

Abstract
In the present study, we examined the ability of estrogen to enhance cholinergic neurite arborization in vitro and identified the signal transduction cascade associated with this effect. Basal forebrain primordia collected from rat pups on postnatal day 1 were cultured for 2 weeks and then treated with 5 nm 17β-estradiol for 24 hr. Cholinergic neurons were identified immunocytochemically with an antibody against the vesicular acetylcholine transporter and digitally photographed. Morphological analysis indicated that female cultures respond to estrogen treatment with an increase in total neurite length per neuron (4.5-fold over untreated controls) and in total branch segment number per neuron (2.3-fold over controls). In contrast, there was no change in total neurite length per neuron in male cultures, and we also observed a decrease in total branch segment number per neuron (0.5-fold below controls). Detailed histograms indicated that estrogen increases primary and secondary branch length and number and also increases terminal neuritic branches to the seventh order in female cultures. In a second set of experiments, we investigated the signal transduction cascade involved in this response, and found that an upstream extracellular signal-regulated kinase (ERK) inhibitor blocked the ability of estrogen to enhance outgrowth in female cultures. Our study provides strong evidence in support of the fact that the ERK pathway is required for estrogen-induced structural plasticity in the cholinergic system of female rats. Understanding the intracellular processes that underlie the response of cholinergic neurons to estrogen provides a necessary step in elucidating how cholinergic neurons can be particularly susceptible to degeneration in postmenopausal women.
- extracellular signal-regulated kinase
- cholinergic system
- basal forebrain
- estrogen
- plasticity
- outgrowth
Introduction
Estrogen (E2) has a wide range of effects on the structure and function of a variety of brain regions, most notably regions not directly linked to reproduction, such as the hippocampus and the basal forebrain cholinergic system (BFCS) (McEwen and Alves, 1999). In the hippocampus, E2 regulates dendritic density and spines (Woolley and McEwen, 1994; Murphy and Segal, 1996; Leranth et al., 2003). An important step in understanding the role of E2 in structural plasticity is the identification of the molecular cascades that may be influenced by it. The mitogen-activated protein kinase (MAPK) pathway (Adams and Sweatt, 2002) has emerged as a major contributor to plasticity in vertebrates. A functional link between E2 and the MAPK pathway has been established in non-neuronal (Migliaccio et al., 1996; Russell et al., 2000; de Jager et al., 2001) as well as in neuronal systems (Watters et al., 1997; Singer et al., 1999; Singh et al., 1999; Kuroki et al., 2000; Zhang et al., 2002). However, a direct involvement of E2 and the MAPK pathway in plasticity has been described only in the hippocampus (Bi et al., 2001). Here we demonstrate a similar functional link in BFCS neurons.
A large portion of BFCS neurons express E2 receptors (ER), providing an anatomical substrate for actions of E2 on this neurotransmitter system (Toran-Allerand et al., 1992; Mufson et al., 1999; Shughrue et al., 2000). It has been known for quite some time (Luine et al., 1975; McMillan et al., 1996) that E2 modulates the cholinergic phenotype, but the specific cellular mechanisms remain unclear. We postulate a direct involvement of E2 in the structural plasticity of basal forebrain cholinergic neurons via the MAPK pathway.
MAPKs are a family of serine–threonine kinases that include the extracellular signal-regulated kinases (ERKs) among its members. ERKs are strongly activated by mitogens and, in the CNS, by neurotrophins and neurotransmitters (Grewal et al., 1999). These kinases are major effectors of signal transduction from the cell surface to the nucleus and are implicated in various forms of memory (Thiels et al., 2002) as well as in cell growth and differentiation. Studies performed predominantly in nonneuronal cells have characterized what could be considered to be the archetypal ERK cascade (Seger and Krebs, 1995; Derkinderen et al., 1999), involving the activation of the Ras family of guanosine triphosphatases, the MAPK-kinase-kinases of the Raf family, the phosphorylation of MAPK kinase (MEK), and finally ERK activation by dual phosphorylation of their Thr-Glu-Tyr motif. ERK1 (44 kDa) and ERK2 (42 kDa) are expressed at high levels in the CNS (Boulton et al., 1991; Flood et al., 1998) and are found in cytoplasmic compartments, including dendrites (Ortiz et al., 1995). They can phosphorylate a wide variety of substrates in the cytoplasm and also in the nucleus, where they can turn on transcription factors and thereby control gene expression. Their targets include microtubule-associated proteins and neurofilaments (Grewal et al., 1999). Phosphorylation of these cytoskeletal proteins by ERKs could arbitrate morphological changes that underlie plastic properties of neurons.
