RasGRF2-mediated active Rac levels. EV lysate incubated with GTP before the pull-downs exhibited the highest active Rac levels (EV+GTP). Representative Western blots show active Rac1 levels and their corresponding total Rac levels. B, p35/Cdk5 does not affect RasGRF2-mediated Ras activity. Transfections are as follows: 1, EV; 2, RasGRF2; 3, RasGRF2+p35+Cdk5; 4, RasGRF2+Cdk5; 5, EV+GTP. CHO cell lysates were incubated with GST-Raf1 and GST beads for 1 hr. Beads with bound active Ras were washed three times before addition of 2× SDS-sample buffer/β-mercaptoethanol and boiling for 5 min. Eluted active Ras samples were resolved by SDS-PAGE and transferred onto nitrocellulose, and Ras was detected with the monoclonal anti-ras Ab provided in the kit. Immunodetected samples are indicated next to the panels. C, CHO cells were cotransfected with the following: 1, EV; 2, RasGRF2; 3, RasGRF2+p35+Cdk5; 4, RasGRF2+Cdk5. Lysates were harvested, and equal amounts were resolved by SDS-PAGE, transferred onto nitrocellulose, and probed for the presence of phospho-MEK1/2, phospho-ERK1/2, total ERK1/2, total Rac1, p35, Cdk5, and RasGRF2 levels (panels are labeled for the detected proteins). Total Rac1 levels are similar in samples, whereas phospho-ERK1/2 levels are reduced after transfection of p35/Cdk5 (sample 3) (confirmed by the level of total ERK1/2 in this sample, which is higher than those of samples 2 and 4). Phospho-ERK1/2 levels are increased when the inactive Cdk5 is present (sample 4). The transfection of RasGRF2 causes an increase in phospho-ERK1/2 levels, whereas the transfection of p35/Cdk5 with RasGRF2 decreased this activation. The cotransfection of the inactive Cdk5 (lacking p35) causes the activated levels of ERK1/2 to return to the levels observed when only RasGRF2 was transfected. Immunodetected proteins are indicated next to the panels. D, Rac activity assays were performed to confirm that the serine737 was indeed the phospho-site targeted by Cdk5 to mediate RasGRF2-mediated Rac activity. Tranfections are as follows: 1, EV; 2, RasGRF2; 3, RasGRF2+p35+Cdk5; 4, RasGRF2S737A+p35+Cdk5; 5, RasGRF2S717A+p35+Cdk5; 6, EV+GTP. The RasGRF2S737A mutant behaved like wild-type RasGRF2; however, the RasGRF2S717A mutant with p35 and Cdk5 showed the reduction in Rac activity demonstrating that serine737 was the site targeted by Cdk5. E, Serine737 phosphorylation by p35/Cdk5 reduces ERK1/2 activity. Transfections are as follows: 1, empty vector; 2, RasGRF2; 3, RasGRF2+p35+Cdk5; 4, RasGRF2S737A+p35+Cdk5. Equal amounts were resolved by SDS-PAGE, transferred onto nitrocellulose, and probed for the presence of phospho-MEK1/2, phospho-ERK1/2, total ERK1/2, p35, Cdk5, and RasGRF2 levels. Immunodetected proteins are indicated next to the panels. The cotransfection of RasGRF2S737A+p35+Cdk5 reversed the downregulation of ERK1/2 activity mediated by RasGRF2+p35+Cdk5, thus confirming that serine737 is the site targeted by p35/Cdk5 to downregulate RasGRF2-mediated ERK1/2 activity.
