

Abstract
Tolerance and dependence result from long-term exposure to opioids, and there is growing evidence linking acute receptor desensitization to these more long-term processes. Receptor desensitization encompasses a series of events leading to the loss of receptor function and internalization. This study examines the onset and recovery from desensitization in locus ceruleus neurons recorded in brain slices taken from animals that have been chronically treated with morphine. After chronic morphine treatment, desensitization was altered as follows. First, the rate of desensitization was increased. Second, recovery from desensitization was always incomplete, even after a brief (1-2 min) exposure to agonist. This contrasts with experiments in controls in which recovery from desensitization, after a brief exposure to agonist, was complete within 25 min. Finally, morphine-6-β-d-glucuronide, a metabolite of morphine that was ineffective at causing desensitization in controls, induced significant desensitization in slices from morphine-treated animals. When brain slices from controls were treated with inhibitors of PKC or monensin, agents known to compromise G-protein-coupled receptor resensitization, desensitization was increased, and recovery was significantly reduced. These results indicate that receptor resensitization maintains signaling during periods of intense and sustained stimulation. After chronic morphine treatment, desensitization is potentiated, and receptor resensitization is compromised.
Introduction
Opioids are important analgesics used clinically for pain management. Long-term use of opioids can, however, result in tolerance and dependence. There are number of studies linking acute receptor desensitization to tolerance and dependence (Bohn et al., 2000; Finn and Whistler, 2001; Ueda et al., 2001; Freye and Latasch, 2003). The process of acute μ-opioid receptor (MOR) desensitization results from a series of events beginning with MOR phosphorylation by protein kinases and ending with receptor internalization (von Zastrow et al., 2003). It is still unclear, however, at which point in the process receptor function is lost.
Previous studies have shown that agonists that can cause significant MOR desensitization also cause MOR internalization (Law et al., 2000; Alvarez et al., 2002). As such, receptor resensitization is thought to be an important step in the regulation of G-protein-coupled receptors. The recycling of the desensitized receptor helps to maintain receptor numbers during sustained agonist stimulation (Law et al., 2000; Whistler et al., 2001; Chen et al., 2003; Tanowitz and von Zastrow, 2003). Similar to the prototypical β2-adrenergic receptor, MOR recycling requires receptor internalization and transport of desensitized receptor to the endosome after acute desensitization (Law et al., 2000; Tanowitz and Von Zastrow, 2003). The acidic environment of the endosome causes the desensitized receptors to change their conformation, enabling β-arrestin 2 to dissociate from the receptors (Zhang et al., 1998). After dephosphorylation by protein phosphatases, functional receptors are transported back to the plasma membrane (Whistler and von Zastrow, 1998; Wolf et al., 1999; Law et al., 2000; Whistler et al., 2002; Chen et al., 2003)
Although the components of MOR regulation have been examined individually in a variety of expression systems, this process has not been carefully examined in native neurons. In this study, the regulation of MOR function was examined in locus ceruleus (LC) neurons. The results show that loss of receptor function results from even a brief exposure to a high agonist concentration. This rapid desensitization occurs before any detectable change in maximal opioid response and recovers completely within 25 min. In slices taken from morphine-treated animals, desensitization is potentiated, and recovery from desensitization is always incomplete. When PKC inhibitors or monensin was used to disrupt receptor resensitization, desensitization was augmented, and recovery was compromised in slices from control animals. The results suggest that chronic morphine treatment impaired MOR signaling by reducing the reactivation of receptor after acute desensitization.
Materials and Methods
Adult (150-250 gm) male Sprague Dawley (Charles River Laboratories, Wilmington, MA) rats were used for all experiments. Brain sections were prepared as described previously (Fiorillo and Williams, 1996). Sections were incubated and perfused with extracellular solution containing (in mm): 126 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11 glucose. Solution was maintained at 37°C and buffered with 95% O2 and 5% CO2. Intracellular recordings were made from locus ceruleus neurons in horizontal brain sections (250-300 μm). Recording electrodes (25-50 MΩ) were filled with KCl (2 m). Holding current (0-200 pA) was applied to inhibit spontaneous activity and hold the membrane potential at approximately -60 mV. Data collection was done with PowerLab (chart version 4.1) sampled at 100 Hz. Data analysis was done with PRISM analysis software. Values are given as mean ± SEM. For all experiments, p < 0.05 was considered as a significant difference. Multiple group comparisons were made with two-way ANOVA analysis or unpaired t test.