Materials and Methods
Cell culture. Animals were treated in accordance with the principles and procedures of the National Institutes of Health Guide for the Care and Use of Laboratory Animals; all protocols were approved by the Institutional Animal Care and Use Committee of California State University Los Angeles. Timed pregnant Fisher 344 rats were obtained from Harlan (San Diego, CA) and kept in the vivarium until delivery, in a temperature- and light-controlled environment with a 12 hr light/dark cycle. On postnatal day 1 (P1; <24 hr), pups were removed from the home cage, separated by sex (done by comparing the distance between the genital papillae and the anus; this distance in males is approximately twice that in the females), and decapitated. The brains were rapidly extracted from the cranium and placed on chilled calcium- and magnesium-free HBSS. The basal forebrain primordium was dissected under a stereomicroscope using the procedure described by Hartikka and Hefti (1988), and tissue was dissociated in 0.1% trypsin and 0.1 mg/ml DNase (Sigma, St. Louis, MO) at 37°C for 30 min. Enzymatic dissociation was halted with the addition of Neurobasal medium-A (Invitrogen, Carlsbad, CA) (Brewer et al., 1993) with 5% horse serum and 10% fetal bovine serum (previously the sera had been treated with charcoal for 24 hr at 4°C). After centrifugation at 1000 × g for 5 min at 4°C, the supernatant was discarded and cells were resuspended by trituration with fire-polished Pasteur pipettes in 2 ml of growth medium (Neurobasal medium-A supplemented with B27, 0.5 mm l-glutamine, 50 ng/ml of 2.5S NGF, and 0.5% streptomycin–penicillin; all from Invitrogen) and filtered through a 40 μm nylon mesh filter (BD Biosciences, Franklin Lakes, NJ) to form a single-cell suspension.
For morphological studies, cells were plated at a density of 105 cells/cm2 (24 well Costar plates; Corning Life Science, Acton, MA), and for protein assays, cells were plated at a density of 106 cells/cm2 (six well Costar plates). Plates were precoated with 0.1 mg/ml poly-l-lysine (Sigma). Although these procedures discourage glial growth, we did not attempt to purify our cultures, and thus we term them “mixed cultures.”
Experimental treatment of cultures. Cells were maintained in culture for 2 weeks. On day 14 in vitro, E2 (17-β-E2; Sigma) was freshly prepared by dissolving in ethanol (1.36 mg/ml). To avoid errors and ensure a final concentration of 5 nm E2 in the medium, treatment was administered by completely replacing the growth medium with a fresh solution containing the treatment. Cell cultures were maintained in treated medium for 30 min (for signaling studies using Western blot analysis) or 24 hr (for morphological studies using immunocytochemistry). Activation of ERK was studied in the presence and absence of the E2 receptor antagonist 7α-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)nonyl]estra-1,3,5(10)-triene-3,17β-diol [ICI 182,780 (ICI); 500 nm; Tocris, Ballwin, MO] (Howell et al., 2000), which inhibits both the α and β isoform of the E2 receptor, and in the presence or absence of the MEK inhibitor 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126; 10 μm; Tocris) (Favata et al., 1998). Stock solutions of 10 mm ICI and 2 mm U0126 were kept at –20°C, and final dilutions were freshly prepared before experimental treatments.
Immunocytochemistry. Neuronal cultures for morphological analyses were fixed with 4% paraformaldehyde in PBS, washed, blocked with 3% horse serum, and incubated with an antiserum against the vesicular acetylcholine transporter (VAChT; 1:1000; Oncogene Research, San Diego, CA) overnight. VAChT is a highly specific marker for cholinergic neurons (Gilmor et al., 1996; Arvidsson et al., 1997). The next day, the plates were washed and incubated sequentially in biotinylated anti-rat IgG (1: 2000; Jackson ImmunoResearch, West Grove, PA), avidin–biotin peroxidase (1:500; Vector Laboratories, Burlingame, CA), and diaminobenzidine (Sigma) to give a brown reaction product. The specificity of staining is well documented but was verified nonetheless in parallel experiments in which either the primary or the secondary antisera were omitted. Because it is still possible that the antiserum may recognize unidentified binding sites, the immunoreactivity detected in this study is more accurately described as VAChT-like immunoreactivity (VAChT-IR).
Morphological analysis. Changes in neurite outgrowth were studied in cell cultures positively stained with the anti-VAChT antiserum. Randomly selected fields, containing readily identifiable neurons, were photographed using an eight bit SPOT RT Slider digital camera (Diagnostic Instruments, St. Sterling Heights, MI) coupled to a phase contrast microscope (Leica, Wetzlar, Germany) or using a 16 bit Photometrics Cool-SNAP digital camera (Roper Scientific, Tucson, AZ) coupled to an inverted Nikon (Tokyo, Japan) microscope. All images were acquired at 200× magnification and saved as color 72 dpi uncompressed transfer-image files. They were then converted to eight bit grayscale images for analysis. Morphometry was performed with NIH Image 1.62 software using the line segment tool and the scaling option.