p35/Cdk5 downregulates Rac-dependent ERK1/2 activation through phosphorylation of serine737 of RasGRF2
Rac is thought to influence cellular morphology through actin reorganization as well as activation of the ERK1/2 pathway (Fam et al., 1997; Fan et al., 1998; Rashid et al., 2001). Here we report that the decrease in Rac activity mediated by p35/Cdk5 and RasGRF2 is coupled to a reduction in phospho-MEK1/2, the upstream regulator of ERK1/2. The decrease in phospho-MEK1/2 levels leads to a decrease in activated–phospho-ERK1/2 levels (Fig. 5C). In CHO cells, phospho-ERK1/2 (activated ERK1/2) is almost undetectable (Fig. 5C, lane 1); however, after introduction of RasGRF2, phospho-MEK1/2 and ERK1/2 levels increase (Fig. 5C, lane 2). p35/Cdk5, when cotransfected with RasGRF2, decreases the phospho-ERK1/2 levels (Fig. 5C, lane 3). This observation is confirmed by phospho-ERK1/2 levels returning to their highest level when inactive Cdk5 is present (Fig. 5C, lane 4). The same pattern is observed with the levels of phospho-MEK1/2. Steady-state levels of Rac are unchanged when RasGRF2 and p35/Cdk5-RasGRF2 are transfected into the cells. We have confirmed that p35/Cdk5 downregulation of Rac activity occurs through the phosphorylation of RasGRF2 on serine737 (Fig. 5D). In these experiments, the cotransfection of RasGRF2S737A with p35/Cdk5 displayed similar Rac activation compared with wild-type RasGRF2 alone (Fig. 5D, lanes 2, 4). Further confirmation of this was afforded through the cotransfection of RasGRF2S717A with p35/Cdk5 (Fig. 5D, lane 5), in which Rac activity was identical to that of wild-type RasGRF2 with p35/Cdk5. The data suggest that phosphorylation of RasGRF2 on serine737 by Cdk5 affects ERK1/2 activity through modulation of Rac activity.
To determine that the phosphorylation of serine737 of RasGRF2 was responsible for the downregulation of ERK1/2 activity, we probed lysates of empty vector, RasGRF2, RasGRF2+p35+Cdk5, and RasGRF2S737A+p35+Cdk5 for phosphorylated or activated ERK1/2. Empty vector lysates displayed low levels of phospho-MEK1/2 and phospho-ERK1/2 (Fig. 5E, lane 1). These increased when RasGRF2 was transfected into the cells (Fig. 5E, lane 2), and as in Figure 5C, lane 2, these levels were reduced with the cotransfection of RasGRF2+p35+Cdk5 (Fig. 5E, lane 3). The site of Cdk5 phosphorylation was confirmed when RasGRF2S737A was cotransfected with p35/Cdk5 and the levels of activated MEK1/2 and activated ERK1/2 remained comparable with RasGRF2 alone (Fig. 5E, lane 4). This showed that serine737 now mutated to alanine and is not phosphorylated by p35/Cdk5, and thus the MEK1/2 and ERK1/2 activation cannot be downregulated by p35/Cdk5.
p35/Cdk5 causes RasGRF2 and MAP1b accumulation in cortical neurons
The modulation of Rac activity and downstream effects on ERK1/2 activity by p35/Cdk5 phosphorylation of RasGRF2 led us to believe that a possible downstream effect of this could be an alteration of microtubule distribution in neurons. To examine this hypothesis, we transfected 3-DIC cortical neurons with RasGRF2 and examined MAP1b localization. We used 3-DIC neurons because at this stage RasGRF2 is undetectable by Western blotting (data not shown), and thus we could specifically determine the effects of RasGRF2-p35/Cdk5 in neurons. When RasGRF2 alone was transfected into the neurons, it was found in the neuronal perikarya and all along the neuronal processes. Additionally, RasGRF2 colocalized with MAP1b in these areas (Fig. 6Aa–Ac). After cotransfection with p35/Cdk5, RasGRF2 was found predominantly “clumped” in the “swollen” neuronal cell body, again colocalizing with MAP1b, with very little staining found along the processes (Fig. 6Ad–Ag). When neurons were transfected with RasGRF2, p35, and dominant-negative Cdk5, the staining pattern of both RasGRF2 and MAP1b reverted to that of RasGRF2 alone, being distributed along the length of the processes (Fig. 6Ah–Ak). We determined that this staining pattern was not caused by p35/Cdk5 because this accumulation of RasGRF2 and MAP1b was not present in p35/Cdk5 transfections (Fig. 6Al–Ao).

