

Abstract
The stress-related neuropeptide corticotropin-releasing factor (CRF) and the serotonin system are both critically involved in the pathophysiology of mental disorders, including anxiety and depression. To understand the potential link between them, we investigated the impact of CRF on 5-HT functions in pyramidal neurons of the prefrontal cortex (PFC), a brain region that is crucial for the control of emotion and cognition. One prominent function of serotonin in PFC is to regulate GABAergic inhibitory transmission, as indicated by a 5-HT-induced large, desensitizing (∼4 min) enhancement of the amplitude and frequency of spontaneous IPSCs (sIPSCs). In PFC slices exposed to CRF treatment, the regulation of sIPSCs by 5-HT was significantly prolonged (8-10 min), and this effect of CRF was blocked by treatment with the competitive CRF receptor antagonist α-helical CRF9-41 and with the CRF-R1-specific antagonist astressin. Inhibiting phospholipase C or protein kinase C (PKC) abolished the prolongation by CRF of the effects of 5-HT on sIPSCs. In PFC slices prepared from animals previously exposed to acute stress (forced swim or elevated platform), the regulation of sIPSCs by 5-HT was significantly prolonged, mimicking the effect of CRF treatment. The stress-induced prolongation of the effects of 5-HT on sIPSCs was diminished by α-helical CRF9-41 treatment, mimicked by direct activation of PKC, and reversed by short-term treatment with drugs that have anxiolytic efficacy. These results show that in response to stressful stimuli, CRF alters the serotonergic regulation of GABA transmission through a mechanism that is dependent on PKC. The interaction between CRF and 5-HT may play an important role in psychiatric disorders, in which both are highly implicated.
Introduction
Corticotropin-releasing factor (CRF), a 41 aa neuropeptide, plays a key role in regulating physiological responses to stressful stimuli (Owens and Nemeroff, 1991). CRF is released from the hypothalamus and from extra hypothalamic neurons after stressor exposure (Tsigos and Chrousos, 2002) and displays a broad distribution in the brain (Sawchenko and Swanson, 1989). The peptide exerts a profound and complex impact on central neurons through its two receptors, CRF-R1 and CRF-R2 (Radulovic et al., 1999; Dautzenberg and Hauger, 2002). Mice deficient in CRF-R1 exhibit an impaired stress response and decreased anxiety-like behavior (Smith et al., 1998), whereas CRF-R2 mutant mice are supersensitive to stress and display increased anxiety-like behavior (Bale et al., 2000). CRF is implicated not only in the patho-physiology of affective and anxiety disorders but also in aversive states associated with drug withdrawal (Heinrichs et al., 1995; Sarnyai et al., 1995).
The serotonin system is also involved in mediating anxiety behaviors and stress responses (Deakin, 1988; Griebel, 1995; Shakesby et al., 2002). Mice lacking serotonin receptors show phenotypes ranging from increased impulsive aggression (Saudou et al., 1994) to elevated anxiety and an antidepressant-like response (Heisler et al., 1998; Gross et al., 2002). One of the main target structures of the serotonergic system is the prefrontal cortex (PFC), a brain region that is highly associated with the control of emotion and cognition (Miller, 1999). Specific changes in the PFC serotonin system found in patients with mental disorders (Stockmeier, 1997; Dean et al., 1999; Meyer et al., 1999) suggest that serotonin plays a crucial and unique role in PFC. One mechanism through which serotonin modulates PFC functions is via the regulation of GABAA receptor-mediated inhibitory synaptic transmission (Zhou and Hablitz, 1999; Feng et al., 2001; Cai et al., 2002; Yan, 2002). This regulation is potentially of clinical significance, because benzodiazepines (which reinforce transmission at GABAA receptors) and 5-HT-interacting agents are currently the principle drugs used in the management of anxiety disorders (Nutt et al., 1990; Taylor, 1990; Griebel, 1995; Gorman, 2003).
Given many of the common functional consequences of CRF and serotonin, we hypothesize that CRF may interact with the serotonin system, thereby aggravating behavioral responses to aversive stressors associated with anxiety and conditioned fear. Several lines of evidence have shown that CRF and stress interfere with serotonin neurotransmission by changing the firing rate of serotonergic neurons in dorsal raphe nuclei (DRN) and serotonin release (Adell et al., 1997; Maswood et al., 1998; Lowry et al., 2000). It is not known whether CRF modulates the synaptic functions of serotonin in limbic forebrain regions. In this study, we investigated the possibility that CRF and acute stress alter the 5-HT regulation of GABA transmission in PFC neurons. Because GABAA receptors are highly involved in anxiety and depression (Macdonald and Olsen, 1994; Lydiard, 2003), and GABAergic inhibition in PFC is a key element in the regulation of “working memory” (Constantinidis et al., 2002), the CRF-induced alteration of the effect of 5-HT on GABA transmission could provide a mechanism for exacerbated deficiencies in emotional and cognitive processes under stressful conditions.
Materials and Methods
Electrophysiological recordings in slices. Young adult rat slices containing PFC were prepared as described previously (Zhong et al., 2003). All experiments were performed with the approval of State University of New York at Buffalo Animal Care Committee. In brief, animals were anesthetized by inhaling 2-bromo-2-chloro-1,1,1-trifluoroethane (1 ml/100 gm; Sigma, St. Louis, MO) and decapitated; brains were quickly removed, iced, and then blocked for slicing. The blocked tissue was cut in 300-400 μm slices with a Vibratome while bathed in a low Ca2+ (100 μm), HEPES-buffered salt solution (in mm: 140 Na isethionate, 2 KCl, 4 MgCl2, 0.1 CaCl2, 23 glucose, 15 HEPES, and 1 kynurenic acid, pH 7.4; 300-305 mOsm/l). Slices were then incubated for 1-6 hr at room temperature (20-22°C) in an NaHCO3-buffered saline bubbled with 95% O2-5% CO2 (in mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 1 pyruvic acid, 0.05 glutathione, 0.1 NG-nitro-l-arginine, and 1 kynurenic acid, pH 7.4; 300-305 mOsm/l.
