

Abstract
Creatine mediates remarkable neuroprotection in experimental models of amyotrophic lateral sclerosis, Huntington's disease, Parkinson's disease, and traumatic brain injury. Because caspase-mediated pathways are shared functional mechanistic components in these diseases, as well as in ischemia, we evaluated the effect of creatine supplementation on an experimental stroke model. Oral creatine administration resulted in a remarkable reduction in ischemic brain infarction and neuroprotection after cerebral ischemia in mice. Postischemic caspase-3 activation and cytochrome c release were significantly reduced in creatine-treated mice. Creatine administration buffered ischemia-mediated cerebral ATP depletion. These data provide the first direct correlation between the preservation of bioenergetic cellular status and the inhibition of activation of caspase cell-death pathways in vivo. An alternative explanation to our findings is that creatine is neuroprotective through other mechanisms that are independent of mitochondrial cell-death pathways, and therefore postischemic ATP preservation is the result of tissue spearing. Given its safety record, creatine might be considered as a novel therapeutic agent for inhibition of ischemic brain injury in humans. Prophylactic creatine supplementation, similar to what is recommended for an agent such as aspirin, may be considered for patients in high stroke-risk categories.
Introduction
Neurological diseases featuring cell death are among the most puzzling and devastating illnesses in medicine. Broadly, these neurodegenerative diseases can be divided categorized as acute (i.e., stroke, traumatic brain injury, and spinal cord injury) and chronic [i.e., amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), Parkinson's disease (PD), and Alzheimer's disease]. Although these diseases have different causes and affect different neuronal populations, they share many pathological mechanisms (Friedlander et al., 1997; Rowland and Shneider, 2001; Hausmann et al., 2002; Zoghbi and Botas, 2002; Friedlander, 2003). Given the paucity of treatments for these diseases, developing novel and effective therapeutics is of utmost importance. Recently, oral supplementation of creatine has been demonstrated to be neuroprotective in a variety of experimental neurological disease models, including ALS, HD, PD, and traumatic brain injury (Klivenyi et al., 1999; Matthews et al., 1999; Ferrante et al., 2000; Sullivan et al., 2000). In addition, studies with cultured rat hippocampal neurons have also shown that creatine protects against glutamate and β-amyloid toxicity, as well as against energetic insults in striatal neurons (Brewer and Wallimann, 2000; Brustovetsky et al., 2001).
Despite its broad neuroprotective properties in vivo and in vitro, the mechanisms of action of creatine are not well understood. Current hypotheses of the mechanisms of creatine-mediated neuroprotection include enhanced energy storage, as well as stabilization of the mitochondrial permeability transition pore by octomeric conformation of creatine kinase (O'Gorman et al., 1997; Wyss and Kaddurah-Daouk, 2000). It is therefore believed that creatine improves the overall bioenergetic status of the cell, making it more resistant to injury. Because caspase-mediated pathways are shared functional mechanistic components in neurodegenerative diseases, as well as in ischemia, we evaluated the effect of creatine supplementation on an experimental stroke model. We demonstrate that oral creatine supplementation inhibits mitochondrial cytochrome c release and downstream caspase-3 activation and results in remarkable ischemic neuroprotection. Associated with inhibition of cytochrome c release and caspase-3 activation and with neuroprotection, creatine administration inhibited ischemia-mediated ATP depletion. This in the first report demonstrating an association between maintenance of cellular ATP levels and inhibition of caspase cell-death pathways. Because creatine is a relatively safe compound, prophylactic administration of creatine to individuals at high risk of stroke might result in significant amelioration of cerebral ischemia-mediated disabilities.