Morphine treatment. Rats were anesthetized with halothane or isoflurane and given one placebo per morphine pellet (75 mg/pellet) on day 1, two pellets on day 3, and two pellets on day 5. Experiments were done on day 6 or 7. Control animals in this study consisted of naive and placebo-treated animals.
Receptor desensitization was assessed in two ways. First, the decline of the hyperpolarization induced by superfusion of a supramaximal concentration of an agonist was measured. Second, the amplitude of the hyperpolarization induced by an EC50 concentration of agonist was measured before (prepulse) and after (test pulse) application of a maximal (desensitizing) concentration of agonist. The prepulse and test pulse were done with [Met] 5enkephalin (ME) (300 nm), and desensitization was induced with ME (10 or 30 μm for 2, 5, 10, or 20 min) or morphine-6-β-d-glucuronide (M6G) (10 μm, 5,10, or 20 min). The test pulse of ME was applied 5 min after the washout of the desensitization treatment and repeated every 5-10 min to measure the extent and recovery from desensitization. The peptidase inhibitors bestatin and thiorphan (10 and 1 μm, respectively) were included in all experiments done with ME.
A concentration-response curve for noradrenaline (NA) was done in the presence of cocaine (1 μm). Data are shown as percentage of maximum response induced by UK14034 (3 μm) from groups of LC neurons of morphine or placebo-treated animals. For all experiments done with NA, slices were pretreated with prazosin (100 nm). Monensin (Sigma, St. Louis, MO) was dissolved in methanol to make a 100 mm stock solution. In experiments in which ME desensitization was 2 min, monensin (2-5 μm) was added to the ME solution before and during the desensitization treatment. In experiments in which desensitization was 10 min, monensin (1-5 μm) was added to the ME (300 nm) during the prepulse treatments. During the 10 min desensitization treatment, monensin (1 μm) was included in the ME (30 μm) solution. When the treatment with monensin caused a depolarization of the membrane potential, the experiment was terminated. A stock solution of chelerythrin (10 mm; Sigma) was made in DMSO and used at 5-10 μm.
Drugs. The drugs used were [Met] 5enkephalin (Sigma), morphine-6-β-d-glucuronide (National Institute on Drug Abuse), bestatin (Sigma), thiorphan (Research Biochemicals, Natick, MA), UK14304 (Research Biochemicals), yohimbine (Sigma), norepinephrine (Sigma), prazosin (Sigma), and staurosporin (Sigma).