To evaluate the regulation of spontaneous IPSCs (sIPSCs) in PFC slices, the whole-cell patch technique was used for voltage-clamp recordings using patch electrodes (5-9 MΩ) filled with the following internal solution (in mm): 100 CsCl, 30 N-methyl-d-glucamine, 10 HEPES, 1 MgCl2, 4 NaCl, 5 EGTA, 0.8 2-(triethylamino)-N-(2,6-dimethylphenyl) acetamide, 12 phosphocreatine, 2 MgATP, 0.2 Na3GTP, and 0.1 leupeptin, pH 7.2-7.3; 265-270 mOsm/l. The slice (300 μm) was placed in a perfusion chamber attached to the fixed stage of an upright microscope (Olympus, Melville, NY) and submerged in continuously flowing oxygenated artificial CSF. For blocking glutamate transmission, the AMPA-kainate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (20 μm) and the NMDA receptor (NMDAR) antagonist d(-)-2-amino-5-phosphonopetanoic acid (20 μm) were added to the recording solution. Cells were visualized with a 40× water-immersion lens and illuminated with near infrared (IR) light, and the image was detected with an IR-sensitive CCD camera. A Multiclamp 700A amplifier (Axon Instruments, Foster City, CA) was used for these recordings. Tight seals (2-10 GΩ) from visualized pyramidal neurons were obtained by applying negative pressure. The membrane was disrupted with additional suction, and the whole-cell configuration was obtained. The access resistances ranged from 13 to 18 MΩ and were compensated 50-70%. Cells were held at -70 mV for the recording of spontaneous IPSCs. Application of the GABAA receptor antagonist bicuculline (10 μm) completely blocked the sIPSCs, indicating that these synaptic currents are mediated by GABAA receptors.
The Mini Analysis Program (Synaptosoft, Leonia, NJ) was used to analyze synaptic activity. Individual synaptic events with fast onset and exponential decay kinetics were captured with threshold detectors in Mini Analysis software. All quantitative measurements were taken 4-6 min after drug application. Neurons used for statistical analysis were required to have synaptic events with stable frequencies and amplitudes during both control and drug application. The detection parameters for analyzing synaptic events in each cell in the absence or presence of drugs were the same. Statistical comparisons of the frequency and amplitude of synaptic currents were made using the Kolmogorov-Smirnov test. Numerical values were expressed as mean ± SEM.
Ligands such as human/rat CRF, α-helical CRF9-41, astressin (Tocris Cookson, Ellisville, MO), and serotonin (Sigma), as well as the second-messenger reagents calphostin C, Gö6850 (i.e., bisindolylmaleimide I), phorbol 12-myristate 13-acetate (PMA), U73122, and myristoylated PKI[14-22] (Calbiochem, La Jolla, CA) were made up as concentrated stocks in water and stored at -20°C. Stocks were thawed and diluted immediately before use.
Stress paradigm. Two stress protocols (forced-swim test and elevated-platform test) that have been described previously (Xu et al., 1997; Price et al., 2002; Roche et al., 2003) were used in our studies. In the swim tests, rats were placed in a cylindrical glass tank (24.5 cm high × 18.5 cm diameter) filled with water to a depth of 20 cm. The 20 cm depth allowed the rats to reach the bottom with their tails only. Rats were forced to swim in warm water (24-26°C) for 30 min. In the elevated-platform tests, rats were placed on an elevated platform (20 × 20 cm) for 30 min in a brightly lit room. The animal showed behavioral “freezing” (i.e., immobility) for up to 10 min. Approximately 30 min after the stress procedures, rats were anesthetized and killed. Experimental groups were matched so that a stressed rat and a control rat were killed on the same day and tissue was processed in parallel. In some experiments, rats were injected intraperitoneally with saline, fluoxetine (20 mg/kg), fluvoxamine (5 mg/kg), or diazepam (3 mg/kg) before they were exposed to the forced-swim stress. One injection was given 12 hr before the stress experiments, and a second shot was sometimes given 45 min before the stress experiments.
Western blot analysis. To detect activated PKC, a phospho-PKC (pan) antibody that recognizes PKCα, PKCβI, PKCβII, PKCϵ, PKCη, and PKCδ isoforms only when phosphorylated at a C-terminal residue homologous to Ser660 of PKCβII was used in the Western blot analysis (Gu et al., 2003). After incubation, slices were transferred to boiling 1% SDS and homogenized immediately. Insoluble material was removed by centrifugation (13,000 × g for 10 min), and the protein concentration of each sample was measured. Equal amounts of protein from slice homogenates were separated on 7.5% polyacrylamide gels and transferred to nitrocellulose membranes. The blots were blocked with 5% nonfat dry milk for 1 hr at room temperature. Then the blots were incubated with the pan antibody (1:2000; Cell Signaling Technology, Beverly, MA) for 1 hr at room temperature. After being rinsed, the blots were incubated with a horseradish peroxidase-conjugated anti-rabbit antibody (1:2000; Amersham Biosciences, Arlington Heights, IL) for 1 hr at room temperature. After three washes, the blots were exposed to the enhanced chemiluminescence substrate. Then the blots were stripped for 1 hr at 50°C, saturated in 5% nonfat dry milk, and incubated with a PKC antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA) recognizing the α, β, and γ isoforms. Quantitation was obtained from densitometric measurements of immunoreactive bands on autoradiograms.
Results
Serotonergic modulation of GABA transmission is prolonged by CRF pretreatment
To test the potential interactions between CRF and serotonin, we examined the impact of CRF on 5-HT regulation of GABAergic inhibitory transmission in PFC. Slices were treated with or without CRF (200 nm) for 1-3 hr, followed by measurement of the effect of 5-HT on sIPSCs in PFC pyramidal neurons. CRF alone did not exhibit any significant effect on sIPSC amplitudes (2.8 ± 2.0%; n = 28; p > 0.01; ANOVA) or frequencies (5.8 ± 5.1%; n = 28; p > 0.01; ANOVA). As shown in Figure 1A-C, bath application of 5-HT (40 μm) caused a reversible increase in the amplitude and frequency of sIPSCs. The increase reached a peak and then started to decline within minutes during extended application of 5-HT. However, in CRF-treated slices, the 5-HT-induced increase in sIPSC desensitized much more slowly (Fig. 1D-F). In a sample of CRF-treated PFC pyramidal neurons that we examined (Fig. 1G), the duration of the effect of 5-HT on the sIPSC was 8.1 ± 0.7 min (n = 50), which was significantly (p < 0.001; ANOVA) longer than that in control cells (4.0 ± 0.5 min; n = 49). In contrast to the prolongation by CRF of the effect of 5-HT, CRF treatment did not significantly alter the enhancing effect of 5-HT on sIPSC amplitudes (Fig. 1H) (control, 113 ± 14.3%, n = 49; CRF-treated, 127.6 ± 15.6%, n = 50; p > 0.01; ANOVA) or sIPSC frequencies (Fig. 1I) (control, 347.8 ± 34.3%, n = 46; CRF-treated, 242.2 ± 40.4%, n = 46; p > 0.01; ANOVA).