p35/Cdk5 phosphorylation of RasGRF2 causes an accumulation of MAP1b in neurons. A, 3-DIC cortical neurons were transfected with RasGRF2 (a–c), p35+Cdk5+RasGRF2 (d–g), p35+dominant-negative Cdk5 (DNCdk5) (h–k), and p35+Cdk5 (l–o). Transfected RasGRF2 localized to the neuronal perikarya and along the neurite (a). MAP1b colocalized with RasGRF2 in these areas (b). After cotransfection of p35/Cdk5 and RasGRF2, RasGRF2 and MAP1b distributions accumulated in the cell bodies of the neurons and were reduced in the processes (e–g). When RasGRF2 was cotransfected with p35/dominant-negative Cdk5, the localization of both RasGRF2 and MAP1b was found in the cell body and along the neurites (i–k), resembling RasGRF2-only transfected neurons (a–c). p35/Cdk5 did not account for the accumulation of MAP1b, because the distribution of the proteins was unaffected in p35/Cdk5-transfected neurons (l–o). Distribution of RasGRF2, Cdk5, and MAP1b was analyzed by immunofluorescence: RasGRF2 was visualized with Texas Red, MAP1b with Oregon Green, and Cdk5 with Alexa-Fluor 633. Overlapping images are shown in c, g, k, and o. Scale bar, 20 μm. B, To determine that the accumulation of MAP1b was caused by a modulation of ERK1/2 activity, the MEK1/2 inhibitor PD98059 was used and visualized as in A. Untreated RasGRF2-transfected neurons (a–c) exhibited RasGRF2 and MAP1b staining in the cell body and along the neurites. RasGRF2-transfected neurons that were treated with PD98059 (d–f) showed the accumulation of MAP1b identical to the results obtained with p35/Cdk5 (Fig. 6 Ad–Ag), confirming that ERK1/2 activity is responsible for the accumulation of MAP1b. C, To confirm that serine737 of RasGRF2 was the p35/Cdk5 phosphorylation site causing the accumulation of RasGRF2 and MAP1b in neurons, we used the RasGRF2S737A and RasGRF2S717A mutants in transfection experiments and investigated the localization of RasGRF2 and MAP1b. Wild-type RasGRF2-transfected neurons (a–c) exhibited MAP1b staining in the cell body and along the neurites. Wild-type RasGRF2+p35+Cdk5 neurons (d–g) exhibited accumulation of MAP1b in the cell body. The transfection of RasGRF2S717A+p35+Cdk5 (h–k) did not change the accumulation pattern of MAP1b compared with wild-type RasGRF2+p35+Cdk5. RasGRF2S737A+p35+Cdk5-transfected neurons (l–o) reversed the MAP1b staining similar to that of RasGRF2+p35+DNCdk5 (Ah–Ak) and of wild-type RasGRF2. Scale bar, 50 μm.
To confirm the involvement of ERK1/2 activity in the distribution of MAP1b, we treated cortical neurons with PD98059 and examined the distribution of MAP1b in RasGRF2-transfected neurons. RasGRF2-transfected neurons that were not treated exhibited MAP1b and RasGRF2 staining along their neurites (Fig. 6Ba–Bc). RasGRF2-transfected neurons treated with PD98059 exhibited MAP1b staining identical to p35/Cdk5-RasGRF2-transfected neurons, in which an accumulation of MAP1b is seen in the neuronal cell body (Fig. 6Bd–Bf). This result shows that the accumulation in the cell body and its absence along the neurites are caused by the inhibition of ERK1/2 activity.