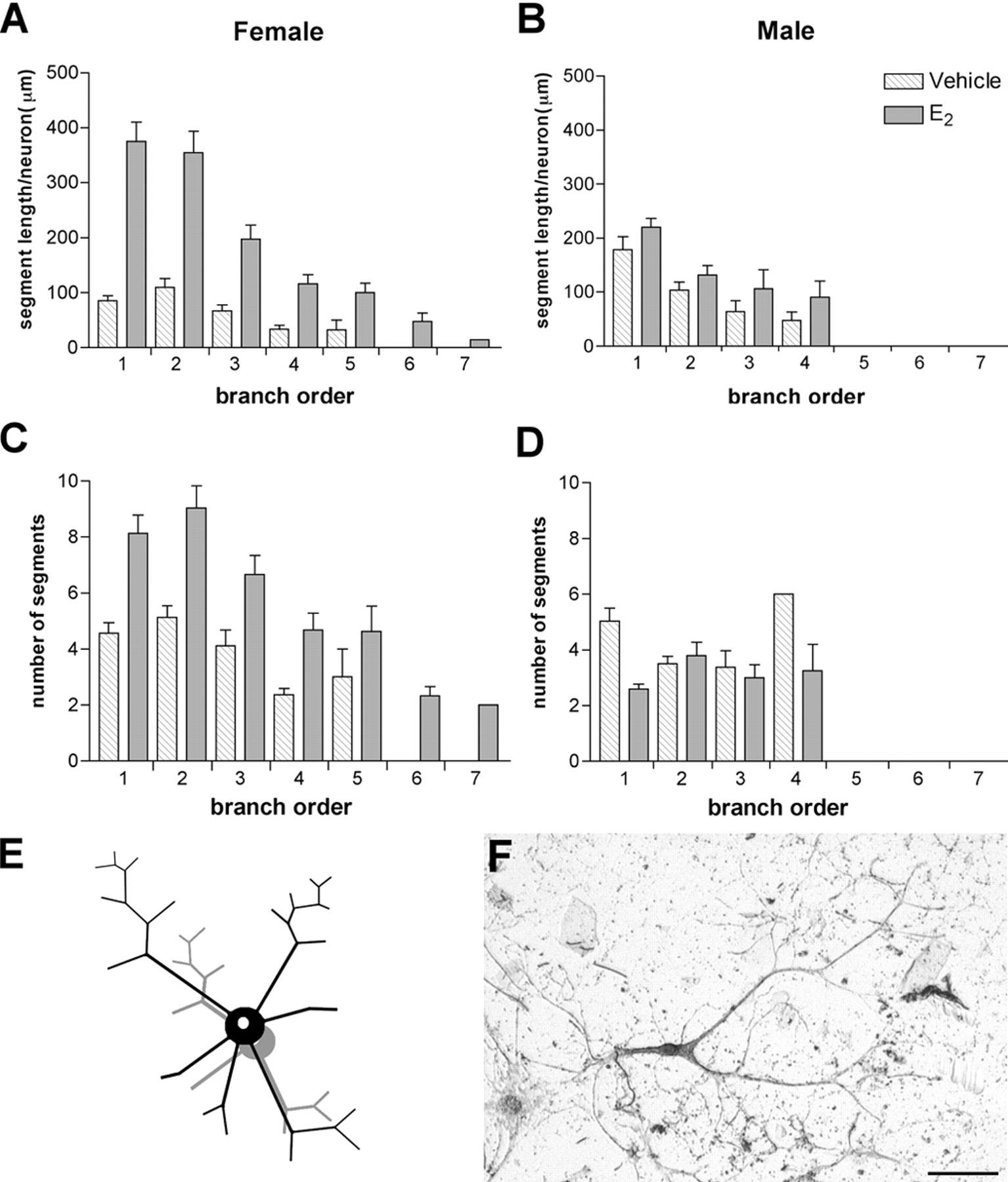
Morphological analysis of VAChT-IR neurons followed a method adapted from a study by Arendt et al. (1986), in which they used branch order to describe morphological differences in human basal forebrain neurons. A centrifugal ordering system was used to establish branch order in a neuron (Fig. 1), and branch order was taken into consideration when calculating mean neurite length per neuron, total neurite length per neuron, and total number of segments (branches) per neuron, as well as the length and number of segments of each order. Measurements were taken from all the neuritic processes of a neuron at the focal plane level. Branch segment length was measured by drawing a segmented line from the point of origin (branch bifurcation or intersection) to its end (either the next bifurcation point or the disappearance of the element from the plane of the image).

Morphological analysis of neurite outgrowth. The diagram shown represents the centrifugal ordering system used to number neurite branches, as described in detail in Materials and Methods.
Before analysis, specific criteria were established to define what would be considered a segment (neurite branch extension) eligible for measurement. Extensions from a soma were considered to be a neurite if they were VAChT-positive and had a diameter no greater than half the diameter of the soma. For bifurcations and subsequent branching, only those segments with a visible origin were included in the measurements. Measurements were calibrated with a micrometer slide, and lengths were expressed in micrometers. Within each treatment group, morphological measurements were taken from 30 neurons that met those conditions (from 12 different culture wells).
For statistical analyses, data were expressed as means ± SEM of individual means from four independent experiments or normalized to the vehicle-treated control. Statistical significance was assessed with unpaired t tests and ANOVA, using StatView Software (SAS Institute, Cary, NC).
Western blot analysis. At the end of the experimental treatments, the medium was aspirated and 400–500 μl of boiling lysis buffer (1% SDS in 100 mm Tris buffer) was added to each well. Culture plates were swirled to facilitate cell detachment from the substratum, and the content of the wells was transferred to microcentrifuge tubes, placed in a boiling water bath for an additional 5 min, and then set in ice. The cells were carefully lysed by passing them 10 times through a 1 ml syringe fitted with a 26 gauge needle. The resulting homogenate was centrifuged at 14,000 rpm for 5 min, and the supernatant was collected. An aliquot from each sample was taken for protein content assay (BCA protein analysis using a Detergent Compatible Protein Assay kit; Pierce, Rockford, IL). The remaining homogenate was diluted in 2× Laemmli buffer with 5% mercaptoethanol (Bio-Rad, Hercules, CA), and 10 μg samples were loaded onto 10% SDS-PAGE gels and separated based on molecular size. Precision unlabeled molecular weight markers and StrepTactin-HRP (both reagents from Bio-Rad) were used to estimate protein weight, and 10 μl of positive (phospho-ERK2; Cell Signaling Technology, Beverly, MA) and negative (ERK2; Cell Signaling Technology) control proteins for the diphosphorylated form (active) of ERK were loaded onto the same gels. The gels were then electroblotted onto nitrocellulose membranes (Osmonics, Minnetonka, MN). All experiments were performed in duplicate.