CRF treatment prolonged the enhancing effect of 5-HT on sIPSC in PFC pyramidal neurons. A, B, Plot of sIPSC amplitude (A) and frequency (B) against time and agonist (5-HT, 40 μm) application. C, Representative sIPSC traces recorded from the neuron used to construct A and B under control conditions (ctl), during early (when the peak response emerged) and late (5 min after peak response) application of 5-HT, and after washing off the agonist (at time points denoted by # for A). Calibration: 100 pA, 1 sec. D, E, Plot of sIPSC amplitude (D) and frequency (E) against time and agonist (5-HT, 40 μm) application in a neuron exposed to CRF (200 nm). Slices were pretreated with CRF for 1-3 hr, and CRF was present throughout the recordings. F, Representative sIPSC traces recorded from the CRF-treated neuron used to construct D and E (at time points denoted by # in D). G, Histograms (mean ± SEM) showing the duration of the effects of 5-HT on sIPSCs in control neurons (-; n = 49) and CRF-treated neurons (n = 50). *p < 0.01; ANOVA. H, I, Histograms (mean ± SEM) showing the percentage increase in sIPSC amplitudes (H) and frequencies (I) by 5-HT in control neurons and CRF-treated neurons.
At the cellular level, CRF activates two types of receptors: CRF-R1 and CRF-R2 (Dautzenberg and Hauger, 2002). To confirm the involvement of CRF receptors in this process, we preincubated PFC slices with the nonspecific CRF receptor antagonist α-helical CRF9-41 (De Souza, 1987; Miyata et al., 1999). As shown in Figure 2A-D, the CRF-induced prolongation of the effect of 5-HT on sIPSC was blocked by α-helical CRF9-41 (3 μm) treatment. To test which type of CRF receptor is potentially involved, we also preincubated PFC slices with the CRF-R1-specific antagonist astressin (Gulyas et al., 1995). As shown in Figure 2, E and F, astressin (1 μm) blocked the CRF-induced prolongation of the effects of 5-HT on sIPSCs. In a group of cells we examined (Fig. 2G), the duration of the effect of 5-HT was increased to 7.8 ± 1.2 min (n = 18) by CRF but was reduced to 4.0 ± 0.5 min (n = 13) in the presence of α-helical CRF9-41 and to 4.1 ± 0.7 min (n = 12) in the presence of astressin, both of which were similar to the effect of 5-HT alone (3.9 ± 0.6 min; n = 17). The enhancement of sIPSC amplitudes (Fig. 2H) or frequencies (Fig. 2I) by 5-HT was not affected by the CRF receptor antagonists. These results suggest that CRF, by activating CRF-R1 receptors, facilitates the functions of serotonin receptors.
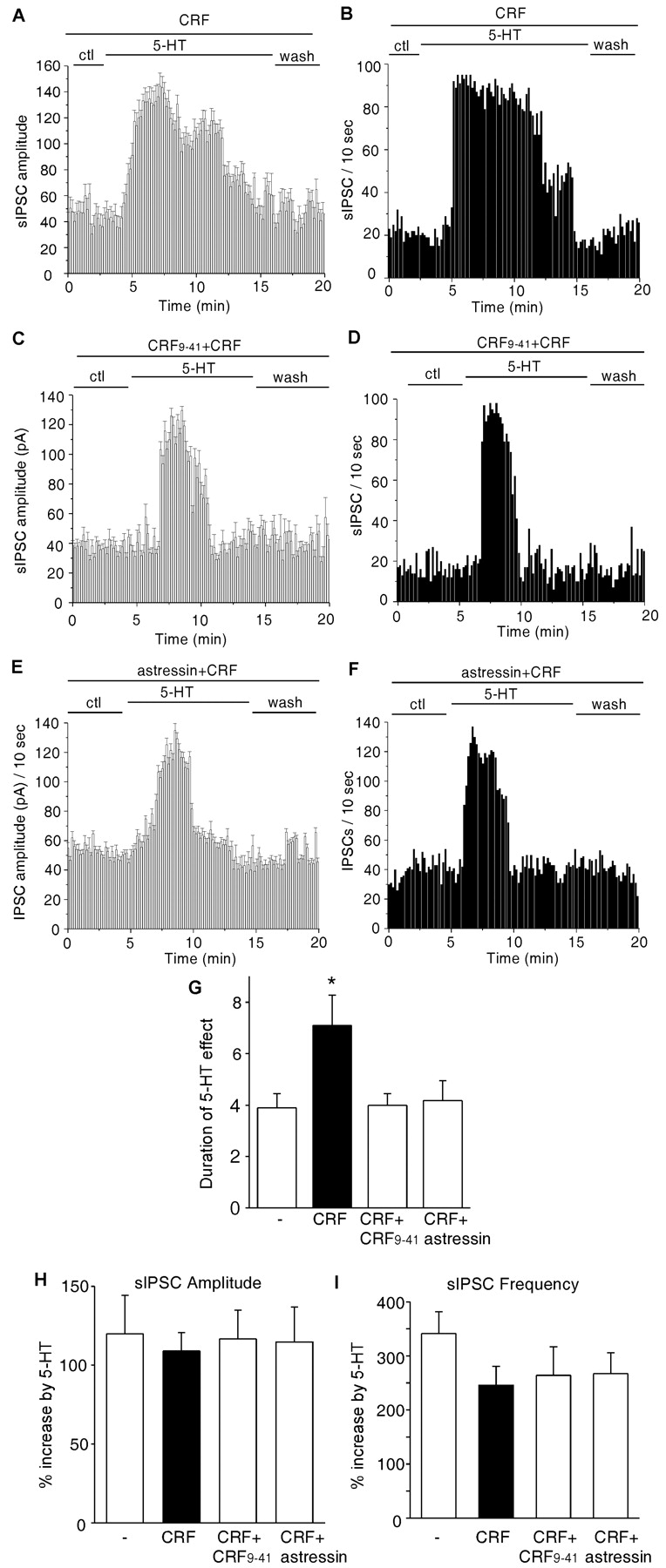
The CRF-induced prolongation of the effect of 5-HT on sIPSC was blocked by CRF receptor antagonists. A-F, Plots of sIPSC amplitude (A, C, E) and frequency (B, D, F) against time and agonist (5-HT, 40 μm) application in a neuron exposed to CRF (200 nm) alone (A, B), the nonspecific CRF receptor antagonist α-helical CRF9-41 (3 μm) plus CRF (C, D), or the CRF-R1-specific antagonist astressin (1 μm) plus CRF (E, F). ctl, Control conditions. G, Histograms (mean ± SEM) showing the duration of the effect of 5-HT on sIPSCs in control neurons (-; n = 17), CRF-treated neurons (n = 18), neurons treated with α-helical CRF9-41 plus CRF (n = 13), and neurons treated with astressin plus CRF (n = 12). *p < 0.01; ANOVA. H, I, Histograms (mean ± SEM) showing the percentage increase in sIPSC amplitudes (H) and frequencies (I) by 5-HT under different treatments.