Materials and Methods
Induction of focal cerebral ischemia. The ischemia protocol was performed as described previously (Zhang et al., 2003b). Briefly, C57BL/6 female mice were fed a 2% creatine-supplemented diet (PMI LabDiet; PMI Nutrition International, Richmond, IN), whereas the control mice were fed a normal diet, for 4 weeks. The weight of the mice was not significantly different in the two groups. The mice (18-22 gm) in both groups then underwent middle cerebral artery (MCA) occlusion for 2 hr, followed by 24 hr of reperfusion. Blood flow was documented with an infrared Doppler (PriFlux System 5000; Perimed, Piscataway, NJ). Regional cerebral blood pressure was monitored with a noninvasive tail cuff (Harvard Instruments, South Natick, MA). Mice (five per group) were tested for neurological deficits 30 min and 24 hr after reperfusion and scored as follows: 0, no observable neurological deficits (normal); 1, failure to extend right forepaw (mild); 2, circling to the contralateral side (moderate); or 3, loss of walking or righting reflex (severe) (Hara et al., 1997). The brains were removed after 24 hr of reperfusion, and the ischemic territory was dissected for caspase-3 activation and cytochrome c release assay. Experiments were conducted in accordance with protocols approved by the Harvard Medical School Animal Care Committee.
Determination of infarct volume. After 24 hr of reperfusion, the mice (five per group) were anesthetized with pentobarbital (60 mg/kg) and killed. The brains were removed quickly and chilled in ice-cold saline for 10 min. Coronal sections (1 mm thick; n = 6) were cut with a tissue slicer, beginning approximately +2.5 mm from the bregma, and the slices were immersed in a saline solution, containing 2.0% (v/v) 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO), at 37°C for 20 min. After staining, each slice was scanned by using a Bio-Rad (Hercules, CA) Imaging Densitometer GS-700. The unstained areas in each image were quantified with the Multi-Analyst 1.02 program (Bio-Rad), and the infarct volume was calculated by summing up the infarct area in the six slices.
Brain tissue cytosolic fractionation and Western blotting. After dissection of ischemic territories, fresh brains were carefully Dounce homogenized in 1 ml of homogenization buffer, containing (in mm): 10 HEPES, pH 7.4, 250 sucrose, 10 KCl, 1.5 MgCl2, 1 EDTA, and 1 DTT. The homogenate was saved, and one-third of each sample was used for caspase-3 Western blotting and the other two-thirds of the sample were centrifuged at 700 × g for 10 min and then at 15,000 × g for 30 min at 4°C, to obtain the cytosolic fraction. Protein concentration was determined by the Bradford assay (Bio-Rad). Total brain lysate and cytosolic fraction samples (50 μg) were loaded for SDS-PAGE and detected by antibodies against cytochrome c (monoclonal antibody at 1:1000 dilution; PharM-ingen, San Diego, CA) and caspase-3 (CM1 antibody at 1:1000 dilution; PharMingen). Tubulin (monoclonal antibody at 1:5000; Sigma) staining was used as a loading control.
HPLC analysis for creatine and ATP. Mouse brains (eight per group) were flash frozen in liquid nitrogen at 30 min after the onset of MCA occlusion. Frozen striatal tissue samples from eight creatine-supplemented mice and eight unsupplemented mice were dissected on a freezing-cold plate (-20°C), placed in 0.4 m perchloric acid (10 ml/mg, wet weight), homogenized, and centrifuged. The supernatant was neutralized with 25 ml of 2 m K2CO3, added to 200 ml of the supernatant, and recentrifuged. Supernatants were stored at -80°C until injected. Standards were prepared in 0.4 m perchloric acid at concentrations of 10 mm for creatine and ATP (based on tissue concentrations). A PerkinElmer Life Sciences (Norwalk, CT) gradient HPLC pump and a Waters Associates (Milford, MA) multiple UV detector were used for analysis of samples, according to methods that have been described previously (Dedeoglu et al., 2003).