To further exclude the involvement of other kinases, we transfected wild-type RasGRF2, RasGRF2+p35+Cdk5, RasGRF2S717A+ p35+Cdk5, and RasGRF2S737A+p35+Cdk5. As expected, and as shown previously, the RasGRF2 and RasGRF2+p35+Cdk5 exhibited the differences in MAP1b and RasGRF2 distributions (Fig. 6Cd–Cg). In Figure 6C, when RasGRF2S717A+p35+Cdk5 was transfected into neurons, the staining pattern was identical to that of wild-type RasGRF2+p35+Cdk5 (Fig.6Ch–Ck).When RasGRF2S737A+p35+ Cdk5 was transfected into the neurons, however, the staining of RasGRF2 and MAP1b was identical to wild-type RasGRF2 only, thus confirming that serine737 was the site targeted by p35/Cdk5 to alter RasGRF2 activity and RasGRF2 and MAP1b distribution (Fig. 6Cl–Co). To confirm that this was a specific effect on MAP1b, we repeated the transfections and examined MAP2, tubulin, and tau staining and found that their distributions were unaffected (data not shown). The results shown in Figures 5 and 6 collectively suggest that RasGRF2 affects ERK1/2 activity and this impacts on MAP1b distribution.
Discussion
The cardinal findings of this study are that the Cdk5 activator, p35, interacts with RasGRF2 in the yeast two-hybrid system, in mammalian CHO and COS-7 cells, in rat brain, and in cortical neurons. p35/Cdk5 phosphorylates RasGRF2 on serine737 to downregulate cellular Rac activity without affecting Ras activity or RasGRF2 steady-state levels. Additionally, p35/Cdk5 decreases the RasGRF2–Rac-dependent ERK1/2 activation, and thus our results show that Cdk5 is involved in a cross-talk relationship with RasGRF2 within the Rac signal transduction pathway. This is the first report to demonstrate that RasGRF2 is phosphorylated to regulate its Rac-activating function and that Cdk5 is responsible for this. Additionally, we found that the cotransfection of p35/Cdk5 and RasGRF2 in neurons led to MAP1b accumulation in the neuronal cell bodies. No such accumulations were observed when the distribution of tau, tubulin, and MAP2 were investigated. This suggested that MAP1b distribution was affected because of the p35/Cdk5 modulation of RasGRF2 activity.
Cdk5 activity is implicated in a number of neuronal functions through the phosphorylation of its substrates. Previous studies from this laboratory have shown that Cdk5 is involved in cross-talk between other signal transduction pathways such as the ERK1/2 and JNK3 pathways (Li et al., 2002; Sharma et al., 2002). Cdk5 phosphorylates MEK1, downregulating the ERK1/2 kinase pathway, and JNK3, also reducing its activity and subsequent phosphorylation of c-Jun. Thus, our results show that p35/Cdk5 mediation of ERK1/2 activity occurs at two distinct control points in the cell: at the MEK1 and upstream at RasGRF2–Rac levels. The involvement of p35/Cdk5 activity in regulating RasGRF2-mediated Rac activity also suggests that under normal physiological conditions, p35/Cdk5 and its membrane-associated functions are localized to controlling a signaling pathway originating from Rac at the membrane, to produce intracellular changes in downstream kinase activities and subsequent changes in cytoskeletal protein distribution. A schematic diagram summarizing our findings is shown in Figure 7. In this figure, all effects attributed to p35/Cdk5 in the present study are indicated by the red arrows.