To examine whether the effect of CRF is specific for 5-HT, we also tested the effect of CRF on muscarinic regulation of sIPSC. Application of the muscarinic ACh receptor agonist carbachol (20 μm for 20 min) enhanced sIPSC amplitude (127.2 ± 31.2%; n = 7) and frequency (354.7 ± 51.2%; n = 7), but no obvious desensitization was found with the muscarinic effect (duration, 16.8 ± 4.5 min; n = 7). CRF treatment did not alter the muscarinic regulation of GABA transmission (amplitude increase, 154.6 ± 27.5%; frequency increase, 390.7 ± 47.5%; duration, 15.6 ± 2.8 min; n = 10).
CRF prolongs the effect of 5-HT on sIPSC through a phospholipase C/PKC-dependent mechanism
We subsequently tried to identify the intracellular pathway through which CRF prolongs the effect of 5-HT on GABA transmission in PFC neurons. Because both CRF receptors are Gs-coupled, the most notable signaling of CRF is the cAMP/PKA pathway (Dautzenberg and Hauger, 2002). However, when PKA was inhibited by bath application of myristoylated PKI[14-22] (1 μm), the CRF-induced prolongation of the effect of 5-HT on sIPSC was still intact (Fig. 3A,B), suggesting the lack of involvement of PKA. In contrast, the phospholipase C (PLC)/PKC pathway has been shown to be involved in CRF potentiation of NMDARs in dopamine neurons (Ungless et al., 2003) and CRF priming of long-term potentiation (LTP) in hippocampal neurons (Blank et al., 2002). Therefore, we tested whether inhibition of this pathway could interfere with the CRF-induced prolongation of the effect of 5-HT on sIPSC.

The CRF-induced prolongation of the effect of 5-HT on sIPSC occurred through a mechanism that was dependent on the PLC/PKC pathway but not on PKA. A-F, Plot of sIPSC amplitude (A, C, E) and frequency (B, D, F) against time and agonist (5-HT, 40 μm) application in neurons exposed to CRF (200 nm) in the presence of the PKA inhibitor PKI[14-22] (1 μm)(A, B), the PLC inhibitor U73122 (1 μm)(C,D), or the PKC inhibitor calphostin C (Calp;1 μm)(E,F).ctl, Control conditions.G, Histograms (mean±SEM) showing the duration of the effects of 5-HT on sIPSCs in control neurons (-; n = 15), CRF-treated neurons (n = 16), and neurons treated with PKI plus CRF (n = 14), U73122 (U731) plus CRF (n = 14), or calphostin C plus CRF (n = 7). *p < 0.01; ANOVA. H, I, Histograms (mean ± SEM) showing the percentage increase in sIPSC amplitudes (H) and frequencies (I) by 5-HT under different treatments.
First, we bath-applied the PLC inhibitor U73122 (1 μm) before and during CRF and 5-HT application. U73122 alone had no significant effect on the regulation of sIPSC by 5-HT (duration, 4.6 ± 0.9 min; amplitude increase, 83.5 ± 13.1%; frequency increase, 218.6 ± 44.9%; n = 10). However, in the presence of U73122, CRF failed to prolong the effect of 5-HT on sIPSC amplitudes and frequencies (Fig. 3C,D). Next, we tested the role of PKC by bath application of the cell-permeable and specific PKC inhibitor calphostin C (1 μm). Treatment with calphostin C did not significantly affect the regulation of sIPSC by serotonin (duration, 3.5 ± 0.6 min; amplitude increase, 78.6 ± 8.5%; frequency increase, 361.2 ± 45.6%; n = 10). However, the CRF-induced prolongation of the effect of 5-HT on sIPSC was diminished by calphostin C treatment (representative examples shown in Fig. 3E,F). Another PKC inhibitor, Gö6850 (5 μm), gave similar results (i.e., blocked the prolongation by CRF of the 5-HT effect) (duration, 3.7 ± 0.8 min; n = 13), whereas Gö6850 alone did not significantly alter the effect of 5-HT on sIPSC (duration, 4.4 ± 0.8 min; amplitude increase, 105.8 ± 24.3%; frequency increase, 242.5 ± 43.2%; n = 17). As summarized in Figure 3G, in a group of cells that we examined, the duration of the effect of 5-HT was increased to 8.8 ± 1.2 min (n = 14) by CRF in the presence of PKI[14-22] but was reduced to 3.1 ± 0.5 min (n = 14) in the presence of U73122 and to 3.4 ± 1.2 min (n = 7) in the presence of calphostin C, both of which were similar to the effect of 5-HT alone (3.9 ± 0.6 min; n = 15). The 5-HT enhancement of sIPSC amplitudes (Fig. 3H) or frequencies (Fig. 3I) was not affected by CRF coapplied with these inhibitors. These results suggest that CRF, by activating the PLC/PKC pathway, prolongs the serotonergic regulation of GABA transmission in PFC neurons.