Results
Onset of desensitization
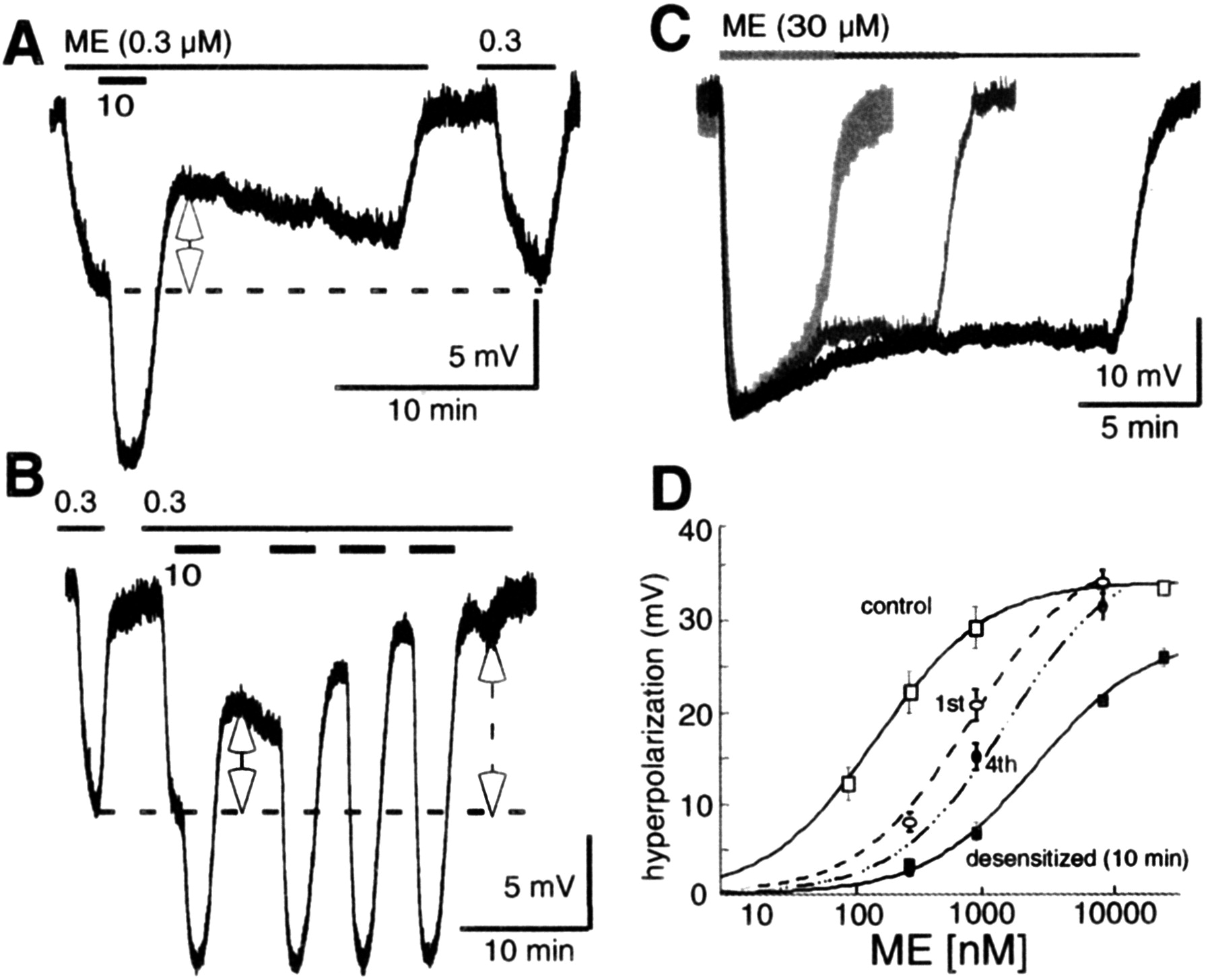
Studies examining the desensitization and recovery of MORs have relied on the decline of the maximal response as the hallmark of desensitization. When LC neurons were exposed to a saturating concentration of ME (30 μm for 5 min), the maximal hyperpolarization gradually declined over time (Harris and Williams, 1991; Osborne and Williams, 1995; Fiorillo and Williams, 1996; Alvarez et al., 2002; Blanchet and Luscher, 2002; Bailey et al., 2003). To determine whether desensitization occurred before the initial decline of the maximum response, slices were given a brief pulse (1-2 min) of ME (10 μm). An EC50 concentration of ME (300 nm) was tested before and immediately after the desensitizing pulse (Fig. 1). After the desensitizing pulse, the hyperpolarization caused by the test concentration of ME was reduced to 50 ± 11% of control (Fig. 1). When additional desensitizing pulses were given, the test response was further desensitized (Fig. 1B). The shift in the concentration-response curve is plotted and shows a decrease in the sensitivity to ME, without a significant decline in the maximum hyperpolarization (Fig. 1D). This experiment shows that the onset of desensitization was rapid and occurred well before the initial decrease of the maximal response.

The onset of MOR desensitization is rapid. A, Treatment with ME (10 μm) for 1-2 min decreased the hyperpolarization induced by an EC50 concentration of ME (300 nm) to 50 ± 11% of control. The EC50 concentration was applied immediately before and 10 min after the desensitizing pulse. B, Additional desensitizing pulses of ME (10 μm) further reduced the amplitude of hyperpolarization induced by ME (300 nm). C, Increasing the desensitization period from 5 to 20 min reduced the maximal hyperpolarization induced by ME (30 μm) to 80 ± 5 (n = 9), 71 ± 5 (n = 9), and 74 ± 3% (n = 7), respectively. D, Concentration-response curve showing the decrease in sensitivity to ME. 1st represents the shift in sensitivity to ME after a single 2 min pulse of ME (10 μm); 4th represents the shift in sensitivity after four pulses; desensitized indicates the change induced by a 10 min desensitization treatment.