Immunodetection of total ERK and diphosphorylated ERK (dpERK) was performed by first blocking the membrane with 5% BSA (Sigma) in borate buffer saline for 30 min at room temperature, followed by the addition of the primary antibody, the polyclonal anti-diphosphorylated ERK1/2 (p44/p42, Thr202/Tyr204; 1:1000; Cell Signaling Technology); the blots were then allowed to react overnight. Antibody binding to protein was detected using a secondary goat anti-rabbit HRP-labeled antibody (Jackson ImmunoResearch) at a concentration of 1:2000 for 1 hr at room temperature and visualized autoradiographically on Hyper-film using enzyme-linked chemiluminescence (Amersham Biosciences, Arlington Heights, IL) according to the manufacturer's instructions. All blots were stripped (2% SDS, 100 mm mercaptoethanol, and 62.5 mm Tris-HCl, pH 6.7), blocked, and reprobed for total ERK protein to verify equal loading, using the same procedures, with a polyclonal anti-ERK1/2 antiserum (1:1000; Cell Signaling Technology) and a goat anti-rabbit peroxidase-conjugated secondary antibody (Jackson ImmunoResearch). The blots were also stained with an antibody against the 51 kDa glial fibrillary acidic protein (GFAP; 1:1000; Sigma) to analyze the effects of treatment on the glial cells present in our cultures.
Densitometric analyses. To quantify protein abundance, films were scanned (CanoScan N656U scanner; Canon, Lake Success, NY) and analyzed by two observers blind to the treatment, using a grayscale at 720 dpi. Band densities were calculated using Scan-It software (Silk Scientific, Orem, UT). Total net intensity (average pixel) values were calculated by subtracting the background within the area measured for each film, to account for any variation in background intensity across films. Optical density (OD) values were normalized by dividing each OD value by the mean OD for the vehicle control, to eliminate variation across experiments. Data are expressed as means ± SEM of individual means from four independent cultures. Statistical significance was assessed with unpaired t tests and ANOVA using StatView Software (SAS Institute). Significance level was set at 0.05.
Results
Estrogen induces neurite outgrowth in basal forebrain cholinergic neurons in vitro: sex-related differences
Morphological analysis of VAChT-IR neurons (n = 30 in each group) in mixed cultures obtained from basal forebrain of male and female P1 Fisher 344 rat pups demonstrated a significant sexual dimorphism. Control cultures were treated with vehicle for 24 hr, fixed, stained, and analyzed. The mean neurite length was 33.3 ± 3.5 μm (SEM) for male VAChT-IR neurons and 18.8 ± 1.6 μm (SEM) in females, a statistically significant difference (unpaired t test; p = 0.0005) (Fig. 2A). To account for this baseline difference, all subsequent morphological data were normalized to reflect fold increase above control. This procedure also was aimed at ensuring that the true potential for E2 action in male versus female neurons was correctly identified.
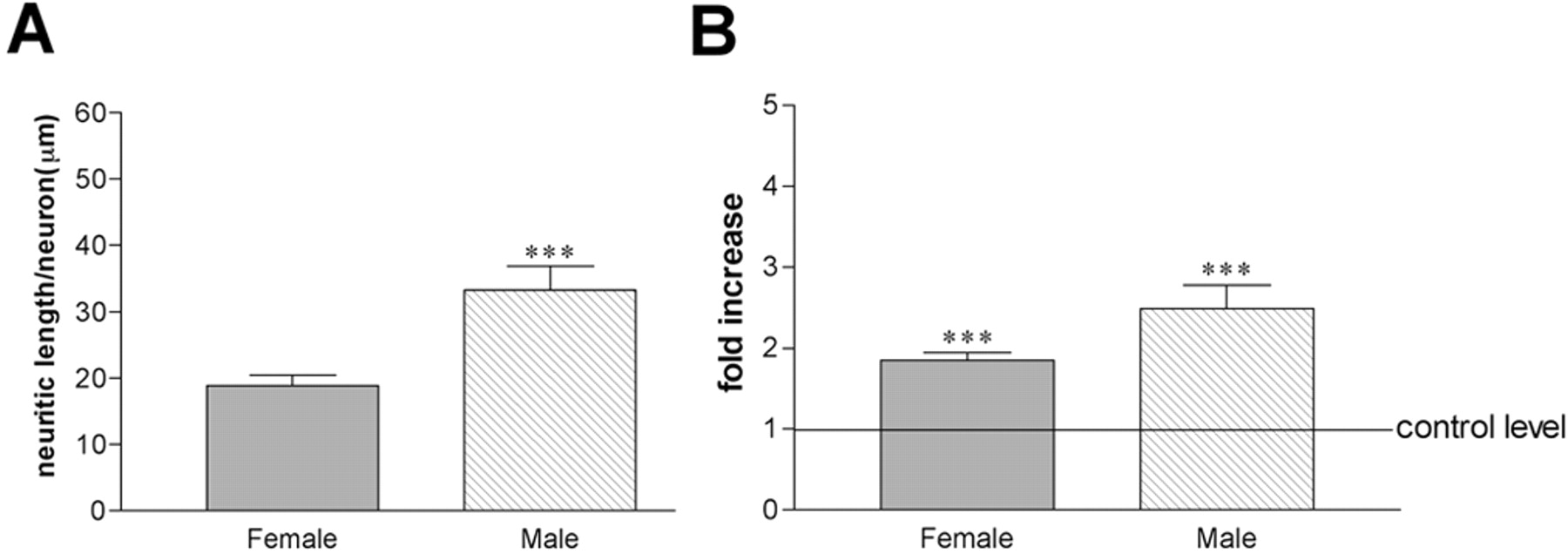
Basal forebrain cholinergic neurons are sexually dimorphic. Bar graphs illustrate the results of the morphometric analysis of basal forebrain neurons in culture, stained with an antibody against VAChT. A, Average arborization size was calculated from a total of 30 neurons in each group of the control (vehicle-treated only) cultures, with males exhibiting significantly larger neuritic arborizations than females (***p < 0.001). B, E2 treatment induced a significant increase in the size of neuritic arborization, calculated in reference to control (untreated) cells (***p < 0.001), an increase more pronounced in male (2.5-fold) than in female (twofold) neurons.