Schematic illustration of p35/Cdk5 involvement in RasGRF2 signaling. In this figure, we summarize our findings that p35/Cdk5 mediates RasGRF2 signaling to the membrane-bound GTPase Rac. Rac is involved in actin dynamics as well as the modulation of ERK1/2 activity. Under normal circumstances, RasGRF2 mediates the transition of inactive GDP-bound Rac to an active, GTP-bound form. This active form of Rac then participates in actin polymerization and dynamics as well as activating ERK1/2 through as yet undetermined intermediary steps. p35/Cdk5 phosphorylates RasGRF2 on serine737, downregulating the transition of inactive to active Rac (solid, lighter-shade arrows). Subsequently, the levels of activated ERK1/2 are also decreased. The role of p35/Cdk5 is particularly specific for this pathway: it does not affect Ras activity or Rac-mediated actin dynamics. The solid black arrow shows p35/Cdk5 involvement in the ERK1/2 pathway, which has been described previously. (Sharma et al. 2002). Thus, p35/Cdk5 is involved at the upstream level of Rac signaling and also at the MEK1 level of the ERK1/2. This cross-talk and ultimate alteration of ERK1/2 activity may be important in maintaining distinct regulation of the ERK1/2 signaling cascade and the distribution of MAP1b under normal physiological conditions. Our results show p35/Cdk5 phosphorylation of RasGRF2, mediating ERK1/2 activity through Rac, leading to MAP1b distribution in neurons. This suggests that the phosphorylation state of MAP1b is important in its distribution along the processes of the neurons as well as its localization within the neuronal cell body. All effects attributed to p35/Cdk5 phosphorylation of RasGRF2, subsequent modulation of Rac and ERK1/2 activity, and MAP1b distribution are indicated by the solid, lighter-shade arrows.
The Rho family of GTPases, of which Rac is a member, is implicated in a number of cellular functions, such as cell migration and neurite outgrowth (Nikolic et al., 1996; Rashid et al., 2001). p35/Cdk5 has been shown to colocalize with Rac in neuronal growth cones, and p35 associates with Rac in a GTP-dependent manner (Nikolic et al., 1998). The active Cdk5 kinase complex causes the hyperphosphorylation of the Rac effector PAK1 kinase, modulating its activity as well as colocalizing with Rac1 itself. Because the Rho GTPases such as Rac and the PAK kinases are implicated in actin polymerization, this modification of PAK1 activity impacts the dynamics of the actin cytoskeleton. From the results reported here, we show that p35/Cdk5 alters active Rac1 levels in the presence of RasGRF2; however, we did not observe any significant changes in its actin levels or its cellular distribution (data not shown).
RasGRF1 and RasGRF2 belong to the RasGRF family of GEFs. RasGRF1 is a GEF that is expressed exclusively in brain, whereas RasGRF2 transcripts are detected in several tissues, including brain and lung. RasGRF1 was initially isolated as a releasing factor for Ras (Shou et al., 1992), in which phosphorylation of RasGRF1 on Ser916 by PKA (Yang et al., 2003) and increases in intracellular calcium concentrations lead to the activation of Ras signaling through RasGRF1 (Farnsworth et al., 1995; Buchsbaum et al., 1996; Freshney et al., 1997). RasGRF1 is also tyrosine-phosphorylated by the nonreceptor tyrosine kinases activated Cdc42-associated kinase 1 and Src to increase RasGRF1 activation of ERK2 and induce Rac–guanine nucleotide exchange activity, respectively (Kiyono et al., 2000a,b). Similar phosphorylation studies of RasGRF2 and correlation of its phosphorylation state to its activity have not been reported until now. The results here show that p35/Cdk5 is involved, in part, in modulating cellular RasGRF2-mediated Rac1 activity; however, it does not rule out the possibility that other kinases may also be involved, because RasGRF2 contains 16 putative proline-directed serine and threonine residues that may well be targets for other proline-directed kinases. Perhaps multiple kinases, acting alone or in coordination with Cdk5, may phosphorylate RasGRF2, having a more robust effect on cellular Rac1 activity and modulation of downstream kinase activities. We do show that the distribution of MAP1b is controlled by ERK1/2 activity and this in turn is mediated by p35/Cdk5, RasGRF2, and Rac activity. In our studies, RasGRF2 is readily detectable in adult brain and in 7-DIC cortical neurons, and thus Cdk5 may be one of the kinases that phosphorylates RasGRF2 in these tissues. In other tissues, kinases such as Cdc2 or Cdk2 may phosphorylate RasGRF2 to perform a similar role as Cdk5.