Given the rapid onset of desensitization, the next set of experiments assessed the extent of opioid receptor desensitization, using different periods of drug application. For these experiments, LC neurons were treated with ME (30 μm) for 5, 10, and 20 min. The decline in the maximal hyperpolarization was not significantly changed between the desensitizing treatments. The responses were 80 ± 5% (n = 9), 71 ± 5% (n = 9), and 74 ± 3% (n = 7) of the maximum hyperpolarization after desensitization for 5, 10, and 20 min, respectively. The concentration-response curve, after a 10 min desensitization period, illustrates the decrease in maximal hyperpolarization and an increase in the EC50, to ∼1.6 μm. These values are similar to that reported in a previous study that used a 5 min desensitization period (EC50: control, 283 nm; desensitized, 1.1 μm) (Osborne and Williams, 1995). Thus, desensitization can be detected after a very short exposure to a saturating concentration of agonist, and the extent of desensitization, measured as the decline in maximal hyperpolarization, reached a limit after a 5 min treatment.
Chronic morphine treatment facilitates desensitization and reduces recovery
The rate and extent of recovery from desensitization, induced by ME, were examined in LC neurons from animals that were treated with morphine for 6-7 d. With a short desensitization treatment (ME, 10 μm for 1-2 min), the hyperpolarization induced by ME (300 nm) was reduced to 29 ± 2% of the prepulse (n = 3) (Fig. 2A). This decrease is significantly larger than that found in controls. Chronic morphine treatment did not alter the magnitude of desensitization measured by the decrease in the maximal hyperpolarization. The maximal hyperpolarization to ME (30 μm) declined to 74 ± 10% of the peak after 10 min, not significantly different from control animals (Fig. 2B).

Morphine treatment potentiates MOR desensitization. A, When the same experiment, as shown in Figure 1 A, was repeated in slices from morphine-treated animals (MTA), a ME (10 μm) desensitization pulse caused a greater reduction of the ME response (300 nm; p < 0.0016). B, Although the desensitization after a single pulse of ME (10 μm) was greater after morphine treatment, 10 min desensitization with ME (10 μm) reduced the maximal hyperpolarization to 74 ± 10%, which is not different from experiments in slices from control animals.
Chronic morphine treatment reduced the recovery from acute desensitization. Recovery after a 2 min desensitization treatment was complete within 25 min when recordings were made in slices from control animals. In these experiments, after a 5 min wash, the hyperpolarization induced by ME (300 nm) was 71 ± 7% of the prepulse and was 94 ± 3% of the prepulse after 25 min (n = 10). After chronic morphine treatment, receptor recovery was reduced after a 2 min desensitization period. After a 5 min wash, the test response was 44 ± 6% of the ME (300 nm) prepulse (n = 4) and was only 60 ± 7% after 25 min (n = 5) (Fig. 3). Recovery after a 10 min desensitization treatment was similarly altered by chronic morphine treatment. After washing for 30 min, the hyperpolarization induced by ME (300 nm) was 59 ± 5% in slices from morphine-treated animals (n = 9), compared with 82 ± 4% in slices from control animals (n = 5-10) (Fig. 4). These results indicate that chronic morphine treatment facilitates acute desensitization and/or decreases receptor resensitization, such that receptor recovery was attenuated and incomplete.

Recovery from a 2 min desensitization treatment. A, After a 2 min desensitization pulse, the loss of receptor function recovers rapidly, and recovery is complete in control animals (94 ± 3% after 25 min). B, After chronic morphine treatment, receptor recovery is incomplete. After 25 min, the ME (300 nm) response was only 60 ± 7% of the prepulse. C, Summary of results showing the difference in the rate and extent of recovery from a 2 min desensitizing pulse of ME (10 μm; 2-way ANOVA, p < 0.0001).

Recovery after a 10 min desensitization treatment. Although a 10 min desensitization treatment with ME (30 μm) caused the same magnitude of desensitization in control and morphine-treated animals, the recovery from desensitization was different. A, In an experiment from a control animal, receptor recovery was faster and more complete (82 ± 4% after 30 min). B, After morphine treatment, recovery was only 59 ± 5% after 30 min. C, Summary of results showing the difference in extent of recovery after a desensitization period of 10 min in slices from control and morphine-treated animals (2-way ANOVA, p < 0.018). An unpaired t test was done at each time point after the wash. n.s., Not significant; p > 0.75; **p < 0.005; ***p < 0.0008; ***p < 0.0004.
