

Abstract
Spinocerebellar ataxia type 1 (SCA1) is an autosomal dominant, polyglutamine-induced neurodegenerative disorder that results in loss of motor coordination caused primarily by a disruption of cerebellar Purkinje cell function. In this study, we developed a conditional SCA1 mouse model to examine whether stopping expression of mutant ataxin-1 alters the disease phenotype. After cessation of SCA1[82Q] transgene expression, mutant ataxin-1, including that in nuclear inclusions, was cleared rapidly from Purkinje cells. At an early stage of disease, Purkinje cell pathology and motor dysfunction were completely reversible. After halting SCA1 expression at later stages of disease, only a partial recovery was seen. Interestingly, restoration of the ability to perform a complex motor task, the accelerating Rotarod, correlated with localization of mGluR1α to the Purkinje cell-parallel fiber synapse. These results show that the progression of SCA1 pathogenesis is dependent on the continuous expression of mutant ataxin-1. Of note, even at a late stage of disease, Purkinje cells retain at least some ability to repair the damage caused by mutant ataxin-1.
Introduction
Disorders caused by expansion of a glutamine tract within the disease-causing protein currently include: spinobulbar muscular atrophy (SBMA), Huntington's disease (HD), dentatorubral pallidoluysian atrophy (DRPLA), and spinocerebellar ataxia (SCA) types 1, 2, 3, 6, 7, and 17 (Zoghbi and Orr, 2000; Nakamura et al., 2001). Spinocerebellar ataxia type 1 (SCA1) is characterized by ataxia, dysarthria, progressive motor dysfunction, and dysphagia with degeneration of cerebellar Purkinje cells, inferior olive neurons, and deep cerebellar nuclei.
The mechanism of SCA1 pathogenesis is being elucidated. Studies in transgenic mice support a dominant, toxic gain of function mechanism caused by expression of the SCA1 encoded mutant ataxin-1 protein. Overexpression of mutant ataxin-1 with 82 glutamines causes ataxia and Purkinje cell degeneration in transgenic mice (Burright et al., 1995; Clark et al., 1997). In contrast, deletion of endogenous murine Sca1 gene did not lead to any signs of overt ataxia or Purkinje cell pathology (Matilla et al., 1998). Development of nuclear inclusions consisting of the polyglutamine protein and other neuronal proteins is a pathological hallmark of most polyglutamine disorders (Ross, 1997; Zoghbi and Orr, 2000). In SCA1 mice, although mutant ataxin-1 must enter the nucleus to cause disease, progressive neuronal dysfunction can develop in the absence of detectable nuclear inclusions (Klement et al., 1998). Alterations in protein folding and degradation pathways also impact disease severity (Cummings et al., 1999, 2001; Fernandez-Funez et al., 2000). Recently, it was shown that phosphorylation of mutant ataxin-1 at serine 776 is critical for pathogenesis by enhancing the stability of ataxin-1 via its interaction with the protein 14-3-3 (Chen et al., 2003; Emamian et al., 2003).
Disease time course is well characterized in the SCA1[82Q] mice (Burright et al., 1995; Clark et al., 1997; Skinner et al., 2001). Importantly, Purkinje cell loss is not significant until after 6 months, well beyond the onset of severe neurological deficits. Thus, the neurological alterations in SCA1 mice are not the result of Purkinje cell death. The long time between the onset of neurological dysfunction and Purkinje cell death in SCA1 mice raises the question of whether the dysfunctional neurons are able to recover if mutant ataxin-1 expression were to be halted.
Previously, Yamamoto et al. (2000) demonstrated that in a conditional mouse model of HD, pathology was halted and perhaps reversed after the cessation of the expression of an N-terminal fragment of mutant huntingtin. Besides detecting a reversal of neuropathology, these investigators found that the inclusions of the mutant polyglutamine peptide disappeared by a proteasomal-dependent pathway (Martin-Aparicio et al., 2001), demonstrating that at least at one stage of disease neurological symptoms were reversible.