RasGRF2 contains regions that are also found in a number of signaling proteins, including two pleckstrin domains, a DH domain, a REM motif, and a motif that is similar to the CDB (Fig. 1A). The regions between the REM and the Cdc25 region in RasGRF2 are rich in proline, glutamic acid, serine, and threonine residues that are thought to target proteins for destruction (de Hoog et al., 2001). This region also contains Ser–Thr–Pro motifs that are characteristic of Cdk and MAPK phosphorylation sites. RasGRF2 has been shown to be ubiquitinated in vivo, and a mutant construct lacking the Cdc25 region, which abolishes the binding of Ras, is not ubiquitinated, suggesting that the binding of Ras is critical for this process (de Hoog et al., 2001). Our results, however, show that RasGRF2 steady-state levels were not altered after introduction of p35/Cdk5, suggesting that p35/Cdk5 does not mediate the destruction of RasGRF2. RasGRF2 has been shown to be an exchange factor for Ras in GTP-nucleotide exchange assays, with the Cdc25 domain found to be responsible for this function (Fam et al., 1997; Fan et al., 1998; de Hoog et al., 2001). The IQ motif in RasGRF2 is a domain that is known to be involved in calmodulin and calcium signaling, and RasGRF2 coimmunoprecipitates with calmodulin in a calcium-dependent manner (Cullen and Lockyer, 2002). From our results, it is clear that RasGRF2 does increase Ras activity, but this is not modulated by p35/Cdk5. This effect may be controlled by other kinases phosphorylating RasGRF2 on other sites.
Recent studies have implicated the deregulation of Cdk5 activity as a contributory factor toward pathogenesis of certain neurodegenerative diseases such as Alzheimer's disease and amyotrophic lateral sclerosis. Neurotoxic insults of neurons cause the activation of the calcium-dependent protease calpain, which cleaves p35 within its myristoylation sequence (Patrick et al., 1999). This produces the more active fragment p25, which not only hyperactivates Cdk5, but also, because of the loss of the membrane attachment, leads to a loss of Cdk5 substrate specificity and localization (Lee et al., 2000). Perhaps in neurotoxic conditions, a loss of localization of Cdk5 activity at the membrane may lead to a loss of regulation of RasGRF2 and Rac to the consequent changes in downstream signaling pathways. This may well have deleterious effects for the neurons affected.
This report provides further evidence of p35/Cdk5 involvement in cross-talk with other signaling pathways. Investigations under neurotoxic conditions may yield more information about the role that a deregulated Cdk5 may play in signal transduction pathways, and this may lead to a better understanding of the cellular events regulated by Cdk5 in normal as well as aberrant conditions.
Footnotes
We thank Dr. Carolyn Smith of the National Institute of Neurological Disorders and Stroke (NINDS) Light Imaging Facility for her assistance with the confocal studies, Dr. Li Huei Tsai (Harvard Medical School, Boston, MA) for p25, p35, and Cdk5 plasmids, and Dr. Mike Moran (MDS Proteomics, Toronto, Ontario, Canada) for the FLAG-RasGRF2 plasmid. We also thank Deborah Kaufmann and Jim Nagle of the NINDS DNA sequencing facility for their help in the sequencing studies.
Correspondence should be addressed to Harish C. Pant, Laboratory of Neurochemistry, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Building 36, Room 4D-28, Bethesda, MD 20892. E-mail: panth{at}ninds.nih.gov.
DOI:10.1523/JNEUROSCI.0690-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244421-11$15.00/0
↵* S.K. and N.A. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}