In a parallel experiment, cultures were treated with 5 nm E2 for 24 hr. We found a significant effect of sex and treatment on mean neurite length per neuron (two-way ANOVA; F(1,116) = 10.159; p = 0.0018). When compared with basal measurements, there was a significant twofold increase in female and 2.5-fold increase in male mean neurite length per neuron (Fig. 2B) (unpaired t test for male cultures, t =–4.838, p < 0.0001; for female cultures, t = –6.979, p < 0.0001). Although this result seemed to indicate a more pronounced effect of E2 treatment on male cholinergic neurons, further analysis showed that this was not the case. We compared independently the two parameters included in the calculation of mean neurite length per neuron [i.e., the total length of the neurite arborization per neuron and the number of neurite segments (or branches) per neuron] (Fig. 3). In female cultures, E2 treatment induced a significant fourfold increase in total neurite length (control, 227.7 ± 25.5 μm; treated, 1029.1 ± 90.4 μm; unpaired t test; t = –8.45; p < 0.0001), whereas in male cultures this measure remained unchanged from controls (control, 301.2 ± 33.8 μm; treated, 320.1 ± 23.5 μm; unpaired t test; t = –0.460; p > 0.05). The number of segments per neuron was also significantly increased in hormone-treated female cultures by twofold (control, 12.2 ± 1.2; treated, 28.5 ± 2.6; unpaired t test; t =–5.595; p < 0.0001). Interestingly, E2 treatment significantly decreased by half the number of segments per neuron in male cultures (control, 9.8 ± 0.8; treated, 5.8 ± 0.9; unpaired t test; t = 2.97; p = 0.0043).

Estrogen has a sexually dimorphic effect on neuritic arborization. Bar graphs illustrate the change in total neurite length and segment number per neuron in cultures treated with E2 for 24 hr, with respect to vehicle-treated controls. In female cultures (A), both parameters are significantly larger than controls, whereas in male cultures (B), there is no change in the total neurite length per neuron and there is a significant decrease in the number of branches. **p < 0.01; ***p < 0.001.
Studies by other groups have shown that E2 induces a plastic reorganization of dendritic spines (Leranth et al., 2003), but to the best of our knowledge there is no information on whether E2 is able to exert outgrowth effects at the axonal level. Although we did not attempt to distinguish between axonal and dendritic processes in our analysis, we began to examine this question by searching for possible differences in proximal versus distal effects of E2. Detailed histograms of segment length and number, according to branch order, revealed a striking difference in the response of male and female neurons to E2, with a significant shift to the right in the values of both parameters. In female cultures, E2 treatment increased fourfold the length of primary neurites (e.g., those arising directly from the cell body) compared with controls (Fig. 4A) and also doubled the number of primary neurites compared with controls (Fig. 4C). In contrast, in male cultures there was no increase in the number of primary neurites per neuron, and the length was reduced by half (Fig. 4B,D). In addition, E2 induced a significant increase in terminal arborization in female cultures, with the appearance of branch segments of the sixth and seventh order that were not present in the female controls or in the male cultures. The overall effect of E2 on female cholinergic neurons is illustrated in Figure 4E, and a representative VAChT-IR neuron is shown in Figure 4F.

Distribution of neurite segment length and number in basal forebrain neurons in culture. A–D, Histograms showing the distribution of segment size and number by order. Branch order 1 corresponds to primary neurites and branch order 7 to the distal-most segments (see Materials and Methods for details). Analysis of these histograms illustrates that in female cultures (A), there is a significant increase in the length of neurite branches in all orders, a change that is more pronounced for primary and secondary neurites, increasing on average from 100 μm (control) to 400 μm (treated). In contrast, in male cultures (B), the segment length remains unchanged. Similarly, the number of segments is significantly higher in females (C) but is reduced in males (D) after E2 treatment. Note also the increased branching in females. The change in neuronal morphology in females is represented schematically in E. F, Abright field photomicrograph of a representative VAChT-IR neuron from female cultures. Scale bar, 100μm.
Estrogen treatment of cholinergic neurons in vitro activates the extracellular signal-regulated kinase cascade
Multiple lines of evidence indicate that E2 can rapidly and transiently activate the MAPK pathway, a signal transduction cascade involved in neuritic growth and synaptic plasticity, and thus a likely candidate to mediate the structural changes described above. We measured the effects of E2 treatment on the phosphorylation of ERK1/2, one of the key proteins in this cascade. Previous reports have also described a significant increase in the levels of activated ERK with 30 min of exposure to the hormone (Singer et al., 1999; Singh et al., 1999).