We then examined whether CRF treatment can indeed increase PKC activity in rat PFC slices. Because the catalytic competence of many PKC isozymes depends on autophosphorylation at the C terminus on a conserved residue (Behn-Krappa and Newton, 1999), a phospho-specific pan PKC antibody that detects PKC isoforms only when phosphorylated at this residue was used to detect activated PKC (Gu et al., 2003). As shown in Figure 4A, application of CRF (200 nm, 1 hr) to cortical slices induced a marked increase in the activated PKC. The levels of total PKC were not changed by the treatment. Quantification data (Fig. 4B) exhibited a 3.5 ± 0.8-fold increase in PKC activity by CRF treatment (n = 4). These results suggest that CRF can elevate the kinase activity of PKC in PFC neurons.

CRF treatment increased PKC activity in PFC slices. A, Immunoblots of phospho-PKC (p-PKC) and PKC. PFC slices were incubated in the absence or presence of CRF (200 nm) for 1 hr. After treatment, slice lysates were blotted with an anti-phospho-PKC antibody. After stripping out signals, membranes were reblotted with an antibody recognizing the total PKC. B, Quantification of p-PKC in PFC slices treated without (-) or with CRF. *p < 0.001; ANOVA.
Acute stress prolongs serotonergic regulation of sIPSC by activating CRF/PKC signaling
Because CRF plays an important role as a mediator of the stress response in the brain (Turnbull and Rivier, 1997; Eckart et al., 1999), we subsequently examined whether acute stress influences the effect of serotonin on GABAergic inhibitory transmission. The stress procedures we used entailed forcing the rat to swim in deep water for 30 min (Price et al., 2002; Roche et al., 2003) or placing the rat on the elevated platform for 30 min (Xu et al., 1997). As shown in Figure 5A-D, the 5-HT-induced increase in sIPSC amplitudes and frequencies was substantially prolonged in the PFC neuron from a swim-stress rat compared with the PFC neuron from a nonswim control rat. As summarized in Figure 5E, the duration of the effect of 5-HT on sIPSC was 9.3 ± 1.2 min (n = 21) in the group of cells from animals exposed to the forced-swimming stress (12 animals) and 10.1 ± 1.5 min (n = 15) in the group of cells from animals exposed to the elevated-platform stress (eight animals), both of which were significantly longer than that in control cells (3.9 ± 0.8 min; n = 26) from non-stressed animals (20 animals). The enhancement by 5-HT of sIPSC amplitudes (Fig. 5F) or frequencies (Fig. 5G) was not affected by these stressors.

Acute stress prolonged the effect of 5-HT on sIPSC. A-D, Plot of sIPSC amplitude (A, C) and frequency (B, D) against time and agonist (5-HT, 40 μm) application in neurons from a nonswim control rat (A, B) or a swim-stress rat (C, D). ctl, Control conditions. E, Histograms (mean ± SEM) showing the duration of the effects of 5-HT on sIPSCs in neurons from non-stressed control rats (n = 26), rats exposed to swim stress (n = 21), or rats exposed to elevated-platform stress (n = 15). *p < 0.01; ANOVA. F, G, Histograms (mean ± SEM) showing the percentage increase in sIPSC amplitudes (F) and frequencies (G) by 5-HT under different conditions.
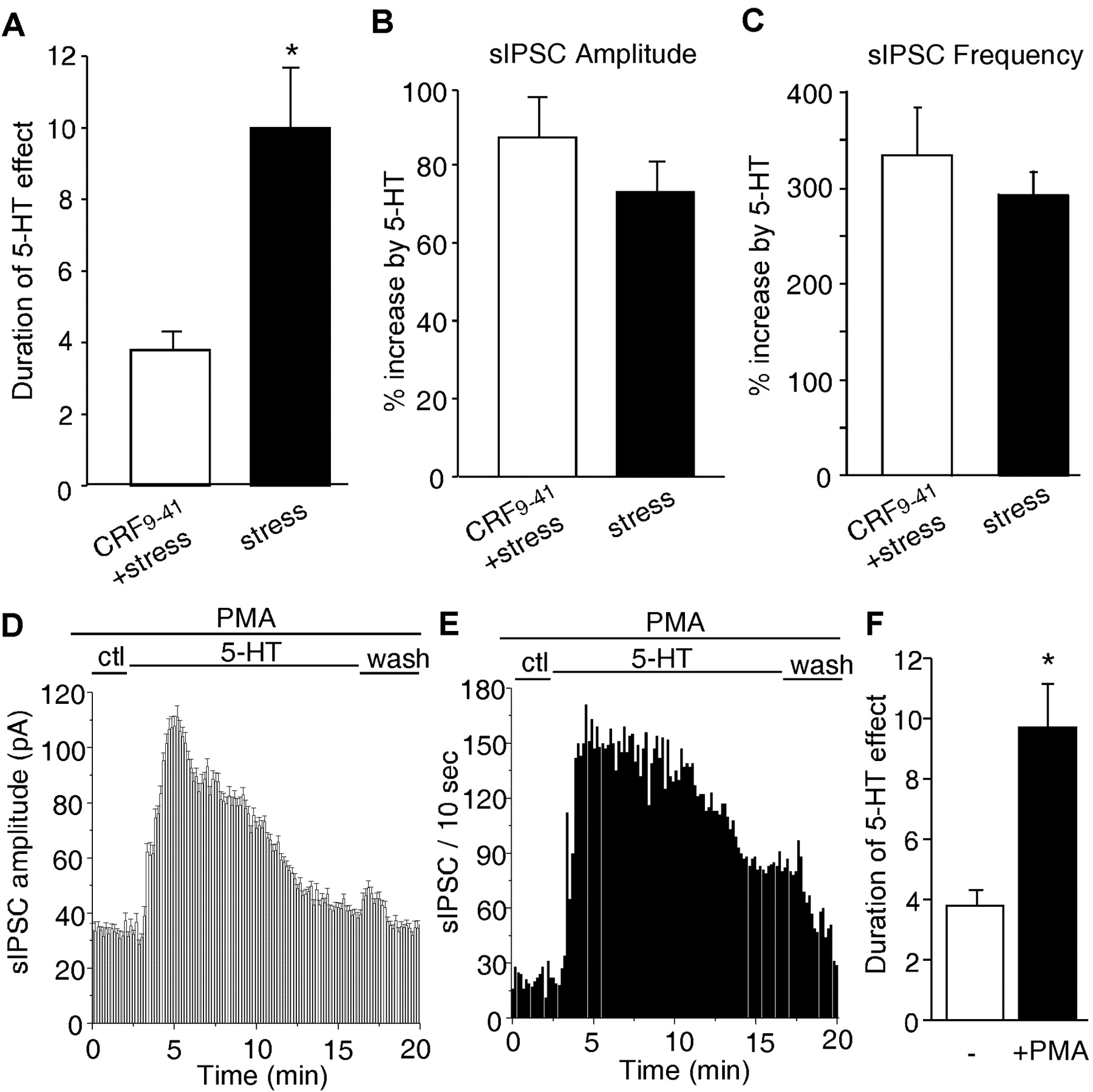
Because acute behavioral stress produces an effect on the serotonergic regulation of GABA transmission that is similar to that seen with CRF treatment of PFC slices, we therefore tested whether the stress-induced change in 5-HT functions occurs through activation of CRF signaling. The CRF receptor antagonist α-helical CRF9-41 was applied to pretreated PFC slices from stressed animals, followed by examination of the effect of 5-HT on sIPSC. As shown in Figure 6A, α-helical CRF9-41 treatment (seven animals) significantly (p < 0.01; ANOVA) blocked the stress-induced prolongation of the effect of 5-HT (3.8 ± 0.5 min; n = 20). The enhancement by 5-HT of sIPSC amplitudes (Fig.6B)