Morphine-6-β-d-glucuronide-induced desensitization
M6G, a metabolite of morphine, does not cause desensitization in LC neurons or receptor internalization in human embryonic kidney 293 cells expressing MOR (Alvarez et al., 2002). This observation was confirmed by comparing the hyperpolarization caused by ME (300 nm) before and after treatment with a saturating concentration of M6G (10 μm) for 5 (n = 5-8), 10 (n = 5-6), and 20 (n = 4-5) min. After washing out the M6G, no significant desensitization was seen in slices from control animals (Fig. 5). In slices from morphine-treated animals, M6G (10 μm,5 min, n = 6) reduced the hyperpolarization induced by ME (300 nm) to 66 ± 5% of control. When the M6G treatment period was increased to 10 min (n = 7-9), the test response was reduced to 55 ± 4%. Recovery from desensitization was not observed even after 45 min (n = 4) (Fig. 5). These experiments further indicate that acute MOR desensitization is facilitated and/or receptor recovery is impaired after chronic morphine treatment.

M6G-induced desensitization. Treatment with M6G (10 μm, 20 min) caused a sustained hyperpolarization. After washout of M6G, ME (300 nm) was used to test for desensitization. To ensure complete washout of M6G, the hyperpolarization induced by ME was measured 30 min after the washout. A, In slices from control animals, the hyperpolarization induced by ME (300 nm) was 94 ± 3% of the prepulse after washout of M6G. B, In slices treated with chelerythrin, to inhibit resensitization, the hyperpolarization induced by ME (300 nm) was only 57 ± 3% of the prepulse after washout of M6G (p < 0.0001). C, After morphine treatment, M6G induced desensitization. The hyperpolarization induced by ME (300 nm) was only 60 ± 7% of the prepulse (p < 0.0045). D, Summary of the results showing the hyperpolarization induced by ME (300 nm) recorded 30 min after the washout of M6G.
Disrupting receptor recycling increases desensitization and attenuates recovery
Monensin and PKC inhibitors have been shown to disrupt G-protein-coupled receptor (GPCR) resensitization (Shih and Malbon, 1994, 1996; Koch et al., 1998; Shih et al., 1999; Law et al., 2000; Marie et al., 2003). To determine the role of receptor resensitization in the process of MOR regulation, experiments were done in the presence of either monensin or PKC inhibitors. The PKC inhibitor chelerythrin (5-10 μm) significantly increased MOR receptor desensitization (see Fig. 7). Treatment of brain slices with chelerythrin alone caused a small reduction of the ME (300 nm)-induced hyperpolarization. Sustained application of chelerythrin (10 μm, 20 min), caused a 9 ± 5% reduction in the ME-induced hyperpolarization. The maximal hyperpolarization induced by ME (30 μm) was not changed by chelerythrin. After a 2 min desensitization period, the hyperpolarization induced by ME (300 nm) was reduced to 31 ± 6% of the prepulse (n = 6-8). After 25 min, recovery was only 68 ± 8%, compared with 94 ± 3% in untreated slices. The same results were seen when staurosporin (100 nm) was used to inhibit PKC. After 5 min, the ME (300 nm)-induced hyperpolarization was 22 ± 6% of the prepulse and after 30 min was 59 ± 5% (n = 6). When monensin was used to disrupt receptor recycling, similar results were obtained. The hyperpolarization caused by ME (300 nm) was reduced to 35 ± 3% immediately after a 2 min desensitization period, and the recovery was only 66 ± 6% after 25 min (n = 4).

Chelerythrin and monensin reduced receptor recovery. A, B, Representative traces in slices using the same experimental protocol. A, Control experiment. B, Experiment done in the presence of chelerythrin. The amount of desensitization induced by ME is larger and the recovery is less in the presence of chelerythrin. C, Summary of results obtained using the protocol illustrated in A and B (2-way ANOVA, p < 0.0001). D, Summary of results measuring the extent and rate of recovery from desensitization induced by ME (30 μm, 10 min). The extent and rate of recovery from desensitization in the presence of monensin and chelerythrin are decreased compared with experiments in untreated slices (2-way ANOVA, p < 0.0001).