Male and female cultures were prepared according to procedures identical to those used in the studies described above, but were exposed for only 30 min to the different treatments. The cells were lysed, and samples were prepared for Western blot analysis of the levels of ERK1/2 and of the activated form of ERK1/2 (dpERK1/2) (Fig. 5). A two-way ANOVA comparing levels of ERK1/2 and dpERK1/2 between sex and treatments showed no effect of sex on the relative levels of these proteins (F(1,5,5,28) = 0.862, p > 0.05 for ERK1/2; F(1,5,5,28) = 0.886, p > 0.05 for dpERK1/2) and no significant interaction of sex on the treatments, thus allowing an independent analysis of the results. In female cultures (Fig. 5A), there was a statistically significant increase in the levels of dpERK1/2 after E2 treatment (ANOVA; F(5,14) = 25.965; p < 0.0001; Fisher's PLSD post hoc test; p = 0.029), an effect that was abolished by the concurrent administration of the MEK inhibitor U0126 (Fisher's PLSD post hoc test; p < 0.0001). There was no statistically significant difference in the levels of dpERK among the vehicle-treated cultures, the E2-only cultures, and the E2 plus ICI-treated cultures (Fisher's PLSD post hoc tests; p > 0.05). In addition, treatment with ICI alone also seemed to raise the levels of dpERK to a similar degree (i.e., slightly above control levels but not enough to reach statistical significance). A dose–response curve gave also similar results. We tested (1) administration of 500 nm ICI 2 hr before E2 treatment, (2) 1 μm ICI alone, (3) 1 μm ICI plus E2, (4) 10 μm ICI alone, and (5) 10 μm ICI plus E2. In all five experiments, levels of dpERK increased above control levels within the same range as shown in lanes 3 and 4 of Figure 5, A and B, with neither reaching the level of activation induced by E2 alone (lane 2) or blocking it completely at control levels (lane 1).

Changes in the expression levels of inactive and diphosphorylated ERK1/2 in female and male basal forebrain cultures. Quantification of the expression of ERK1/2 and dpERK1/2 in female (A) and male (B) cultures is shown. The bar graph values represent the mean of four independent experiments. The asterisks in the bar graphs indicate a highly significant difference with respect to control levels. Statistical significance between the groups is described in Results. The representative Western blots detecting ERK1/2 and dpERK1/2 across the six experimental conditions are placed under the corresponding bar graphs, except the control lane (the first lane on the left), which is placed under the y-axis.
The pronounced decrease in the levels of dpERK1/2 after treatment with U0126, with and without E2, was statistically significant not only when compared with the control levels but also when compared with dpERK1/2 levels in all other groups. The levels of ERK1/2 were proportionally elevated in the U0126-treated cultures, an increase that was also statistically significant compared with controls and with the other treatment groups (ANOVA; F(5,14) = 10.006; p = 0.0003; Fisher's PLSD post hoc test).
In male cultures (Fig. 5B), treatment with E2, treatment with ICI, or the combination of both were unable to increase dpERK1/2 significantly beyond the control levels. As expected, there was a significant effect of U0126, both alone and in combination with the hormone, that reduced dpERK1/2 well below the control levels in a highly significant manner (ANOVA; F(5,14) = 14.435; p < 0.0001; Fisher's PLSD post hoc test). In the same experiments, the decrease in dpERK1/2 was accompanied by an elevation in ERK1/2, significantly different from controls (ANOVA; F(5,14) = 4.319; p = 0.0138; Fisher's PLSD post hoc test).
Because our cultures contained non-neuronal cells, we wondered whether some of the increase in phosphorylated ERK1/2 after E2 treatment could be the result of glial activation. To address this question, albeit indirectly, we stripped and reprobed the same blots used to quantify ERK1/2 with an antibody against GFAP. GFAP, the intermediate filament specific to astrocytes, is believed to be essential to astrocyte motility and morphology, because it provides structural stability in astrocytic processes (Eng et al., 2000). Although E2 has been described as a modulator of GFAP expression (Mong et al., 1996), our results (Fig. 6) indicate that GFAP protein levels in the basal forebrain cultures were not affected by any of the treatments (two-way ANOVA comparing sex and treatment; p > 0.05).

GFAP levels in basal forebrain cultures. GFAP immunostaining in vehicle-treated (A) and E2-treated (B) female cultures did not reveal any overt differences in the morphology of glial cells. Scale bar, 100 μm. C, Quantification (in average pixel value) of the levels of expression of GFAP in female and male basal forebrain cultures across different treatments. Representative Western blots are placed under the corresponding bar graphs. No change in the expression of this astrocytic marker was found.