Stress-induced prolongation of the effect of 5-HT on sIPSC was blocked by a CRF receptor antagonist and mimicked by a PKC activator. A, Histograms (mean ± SEM) showing the duration of the effect of 5-HT on sIPSC in neurons from stressed animals treated with (n = 20) or without (n=24) α-helical CRF9-41. *p<0.01;ANOVA. B,C, Histograms (mean ± SEM) showing the percentage increase in sIPSC amplitudes (B) and frequencies (C) by 5-HT under different treatments. D, E, Plot of sIPSC amplitude (D) and frequency (E) against time and agonist (5-HT, 40 μm) application in aneuron exposed to the PKC activator PMA (0.5 μm). Slices were pretreated with PMA for 1hr, and PMA was present throughout the recordings. ctl, Control conditions. F, Histograms (mean ± SEM) showing the duration of the effects of 5-HT on sIPSCs in control neurons (-; n = 8) and PMA-treated neurons (n = 14). *p < 0.01; ANOVA.
or frequencies (Fig. 6C) was essentially unaltered by α-helical CRF9-41 in PFC slices from stressed animals.
Because CRF prolongs the serotonergic regulation of GABA transmission by activating the PLC/PKC pathway (Fig. 3), we further examined the effect of the PKC activator PMA (0.5 μm) on the regulation of sIPSC by 5-HT. As shown in Figure 6, D and E, PMA treatment prolonged the effect of 5-HT on sIPSC amplitudes and frequencies (duration, 9.4 ± 1.5 min; n = 14) (Fig. 6F), similar to the impact of CRF and acute stress. Together, these results suggest that stress, by activating the CRF/PKC pathway, prolongs the serotonergic regulation of GABA transmission in PFC neurons.
Acute stress-induced alteration of the 5-HT regulation of sIPSC is reversed by treatment with anti-anxiety drugs
We then examined whether the stress-induced prolongation of the regulation by 5-HT of GABA transmission in PFC neurons can be reversed by anxiolytic drugs. Selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine are used to treat anxiety and panic disorders, in addition to treating depression (Zohar and Westenberg, 2000). In the animal model of learned helplessness, which is a biobehavioral model for stress-induced anxiety causing depression, SSRIs prevent helplessness (Petty et al., 1996). Therefore, we injected rats with fluoxetine before exposing them to the forced-swim stress procedure. As found commonly, changes in immobility in the forced-swim test (a predictive measure of antidepressant/anxiolytic efficacy) can be observed when fluoxetine is administered relatively acutely (within 24 hr before the swim assessment).
Acute fluoxetine treatment did not significantly alter basal GABA transmission, as reflected by sIPSC mean amplitude (saline, 35.0 ± 2.3 pA, n = 20; fluoxetine, 39.2 ± 4.1 pA, n = 5) and frequency (saline, 267 ± 31.2 events/min, n = 20; fluoxetine, 283.8 ± 51.6 events/min, n = 5). Moreover, acute treatment with fluoxetine did not alter 5-HT regulation of GABA transmission, as shown by the 5-HT-induced increase in sIPSC amplitude (saline, 108.7 ± 13.4%, n = 20; fluoxetine, 101.2 ± 15.9%, n = 5) and frequency (saline, 283.5 ± 38.6%, n = 20; fluoxetine, 265.5 ± 40.6%, n = 5) and the duration of the 5-HT effect (saline, 4.2 ± 0.5 min, n = 20; fluoxetine, 4.1 ± 0.6 min, n = 5). However, in the neuron from a fluoxetine-injected rat, acute stress failed to prolong the serotonergic regulation of sIPSC (Fig. 7A,B).