Results
Protection from MCA occlusion-mediated injury in creatine-treated mice
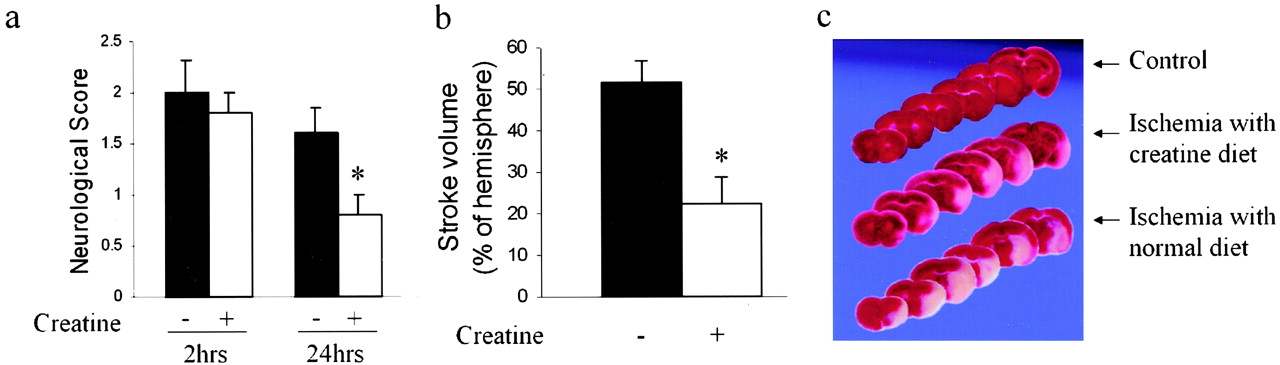
We evaluated whether creatine may mediate neuroprotection after cerebral ischemia. To test this hypothesis, we fed mice with food supplemented with 2% creatine for 1 month, and thereafter subjected them to 2 hr of MCA occlusion followed by 24 hr of reperfusion. Cerebral blood flow and systemic blood pressure were not different between the two groups. Parameters that were evaluated included neurological outcome and infarct size. All mice were hemiparetic after 2 hr of MCA occlusion. However, a significant improvement in the neurological score at 24 hr after reperfusion was seen in the creatine-fed group compared with control mice (n = 5; p < 0.01) (Fig. 1a). Brain lesion volume was determined in creatine-fed and control groups 24 hr after reperfusion. Correlating with improved behavior score, cerebral infarction of creatine-fed mice was reduced by 56% compared with that of mice fed unsupplemented food (51.4 ± 5.5 vs 22.0 ± 6.3 mm3; n = 5; p < 0.01) (Fig. 1b,c). In contrast to mice fed with a creatine-supplemented diet for 1 month, we did not find ischemic neuroprotection in mice fed with a creatine-supplemented diet for only 1 week (data not shown), suggesting that creatine-mediated neuroprotection in vivo requires a time-dependent metabolic alteration.

Protection from MCA occlusion-mediated injury in creatine-treated mice. a, After 2 hr of MCA occlusion, neurological grading was performed after 30 min and 24 hr of reperfusion [0, no observable neurological deficits (normal); 1, failure to extend right forepaw (mild); 2, circling to the contralateral side (moderate); 3, loss of walking or righting reflex (severe)]. b, Infarct volume of mice fed with creatine for 4 weeks, assessed 24 hr after ischemia (n = 5; *p < 0.01). c, Representative images of TTC-stained coronal brain slices. White represents infarcted tissue, and red represents live tissue.
Inhibition of cytochrome c release and caspase-3 activation in ischemic brain territories
Caspase activation has been mechanistically implicated to play a key functional role in mediating ischemia-mediated brain injury (Friedlander et al., 1997; Hara et al., 1997; Ferrer and Planas, 2003; Zhang et al., 2003b). Although the precise mechanism by which ischemia and reperfusion result in caspase activation is not clear, recent studies provide evidence suggesting that the mitochondria play an important role in transmitting apoptotic signals during ischemia to induce caspase activation (Plesnila et al., 2001; Blomgren et al., 2003; Zhang et al., 2003b). Western blot analysis of unfractionated brain lysates and cytosolic fractions demonstrated inhibition of cerebral ischemia-mediated caspase-3 activation and cytochrome c release, respectively, in creatine-fed mice compared with control mice (Fig. 2). These data provide in vivo confirmation that creatine-mediated neuroprotection results, at least in part, from directly or indirectly inhibiting cytochrome c release and downstream caspase-3 activation.