Estrogen-induced outgrowth of cholinergic neurons in vitro is blocked by the U0126 inhibitor of the extracellular signal-regulated kinase cascade
It is well known that ERK1/2 is directly phosphorylated by MEK. To confirm a direct involvement of ERK on the increased neuritic arborization induced by E2 treatment, we repeated the experiments described above, treating female cultures with E2 for 24 hr in the presence or absence of the MEK inhibitor U0126. We verified that neurite outgrowth was blocked in the presence of the inhibitor (Fig. 7). The total neurite length per neuron (Fig. 7A) increased significantly by twofold in cultures treated with E2 (ANOVA; F(2,87) = 12.380; p < 0.0001), but post hoc Fisher's PLSD test indicated no significant difference between the control and the inhibitor-treated cultures. Comparison of the total number of segments in these experiments showed significant differences among the treatment groups (ANOVA; F(2,87) = 9.603; p = 0.0002), reflecting a significant twofold increase in the E2-treated group, and no differences between the control and the inhibitor-treated groups (Fisher's PLSD post hoc test; p > 0.05) (Fig. 7B). Regarding the distribution of branch length and number according to order (Fig. 7C,D), there was no difference between the histograms for the inhibitor-treated cultures and the vehicle-treated ones, thus providing further confirmation that the addition of the MEK inhibitor to the E2 treatment blocked the outgrowth effect at every level.

Effect of the MEK inhibitor U0126 on estrogen-induced neurite outgrowth in female basal forebrain cultures. The MEK inhibitor U0126 blocked E2-induced neurite outgrowth in female basal forebrain cultures. There was a significant twofold increase in the total length (A) and in the number of segments (C) after E2 treatment, but this effect was abolished (no outgrowth beyond control values) when U0126 was administered simultaneously. Statistical values are given in Results. B, D, Histograms showing the distribution of segment size and number by order. Branch order 1 corresponds to primary neurites and branch order 7 to the distal-most segments. Analysis of these histograms indicated that the effect of U0126 showed no preference for proximal versus distal segments and reduced both segment length and number to control values.
Discussion
Major findings in our study include E2-induced neurite outgrowth and branching in cholinergic neurons obtained from female rats. The differential effect of E2 in male and female cultures further demonstrated a sexual dimorphism in signal transduction in the CNS. E2 triggered ERK1/2 phosphorylation without affecting ERK1/2 expression, an effect blocked by the MEK inhibitor U0126, which also blocked E2-induced structural changes. These results suggest that E2 promotes the formation of neurite branches through a mechanism involving ERK1/2 phosphorylation regulated by MEK. It is interesting to note that phosphorylation of MAPK is similarly required for basic FGF-mediated axonal branch formation in cultured rat hippocampal neurons (Abe et al., 2001).
Neuroendocrine regulation of sexually dimorphic brain structures
Although some of the sexual dimorphism in the brain has been presumed to be attributable to differences in plasma levels or local brain concentrations of E2, evidence also exists for sexual dimorphisms in the cellular response to the hormone. For example, Leranth et al. (2003) recently reported that gonadectomy reduced spine density in male rats, an effect reversed with testosterone treatment but not with E2.
The importance of sex steroids during specific periods of brain development is well established (MacLusky and Naftolin, 1981; Arnold and Gorski, 1984), and gender-related differences in neuroanatomy have been described in many species. Recently, Agate et al. (2003) have provided a striking example to illustrate that sexual differentiation of nongonadal tissues in birds is attributable to differences in the local actions of sex chromosome genes. Other sexually dimorphic phenotypes influenced by the genetic sex of cells have been described previously (Reisert and Pilgrim, 1991; Carruth et al., 2002; De Vries et al., 2002). Of direct relevance to our study, the ventral paleostriatum, the origin of the cholinergic innervation of the song control nuclei in the zebra finch, contains cholinergic neurons that are of bigger size and higher density in males than in females (Sakaguchi et al., 2000). In mice, basal forebrain lesions at birth result in sex-specific changes in binding to cortical glutamatergic receptors (Hohmann et al., 1998). It is also known that endocrine regulation of sexually dimorphic brain structures is genetically controlled (Lephart et al., 2001), particularly by E2. E2 both defeminizes and masculinizes the brains of rodents when given systemically (Gorski, 1963) or intracranially (Sutherland and Gorski, 1972), and these actions are region-specific. The effect of E2 on the regulation of muscarinic receptors and ChAT activity was first described >25 years ago (Luine et al., 1975; McEwen et al., 1982; Luine, 1985), and other studies have reported sex differences in cholinergic markers both in developing and in adult animals (Luine and McEwen, 1983; Loy and Sheldon, 1987; Kornack et al., 1991; McMillan et al., 1996; Ricceri et al., 1997). This critical period for sexual differentiation of the rodent brain is demarcated by sex differences in the level of circulating gonadal steroids, which in turn determines synaptic connectivity. Although several reports have established a role for E2 in neurite growth in cultures from the hippocampus (Brinton, 1993), the neocortex (Brinton et al., 1997), the medial amygdala (Lorenzo et al., 1992), and the hypothalamus (Diaz et al., 1992; Cambiasso and Carrer, 2001), the molecular and cellular mechanisms of E2-induced structural changes remain unclear.
The MAPK signaling cascade and outgrowth
Low levels of phosphorylated ERK1/2 were detected in our control (untreated) cultures. Yet, E2-induced phosphorylation of ERK1/2 in the presence of U0126 was well below the basal levels (Fig. 5), and E2-induced neurite outgrowth and branching was abolished under these conditions (Fig. 7). This suggests that low levels of ERK1/2 phosphorylation in control cultures do not contribute to neuronal morphology. Several possible explanations can be considered. Perhaps E2-induced phosphorylation of ERK1/2 activates some specific mechanism that does not work without E2 signaling. Or, if there is a threshold for ERK1/2 to induce morphological changes, levels of ERK1/2 under control conditions may not be enough to affect neuronal morphology. Finally, ERK1/2 phosphorylation may be important but not sufficient to induce morphological changes by itself. Perhaps neurite outgrowth and branching require additional signals triggered by E2. Future work will address these questions.