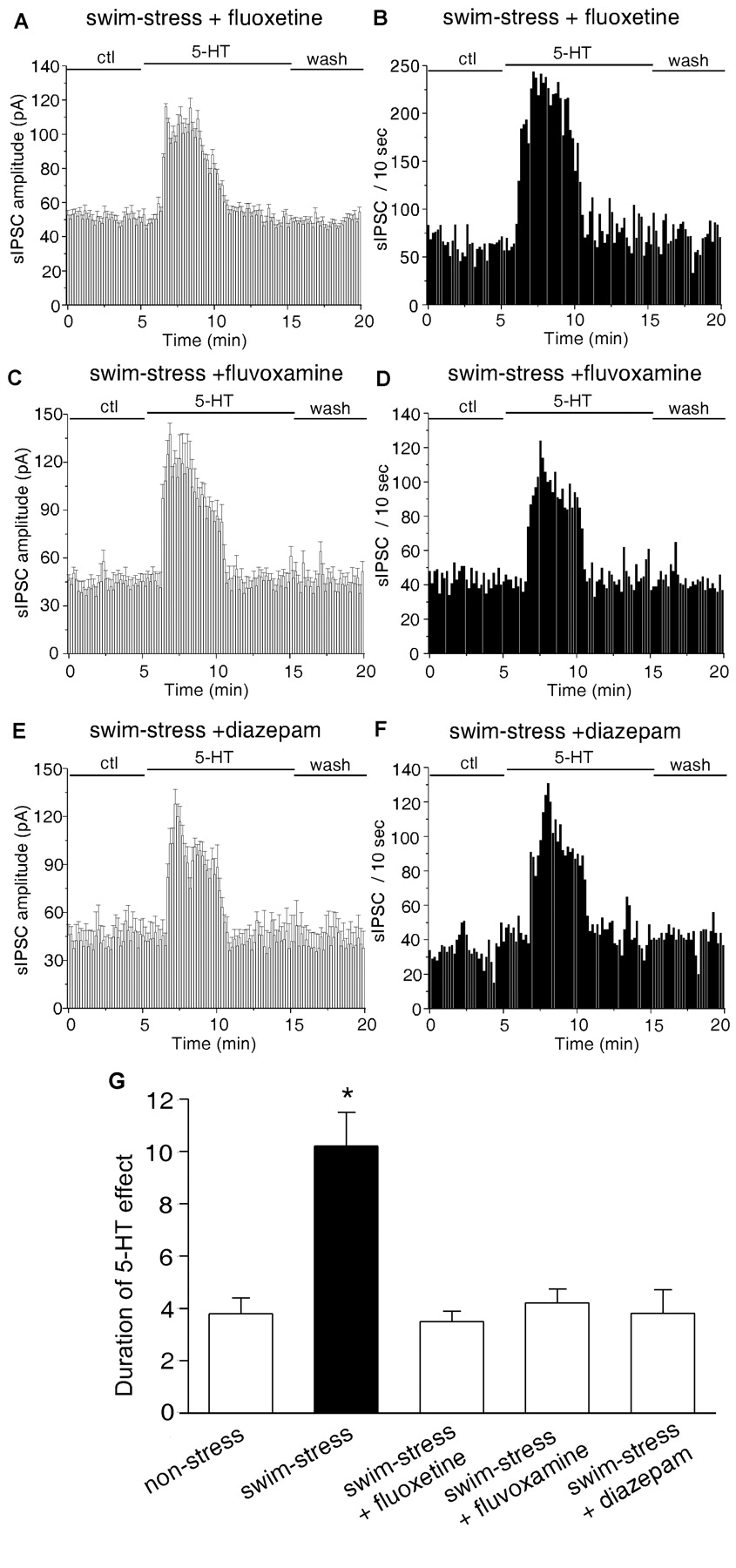
Short-termtreatment with anti-anxiety drugs reversed the stress-induced prolongation of the 5-HT effect on sIPSC. A-F, Plots of sIPSC amplitude (A, C, E) and frequency (B, D, F) against time and agonist (5-HT, 40 μm) application in a neuron from a swim-stress rat that has been injected intraperitoneally with fluoxetine (20 mg/kg; A,B), fluvoxamine (5 mg/kg; C, D), or diazepam (3 mg/kg; E, F). ctl, Control conditions. G, Histograms (mean ± SEM) showing the duration of effects of 5-HT on sIPSCs in neurons from nonstressed control rats (n = 10), saline-injected rats exposed to swim stress (n = 13), fluoxetine-injected rats exposed to swim stress (n = 16), fluvoxamine-injected rats exposed to swim stress (n = 5), or diazepam-injected rats exposed to swim stress (n = 7). *p < 0.01; ANOVA.
Fluvoxamine, another potent SSRI, has demonstrated short-term efficacy in the treatment of a number of anxiety disorders (Figgitt and McClellan, 2000). Therefore, we also tested the capability of fluvoxamine to reverse the acute stress-induced alteration of serotonin functions. Animals were injected intraperitoneally with fluvoxamine (5 mg/kg) 12 hr before exposure to the forced-swimming stress. As shown in Figure 7, C and D, in the neuron from a fluvoxamine-injected rat, acute stress failed to prolong the serotonergic regulation of sIPSC.
To test whether other nonserotonergic anti-anxiety compounds are also able to restore serotonergic functions in stressed animals, we examined the effect of benzodiazepines, which have long been used to treat anxiety (Gorman, 2003). Animals were injected intraperitoneally with diazepam (3 mg/kg) 12 hr before exposure to the forced-swimming stress. As shown in Figure 7, E and F, in the neuron from a diazepam-injected rat, acute stress lost the capability to prolong the serotonergic regulation of sIPSC.
Data from stressed animals treated with or without different anti-anxiety drugs are summarized in Figure 7G. In the group of cells from saline-injected rats (seven animals), the duration of the effect of 5-HT was increased to 10.2 ± 1.3 min (n = 13) by acute stress, whereas in the group of cells from fluoxetine-injected rats (seven animals), the duration of the effect of 5-HT was 3.5 ± 0.4 min (n = 16) in response to acute stress, which was similar to the effect of 5-HT in neurons from nonstressed animals (3.8 ± 0.6 min; n = 10). The enhancement of sIPSC amplitudes and frequencies by 5-HT in these stressed animals was not significantly altered by fluoxetine (amplitude increase, 113.7 ± 20.0%; frequency increase, 223.4 ± 39.1%; n = 16). In the group of cells from fluvoxamine-injected rats (three animals) or diazepam-injected rats (four animals), the duration of the effect of 5-HT was 4.2 ± 0.5 min (n = 5) or 3.8 ± 0.9 min (n = 7), respectively, in response to acute stress, similar to the effect of 5-HT in neurons from nonstressed animals (3.8 ± 0.6 min; n = 10). The enhancement of sIPSC amplitudes and frequencies by 5-HT in these stressed animals was not significantly altered by fluvoxamine (amplitude increase, 116.4 ± 31.1%; frequency increase, 246.7 ± 44.1%; n = 5) or diazepam (amplitude increase, 112.1 ± 35.1%; frequency increase, 265.2 ± 77.5%; n = 7). These results suggest that short-term treatment with anti-anxiety drugs can prevent acute stress from prolonging the serotonergic regulation of GABA transmission.
Discussion
Stress has been recognized to strongly influence cognitive and emotional processes subserved by PFC, including working memory, attention, and inhibition of inappropriate responses (Arnsten, 1998). Elucidation of the functional role of key neuromodulators, such as serotonin and dopamine, is central to understanding why prefrontal cortical deficits are so prominent in many mental illnesses that are exacerbated by stress (Mazure, 1995). The involvement of dopamine D1 receptors in stress-induced impairment of PFC function has been documented (Murphy et al., 1996; Zahrt et al., 1997), whereas the involvement of serotonin receptors is essentially unknown.