Given that disrupting receptor resensitization increased desensitization and reduced recovery, the magnitude of desensitization would be predicted to increase. Treatment with either chelerythrin or monensin increased the magnitude of desensitization. In chelerythrin, desensitization with ME (30 μm, 10 min) reduced the maximal hyperpolarization to 58 ± 4% of maximum, compared with 71 ± 5% (n = 6) in controls. In the presence of monensin, the hyperpolarization induced by ME (30 μm) declined to 54 ± 12% of the maximum (n = 5). In LC slices from morphine-treated animals, this effect of monensin was even greater, decreasing the hyperpolarization to 39 ± 5% of the maximum after 10 min (n = 6) (Fig. 6).

Monensin increased the extent of desensitization in slices from control and morphine-treated animals. A, B, Recordings in slices taken from morphine-treated animals. A, In the absence of monensin. B, After treatment with monensin. The maximal hyperpolarization induced by ME (30 μm, 10 min) had a greater decline in slices treated with monensin. C, Summary of results obtained with experiments taken from untreated (control) and morphine-treated animals (MTA) in the absence (-MO) and presence (+MO) of monensin. The results show that monensin increased the extent of desensitization and that the effect was larger in slices from morphine-treated animals (*p < 0.01, 0.02; **p < 0.004).
Receptor recovery, after a 10 min desensitization period in the presence of monensin or chelerythrin, was also significantly reduced (Fig. 7). In monensin-treated slices, the hyperpolarization induced by ME (300 nm) was only 37 ± 13% of the prepulse 30 min after washout (n = 5). Recovery in chelerythrin-treated slices was only 56 ± 7% after 30 min (n = 8).
PKC inhibitors unmask M6G-induced MOR desensitization in control animals
It was difficult to determine whether M6G caused desensitization in control animals because of the slow washout of M6G from the brain slice. It took between 10 and 20 min for the membrane potential to return to baseline after washing M6G (10 μm). There was no significant decline in the response induced by ME (300 nm) 30 min after washing M6G. It was possible, however, that M6G caused desensitization that recovered during the slow wash. To determine whether M6G caused desensitization, experiments were conducted in the presence of chelerythrin. By disrupting receptor resensitization, any desensitization that occurred would be detectable. After treatment of the slice with chelerythrin (5 μm), M6G (10 μm, 10 min) caused a significant decrease of the hyperpolarization caused by ME (300 nm) (Fig. 5B). The hyperpolarization induced by ME (300 nm) was only 57 ± 3% of the prepulse 30 min after the washout of M6G. Thus, as was observed in slices from morphine-treated animals, treatment with chelerythrin reveals M6G-induced desensitization.
Homologous desensitization
A recent report described heterologous desensitization of α2-adrenoceptors after MOR desensitization (Blanchet and Luscher, 2002). In the present study, heterologous desensitization of α2-adrenoceptors was tested by measuring the hyperpolarization caused by a submaximal concentration of NA (30 μm) before and after MOR desensitization. NA (30 μm) induced 24 ± 2 mV hyperpolarization before and 20 ± 3 mV (n = 5) after a desensitization treatment with ME (30 μm, 10 min, in the presence of monensin, in which acute desensitization was the greatest). In addition, the hyperpolarization induced by a maximal concentration of UK14304 (3 μm), a full agonist at α2-adrenoceptors, did not change after desensitization with ME (30 μm for 20 min). The hyperpolarization induced by UK14304 was 26 ± 1 mV after desensitization, which is 83 ± 5% of the maximal opioid response. This observation is consistent with previous reports indicating that heterologous desensitization makes up only a small component of the total amount of desensitization using the same recording condition (Christie et al., 1987; Fiorillo and Williams, 1996).
Opioid tolerance is homologous after chronic morphine treatment
To determine whether the cellular adaptations responsible for the altered MOR responsiveness would extend to other G-protein-coupled receptors, the hyperpolarization induced by α2-adrenoceptors was examined. There was no significant difference in the EC50 or the maximal hyperpolarization induced by NA (Fig. 8). In naive/placebo-treated animals, NA (10 μm) caused a 12 ± 3 mV hyperpolarization (n = 5). The maximal hyperpolarization was 19 ± 3 mV (NA, 50 μm). After chronic morphine treatment, NA (10 μm) caused a 15 ± 2 mV hyperpolarization, and the maximal response was 25 ± 2 mV (n = 4).