Caspase-3 activation and cytochrome c release in ischemic brain territories after 2 hr of ischemia and 24 hr of reperfusion. a, Activation of caspase-3 after ischemia and its inhibition by creatine pretreatment (whole-brain lysates; each lane represents a separate mouse). cyto. c, Cytochrome c. b, Cytochrome c release after ischemia in mice with or without creatine pretreatment (cytosolic extracts; each lane represents a separate mouse).
Creatine treatment reduced the drop in brain ATP concentrations after ischemia
The fact that neuroprotection is dependent on longer treatment of creatine led us to measure the bioenergetic status of brain tissue, including creatine and ATP concentrations after ischemia. There was no significant difference in creatine concentration in the contralateral brain hemisphere between creatine-treated and untreated mice. Compared with the contralateral hemisphere, the creatine level of the ischemic territory within the ipsilateral brain hemisphere in mice fed with unsupplemented chow was significantly reduced at 30 min after ischemia from 11.2 ± 0.4 to 9.3 ± 0.7 μmol/gm (n = 8; p < 0.01). In the creatine-fed mice, the creatine level in the ischemic territory was not significantly reduced (11.4 ± 0.4 vs 10.8 ± 0.5 μmol/gm; n = 8; p = 0.18). We found a significant reduction in creatine levels in control mice after ischemia compared with creatine-fed mice (∼13.9%; n = 8; p < 0.01) (Fig. 3a). The buffering of postischemic ATP depletion was significantly more notable. Compared with the contralateral hemisphere, the ATP level of the ischemic territory within the ipsilateral brain hemisphere in mice fed with unsupplemented chow was significantly reduced by 30 min after ischemia from 1.82 ± 0.22 to 0.73 ± 0.36 μmol/gm (n = 8; p < 0.01). In the creatine-fed mice, ATP levels in the ischemic territory were not significantly reduced (2.08 ± 0.19 vs 1.67 ± 0.39 μmol/gm; n = 8; p = 0.23). Therefore, compared with mice fed with unsupplemented food, the reduction in ATP after ischemia in the ipsilateral hemisphere was remarkably reduced by ∼56.3% in the ischemic territory of creatine-fed mice (n = 8; p < 0.001) (Fig. 3b). The above data describe a model correlating inhibition of ischemia-induced reduction of ATP levels with inhibition of cytochrome c release and caspase-3 activation as well as neuroprotection.

Brain creatine and ATP concentrations after ischemia with or without creatine treatment. Brain samples were analyzed after 30 min of MCA occlusion. a, Creatine levels in ischemic (ipsilateral) and nonischemic (contralateral) brain tissues. n = 8; *p < 0.01. b, ATP levels in ischemic (ipsilateral) and nonischemic (contralateral) brain tissues. n = 8; **p < 0.001.
Discussion
This is the first report providing evidence of creatine-mediated protection and inhibition of caspase cell-death cascades in cerebral ischemia. Our results demonstrate that creatine may exert its protective effect, at least in part, by inhibiting cytochrome c release and subsequent caspase-3 activation. Opening of the mitochondrial permeability transition pore or rupture of the outer mitochondrial membrane has been proposed to be a trigger of postmitochondrial cell-death cascades by the release of cytochrome c and other apoptogenic mediators (Kristal and Brown, 1999; Friedlander, 2003; Mattson and Kroemer, 2003). We have demonstrated previously that minocycline directly inhibits permeability transition-mediated cytochrome c release (Zhu et al., 2002). This finding was demonstrated in isolated mitochondria preparations, as well as in intact cells and in vivo. However, unlike minocycline, creatine does not seem to directly inhibit cytochrome c release in purified mitochondria (data not shown), suggesting that the mechanism of inhibition of cytochrome c release is different in these two drugs. Consistent with the differing mechanisms of action of creatine and minocycline, these two compounds demonstrate additive neuroprotection in a mouse model of ALS (Zhang et al., 2003a). Given that it is becoming increasingly apparent that a multidrug approach will be required for the treatment of this set of complex and devastating diseases, understanding intracellular targets and the mechanisms of action of the different compounds will assist in targeted combinatorial therapeutic design.