In addition to affecting structural plasticity, including dendritic atrophy and neurogenesis (McEwen, 1999), stress also dramatically affects synaptic plasticity at excitatory synapses (Saal et al., 2003), a putative learning and memory mechanism. Acute stress blocks LTP induced by high-frequency stimulation (Shors et al., 1989; Xu et al., 1997; Maroun and Richter-Levin, 2003) and facilitates long-term depression induced by low-frequency stimulation (Kim et al., 1996; Xu et al., 1997). The effects of stress on synaptic plasticity can be overcome by lowering endogenous 5-HT levels (Shakesby et al., 2002), suggesting the involvement of serotonergic mechanisms in mediating stress responses. In agreement with this, many stressors produce a large accumulation of extracellular 5-HT within the DRN and forebrain regions, including PFC (Yoshioka et al., 1995; Adell et al., 1997; Maswood et al., 1998). This is probably attributable to the CRF-induced increase in the firing rates of 5-HT neurons in the caudal DRN and activation of mesolimbocortical serotonergic pathways during uncontrollable stress (Lowry et al., 2000). To understand the interactions between the stress-related neuropeptide CRF and the serotonin system, most studies have focused on the influence of CRF and stress on serotonin release and serotonergic neuron excitability (Adell et al., 1997; Maswood et al., 1998; Lowry et al., 2000; Price and Lucki, 2001). Little is known about the impact of CRF and stress on the function of serotonin in its target areas.
The findings of the present study are the first to show that acute stress alters the serotonergic regulation of GABA transmission (assayed by exogenous application of 5-HT) in PFC pyramidal neurons. This stress-induced alteration of the effect of 5-HT occurs through a mechanism that is dependent on CRF, which is demonstrated by two lines of evidence. First, CRF mimics the effects of acute stress on 5-HT regulation of GABA transmission. Second, the stress-induced alteration of the 5-HT regulation of GABA transmission is prevented by a CRF receptor antagonist. These results suggest that stress and CRF can affect not only 5-HT release but also the actions of 5-HT on its target neurons.
Serotonin, by activating different 5-HT receptors, has a powerful and complex impact on GABAergic synaptic transmission in PFC neurons (Zhou and Hablitz, 1999; Feng et al., 2001; Cai et al., 2002; Yan, 2002). A prominent effect of 5-HT is the large increase in the amplitude and frequency of spontaneous IPSC (Zhou and Hablitz, 1999). One interesting feature of the 5-HT regulation of sIPSC is that the effect of 5-HT declines within 3-4 min in the continued presence of the agonist. However, in PFC slices pretreated with CRF or from stressed animals, the regulation of sIPSC by 5-HT lasts much longer (8-10 min). Therefore, after stressor exposure, more 5-HT will be released and 5-HT signaling will be prolonged, which could synergistically lead to potentiated and sustained regulation of GABAergic inhibition by 5-HT. Given the key role of PFC/GABAergic inhibition in shaping the temporal flow of information and thus the regulation of working memory (Constantinidis et al., 2002), the altered regulation of GABA transmission by serotonin in response to stressful stimuli could lead to disturbed PFC functions. In support of this possibility, the stress-induced alteration of the effect of 5-HT on GABA transmission is reversed by fluoxetine, fluvoxamine, and diazepam, all of which have behavioral anxiolytic effects against stress-induced anxiety (Petty et al., 1996; Figgitt and McClellan, 2000; Zohar and Westenberg, 2000; Gorman, 2003). In cells from acute SSRI-treated animals, the exogenously applied 5-HT induced a transient enhancing effect on spontaneous GABA transmission, similar to what was found in control cells. This suggests that the functions of 5-HT receptors are not significantly altered by acute SSRI treatment. Although SSRIs can block reuptake of 5-HT, they are not able to change the action of an exogenously applied high concentration of 5-HT (40 μm).
The cellular mechanism that may underlie the prolongation by CRF of the 5-HT effect on GABA transmission in PFC neurons was also investigated. It has been shown that CRF receptors couple to multiple G-proteins to activate diverse intracellular signaling pathways in mouse hippocampus (Blank et al., 2003). We found that the PLC/PKC pathway is involved in the CRF-induced alteration of the effect of 5-HT on sIPSC in rat PFC neurons, similar to the signaling involved in CRF potentiation of NMDARs in dopamine neurons (Ungless et al., 2003) and CRF facilitation of LTP in hippocampal neurons (Blank et al., 2002). How CRF/PKC prolongs the regulation of sIPSC by 5-HT is unclear. Two mechanisms could account for the effect of 5-HT on GABA transmission. First, serotonin elevates the excitability of GABAergic interneurons, therefore leading to the increased GABA release. Second, 5-HT enhances the probability of action potential-dependent GABA release from axon terminals, thereby leading to an increase in the contribution of large-size (multiquantal) sIPSCs to the overall population of synaptic events. Because the desensitizing effect of 5-HT on sIPSCs is not affected by PKC inhibitors, it is therefore unlikely to be mediated by a PKC-mediated phosphorylation, desensitization, or internalization of 5-HT receptors (Raymond, 1991; Bhattacharyya et al., 2002). For the prolongation by CRF of the 5-HT effect on sIPSC, we speculate that released CRF in response to stressful stimuli potently activates PKC, and PKC phosphorylation of an unidentified regulatory protein causes a prolonged serotonin excitation of GABAergic interneurons or serotonin increase of GABA release at presynaptic terminals.
Together, the central finding of this study is that CRF and acute stress can alter the functions of 5-HT in PFC pyramidal neurons (i.e., prolong the enhancement by 5-HT of spontaneous GABA transmission through a mechanism involving PKC). This finding provides a possible mechanism for the stress-induced exacerbation of psychiatric disorders that are associated with aberrant actions of serotonin.
Footnotes
This work was supported by National Institutes of Health Grants MH63128 and AG-21923 and National Science Foundation Grant IBN-0117026 (Z.Y.). We thank Xiaoqing Chen for technical support.
Correspondence should be addressed to Dr. Zhen Yan, Department of Physiology and Biophysics, State University of New York at Buffalo, 124 Sherman Hall, Buffalo, NY 14214. E-mail: zhenyan{at}buffalo.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245000-09$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}