Whether E2 receptors are directly involved in the activation of signaling cascades is still controversial. Extensive information on the structures of the two known ER forms clearly indicates that neither is a transmembrane protein. A variety of studies, such as the preservation of rapid E2 actions on kainate-induced neuronal currents in the hippocampus of the ERα knock-out mouse or wild-type mice treated with ICI (Gu et al., 1999), also suggest the existence of a functional membrane ER, distinct from the intracellular nuclear forms. In contrast, ER-transfected rat fibroblasts rapidly phosphorylated MAPK in response to E2, and this activation was completely blocked by ICI in ERα-transfected cells only (Wade et al., 2001).
In our cells, the effect of ICI 182,780 in MAPK cascade signaling is unclear. Because dpERK levels in E2 plus ICI-treated cultures were not significantly different from controls, it could be argued that the addition of ICI blocks E2-induced upregulation. However, for this result to be conclusive, it should be accompanied by a significant difference in dpERK levels between E2 and E2 plus ICI treatments, which was not the case. Furthermore, because dpERK levels in E2 plus ICI-treated cultures were not significantly different from those obtained after E2 treatment alone, it could be argued that ICI did not mediate inhibition, suggesting a mechanism independent of the one currently described for ERα and ERβ. This second interpretation would be in agreement with Singh et al. (1999), who reported that the rapid phosphorylation of ERK1/2 in organotypic explants of cerebral cortex treated with E2 could be blocked with a MEK inhibitor but not with ICI, and who also provided additional evidence suggesting that ERK1/2 activation by E2 is ER independent (Singh et al., 2000). A study by Cambiasso and Carrer (2001) also indicated that ICI was ineffective in blocking the E2 neuritogenic effect. Similar results have been obtained in a human neuroblastoma cell line (Watters and Dorsa, 1998). In contrast, using the SK-N-BE cell line transfected with ERα or ERβ, Patrone et al. (2000) reported that E2 induces two distinct morphological phenotypes. Neurons in the basal forebrain express both receptors (Shughrue et al., 1997), but ERα is the predominant form that colocalizes with ChAT expression (Shughrue et al., 2000). Although we have evidence for the presence of ERα and ERβ in our cell-culture lysates (R. Dominguez, personal communication), the present experimental paradigm does not lend itself to conclusions pertaining to the involvement of the intracellular receptors. Furthermore, the relative levels of both receptors were similar in our cultures of female or male origin. It is interesting to note that in explant cultures prepared from preoptic area, a sex difference in receptor density has been described, with ∼50% of the neurons containing binding sites for [3H]estradiol in females and only 20% in males (Hosli and Hosli, 1999). This difference could account perhaps for the sprouting response in female but not in male cultures, but it will require additional work to elucidate the role of ERs in cholinergic neurite outgrowth.
Is the effect of estrogen direct or mediated?
Finally, the validity of our results could be questioned because of the cellular heterogeneity of our system. We did not rule out the presence of noncholinergic neurons within our cultures, so we cannot exclude the possibility that the effects of E2 on VAChT-IR elements are mediated indirectly. In addition, astrocytes, which modulate neuronal activity (Theodosis and Poulain, 1999), are also a target of E2 (Garcia-Segura et al., 1989; Tobet et al., 1994) and contain E2 receptors (Hosli et al., 2001). Surprisingly, in contrast to the proliferative and MAPK-stimulating effects of E2 in neurons, Zhang et al. (2002) observed that E2 decreased cell proliferation in astrocytes, associated with downregulated ERK1/2, and increased cell death. If E2 were to have the same effect in our mixed cultures, this may have led us to underestimate the extent of ERK1/2 activation. We have some evidence to suggest that this is not the case, because experiments using pure glial cultures from the basal forebrain showed no additional activation of ERK1/2 beyond baseline when treated with 5 nm E2 (results not shown). In addition, acute 30 min exposure of astrocytes in culture to 50 mm ethanol has no significant effect on MAPK activity (Smith and Navratilova, 2003), allowing us to conclude that our data were not the result of different effects of ethanol (our dilution medium) on astrocytes versus neurons.
What is the physiological meaning of E2-induced neuritic outgrowth? Neuroprotective effects of estrogen are being actively investigated (Wise, 2003), and cytoarchitectural modifications by E2 may participate in neuronal regeneration under pathological conditions. In particular, E2 may play a role in supporting the survival of cholinergic neurons in neurodegenerative disorders, and newly formed neurite branches may lead to the formation of new synapses. Moreover, structural enhancement of neuronal morphology has been postulated to be an integral step in cellular processes, leading to information storage in the nervous system, and perhaps E2-induced sprouting within cholinergic neurons could underlie the cognitive enhancement effects of E2 treatment.
Footnotes
This work was supported in part by United States Public Health Service Grant AG17605 to S.L. and a Minority Biomedical Research Support-Research Initiative for Scientific Enhancement fellowship to R.D. We thank Jamil Momand (California State University Los Angeles) and Christian Pike (University of Southern California) for helpful discussions.
Correspondence should be addressed to Sonsoles de Lacalle, California State University Los Angeles, 5151 State University Drive, Los Angeles, CA 90032. E-mail: sdelacalle{at}mac.com.
DOI:10.1523/JNEUROSCI.2586-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/240982-•$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}