Morphine treatment did not alter α2-adrenoceptor function. A, Concentration-response curve for NA slices from animals treated with placebo and morphine pellets. The amplitude of the hyperpolarization is plotted as the percentage of hyperpolarization induced by a maximal concentration of UK14034 (3 μm). B, Morphine treatment did not change the desensitization caused by NA. After a desensitizing pulse of NA (50 μm, 5 min), the hyperpolarization induced by NA (10 μm) was the same in the morphine- and placebo-treated animals.
Given that desensitization of MORs was increased after chronic morphine treatment, it was possible that desensitization of other, similarly coupled receptors may also be affected. This possibility was tested with the use of a saturating concentration of NA to desensitize the α2-adrenergic receptors. The hyperpolarization induced by NA (10 μm) was tested before and after treatment with a saturating concentration of NA (50 μm, 5 min). The hyperpolarization induced by the test pulse, after the 5 min desensitizing treatment, was 77 ± 7% and 74 ± 9% of the control, in slices from placebo-treated animals (n = 4) and morphine-treated animals (n = 5), respectively. Thus, chronic morphine treatment did not result in a generalized increase in the desensitization of the α2-adrenoceptor.
Discussion
The results from this study indicate that acute desensitization of the MOR could be detected within 1-2 min. After chronic morphine treatment, this process was facilitated, and recovery from acute desensitization was inhibited. In control, recovery from the 1-2 min desensitization was complete within 25 min. After chronic morphine treatment, desensitization was facilitated, and complete recovery was not observed. The results also indicate that the receptor reserve and the process of receptor resensitization were impaired by morphine treatment. When receptor resensitization was disrupted in slices taken from control animals, MOR desensitization was potentiated, and recovery from desensitization was reduced. As such, one mechanism by which chronic morphine treatment altered receptor function was by reducing receptor reserve and/or disrupting the normal resensitization of MOR.
Desensitization and internalization of receptors
The most widely studied mechanism underlying the regulation of GPCRs, and specifically MORs, comprises a stepwise process that begins with phosphorylation of the receptor by G-protein receptor kinase (Zhang et al., 1998; von Zastrow, 2001; Claing et al., 2002). The phosphorylated receptor has an increased affinity for β-arrestin, resulting in the displacement of G-proteins (Zhang et al., 1998). The ligand-β-arrestin-receptor complex is then thought to enter the endocytic pathway. Like the β-adrenoceptor, MORs have been shown to recycle rapidly, from the endosomal compartment back to the plasma membrane. Although these studies showed that this process is important for receptor recycling, there was no evidence directly linking this process to the observed loss of receptor function (El Kouhen et al., 1999; Law et al., 2000).
Complete recovery, from desensitization found after a short desensitizing treatment, indicates that receptor resensitization can be a fast process. The observation that recovery was compromised by monensin, an agent shown to interfere with receptor recycling, suggests that the rapid turnover includes receptor sequestration and reinsertion of functional receptor back to the plasma membrane. Therefore, the initial steps of acute desensitization rapidly decrease receptor coupling and/or number, resulting in a shift of the concentration-response curve to the right. With continued depletion of receptors, the maximum response declines and reaches a steady state after ∼5 min. At this point, the rate of receptor resensitization/recycling and the rate of receptor desensitization are at equilibrium, such that there was no further decline in the hyperpolarization when the desensitization period was increased to 10 or 20 min.
Another possibility is that different populations of the receptors are responsible for each of the components of desensitization and recovery. There are two splice variants of rat MOR (MOR-1 and MOR-1B) with different C termini (Koch et al., 1998, 2001). These splice variants have different rates for receptor desensitization and recovery. Phosphorylation of threonine 394 on MOR-1 slowed internalization and delayed recovery after desensitization (Wolf et al., 1999). The short C terminus found on MOR-1B enhances its interaction with clatherin-coated pits, enabling a faster rate of receptor internalization and recovery (Koch et al., 1998, 2001). Thus, the rapid recovery from desensitization may result from one splice variant, and the slow or incomplete recovery may result from a second form of the receptor. There are, however, no studies that have examined the expression of these splice variants in the LC.
It is also possible that a completely different mechanism is responsible for the slower rate of recovery found after a more prolonged period of drug exposure. A recent study in locus ceruleus neurons suggested that acute desensitization, induced by the stable opioid peptide ligand [d-Ala 2, N-MePhe 4, Gly-ol 5]enkephalin (DAMGO), resulted in desensitization that was manifested at the G-protein-gated potassium channel (Blanchet and Luscher, 2002). Previous work and the present results indicate that neither acute desensitization nor chronic morphine treatment caused a dramatic decrease in the potassium conductance activated by the α2-adrenoceptor.