Although we do not demonstrate directly the definitive target of creatine-mediated ischemic neuroprotection, two models may provide an explanation for our findings. One mechanism is that creatine supplementation may result in improved ability of the neuron to withstand ischemia-mediated energy depletion. In this manner, the neuron preserves ATP levels, which thereafter leads to inhibition of cytochrome c release, caspase-3 activation, and cell death. An alternative mechanism is that creatine treatment results in an increased resistance to ischemia-mediated cell death by a mechanism independent of mitochondrial cell-death pathways, unrelated to primary buffering of ATP levels. Tissue resistant to ischemia-mediated cell death would thereafter be functional and would also, as a secondary effect, be able to maintain levels of ATP. Therefore, preserved ATP may result as a consequence of tissue preservation and not as a primary mediator of neuroprotection. The data presented in this report do not distinguish between these two possibilities, and furthermore, these possibilities are not mutually exclusive.
In ischemia and reperfusion, neurons in the brain tissue region that is most severely affected by hypoxic injury die rapidly by necrosis (ischemic core), whereas neurons exposed to lesser degrees of hypoxia die by apoptosis (ischemic penumbra). An association between a shift from necrotic cell death to apoptotic cell death and increasing levels of cellular ATP has been described recently (Tatsumi et al., 2003). As an extension of this finding, our model is consistent with a mechanism in which creatine supplementation results in greater ability to buffer ATP levels and reduce cell death. As seen in the coronal brain sections in Figure 1c, creatine-mediated reduction of tissue injury occurs in the ischemic penumbra, indicating that this therapeutic manipulation is reducing cell death of neurons that are closer to the survival threshold.
The magnitude of creatine-mediated neuroprotection compares positively with that of other effective experimental neuroprotective strategies, including caspase inhibition, as well as transgenic overexpression of Bcl-2 in neurons in mice (Martinou et al., 1994; Friedlander et al., 1997; Hara et al., 1997; Yaoita et al., 1998; Alkayed et al., 2001). However, to demonstrate neuroprotection, not only was pretreatment required, but a month of creatine supplementation was also necessary (1 week of creatine administration did not confer neuroprotection; data not shown). The reason for this requirement is not clear at present. The length of pretreatment needed is likely related to the level of energetic buffering needed to achieve neuroprotection. In addition to being neuroprotective in vivo, creatine has been reported to inhibit neuronal cell death in a variety of paradigms mediated by a variety of neurotoxins (Brewer and Wallimann, 2000). The increased ability to resist ischemic injury mediated by creatine supplementation correlates directly or indirectly with the ability to maintain cellular homeostasis and prevent hypoxia-mediated release of cytochrome c and downstream caspase-3 activation. Given that creatine is a relatively safe compound, people at high risk of cerebral ischemic injury might be good candidates to receive creatine supplementation. Prophylactic therapeutics for people in high stroke-risk categories are similar to the indications for the recommendation of aspirin treatment in this same group of patients. The evidence described above provides support for additional preclinical evaluation of creatine with the aim of evaluating it in human clinical stroke trials.
Footnotes
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (R.M.F., R.J.F., M.F.B.), the National Institute on Aging (B.S.K.), the Huntington's Disease Society of America (R.M.F., R.J.F, M.F.B.), the Hereditary Disease Foundation (B.S.K., R.J.F), and the Veterans Administration (R.J.F.). We thank E. Friedlander for editorial assistance and Dr. D. Ulmann for consultation with the HPLC analysis.
Correspondence should be addressed to Robert M. Friedlander, Department of Neurosurgery, Harvard Medical School, 75 Francis Street, Boston, MA 02115. E-mail: rfriedlander{at}rics.bwh.harvard.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245909-04$15.00/0
↵* S.Z. and M.L. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}