Experiments done in vivo also indicate that there are alternate pathways for the regulation of MORs. For instance, in β-arrestin-2 knock-out animals, the development of morphine tolerance was initially delayed but became apparent later in treatment. When these mice were treated with chelerythrin, a PKC inhibitor, morphine tolerance was attenuated (Bohn et al., 2002). These results suggest that there are two pathways mediating the effect of morphine tolerance, one that is β-arrestin-2-dependent and one that is PKC-dependent. Although it is not clear how the results obtained in vivo in the knock-out animals relate to the present results, it is clear that tolerance to morphine is regulated at multiple levels.
Desensitization and receptor reserve
Chronic morphine treatment results in a rightward shift of the concentration-response curve to DAMGO (Christie et al., 1987). A similar rightward shift is seen in LC neurons from control animals after a brief application of ME (10 μm). With repeated short applications, the rightward shift is increased without a change in the maximal response. Sustained applications of ME (30 μm) produced a gradual decline of the maximum hyperpolarization. This gradual decline in responsiveness as well as the reduction of the maximal response is similar to that seen in experiments in which receptor reserve has been depleted by treatment with β-chlornaltrexamine or β-funaltrexamine (Christie et al., 1987; Law et al., 2000; Borgland et al., 2003). These results suggest that the process of receptor desensitization is rapid and occurs as a continuum. The loss of receptor function closely correlates with the loss of receptor reserve. As such, the rightward shift in the concentration-response curve, to ME, is a sensitive assay for the initial decline in receptor reserve. After chronic morphine treatment, the rightward shift of the concentration-response curve persists. This observation indicates that the receptor reserve was compromised, and resensitization was impaired such that a subset of internalized receptors were either not recycled or recycled at a considerably slower rate. The reduced receptor reserve, in addition to the lack of recovery from acute desensitization, would increase the degree of receptor-dependent tolerance.
The regulation and trafficking of μ-opioid receptors has been shown to be agonist-dependent (Sternini et al., 1996; Whistler and von Zastrow, 1998; Alvarez et al., 2002; Patel et al., 2002; Borgland et al., 2003). Most agonists, when applied at saturating concentrations, cause receptor desensitization and internalization. Morphine and its metabolite M6G are the exceptions, in that they are ineffective at inducing receptor desensitization and internalization under basal conditions (Kovoor et al., 1998; Alvarez et al., 2002; Borgland et al., 2003). When GRK2 and/or β-arrestin are overexpressed, on the other hand, internalization of MORs can be induced by morphine (Whistler and von Zastrow, 1998; Zhang et al., 1998). Chronic morphine treatment has been shown to elevate the expression level of GRK2 and β-arrestin in the locus ceruleus (Terwilliger et al., 1994). These molecular adaptations can contribute to the increase in desensitization caused by M6G in slices from morphine-treated animals.
When brain slices from control animals were treated with chelerythrin, M6G-induced desensitization was detected. This observation suggests that M6G can induce desensitization of MOR under basal conditions. There are two possible explanations for the inability to detect the loss of receptor function under control conditions. One is that M6G-induced MOR desensitization recovers rapidly; therefore, the loss of receptor function was not seen because of the slow washout. It is also possible that M6G is not as efficient as ME at inducing MOR desensitization. As a result, the rapid rate at which receptor resensitization occurred was sufficient to maintain normal receptor function. When receptor resensitization was disrupted by treatment with chelerythrin, the loss of receptor function was apparent.
The results from this study show that the regulation opioid receptor signaling is a dynamic process. The onset of desensitization is as rapid as the initiation of the acute response. Under control conditions, receptor resensitization is a fast process. In other experiments, extended desensitization, hours or days of drug exposure (as with morphine treatment), alters the rate and extent of recovery from desensitization. The overlapping time courses and range of processes that alter receptor activity suggest that multiple mechanisms underlie the regulation of these receptors.
Footnotes
This study was supported by National Institutes of Health Grant DA08163. We thank Drs. V. Alvarez, M. Connor, H. Morikawa, and M. Torrecilla for comments on this manuscript.
Correspondence should be addressed to J. T. Williams, Vollum Institute, Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: williamj{at}ohsu.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247699-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}