To address the general point of whether disease reversal is a property shared by the polyglutamine disorders, we created a conditional mouse model of SCA1 expressing intact mutant ataxin-1. We found that the ability of Purkinje cells in conditional SCA1[82Q] mice to recover decreased with duration of disease. At an early stage, disease was completely reversible. In contrast, halting ataxin-1 expression at a later stage of disease yielded a partial recovery.
Materials and Methods
Conditional SCA1 transgenic mice. Transgenes were purified as described (Burright et al., 1995). The transgene was injected into FVB/N embryos, which were implanted into pseudopregnant FVB/N females. Offspring were screened for presence of the transgene by Southern blot or PCR analysis of tail DNA. Mice were given 2 mg/ml doxycycline (dox) in a 5% sucrose solution in their drinking water.
Immunostaining of mouse cerebellum. The extent of Purkinje cell dendritic arborization was assessed by calbindin immunostaining and measurement of the thickness of the molecular layer. Immunofluorescence was done as described (Klement et al., 1998). After blocking with 2% normal goat serum, sections were incubated for 48 hr at 4°C with anti-ataxin-1 11750 (1:1000) or 11NQ (1:1000), or anti-calbindin (1:1000) antibodies, and incubated with anti-mouse-conjugated Alexa Fluor 488 (Molecular Probes, Eugene, OR) or anti-rabbit-conjugated Cy3 (Jackson ImmunoResearch, West Grove, PA) followed by washing and mounting. Images were collected on a confocal microscope (MRC 1000; Bio-Rad, Hercules, CA).
Western blotting analysis. Cerebella were homogenized in 0.25 m Tris-HCl, pH 7.5, containing protease inhibitors (protease inhibitor mixture tablet; Roche, Mannheim, Germany). The homogenate was frozen in liquid nitrogen and then thawed at 37°C. Freeze-thaw cycle was repeated two more times. The lysate was centrifuged at 6000 × g for 1 min at 4°C, and the supernatant was collected. Ten micrograms of protein were separated in a 3-8% NuPAGE Tris-acetate gel (Invitrogen, San Diego, CA) and transferred to nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). The membrane was blocked in 5% dry milk in PBS containing 0.05% Tween 20 and probed with the anti-mGluR1α antibody (Calbiochem, La Jolla, CA) or anti-α-tubulin antibody (Sigma, St. Louis, MO) in blocking solution. After incubating the membrane with anti-rabbit HRP or anti-mouse HRP-conjugated secondary antibody (Amersham Biosciences, Arlington Heights, IL), bands were visualized by chemiluminescence (Renaissance kit; NEN, Boston, MA). The amount of mGluR1α was quantified by Western blotting using the Molecular Analyst software (Bio-Rad). The amount of mGluR1α present (monomer and dimer together), relative to the level of tubulin, is expressed as the percentage of the mGluR1α in wild-type mice.
RNA analysis. For Northern blot analysis, RNA was isolated from one-half of a cerebellum using Trizol reagent (Invitrogen) following the manufacturer's recommended protocol. RNA was electrophoresed in the presence of glyoxal, blotted to a nylon membrane, and probed with an ataxin-1 cDNA. Blots were subsequently probed with a GAPDH cDNA probe to ascertain the amount of RNA loaded per lane. The amount of transgene RNA was normalized to the amount of GAPDH RNA in each lane.
Assessment of motor ability. To assess motor skill by home cage behavior, three mice from each group were videotaped for 10 min. Two observers, blinded to whether the mice were treated with dox viewed the videotapes, and scored the mice on the severity of their motor phenotype. Criteria used were the ability to rear and maintain balance when standing on their hindlegs, amount of head swaying, and alterations in gait. Motor ability was also assessed using an accelerating Rotarod apparatus, as described (Clark et al., 1997). Mice were tested using four trials per day (10 min rests between trials) on 4 consecutive days. During a trial, the rod accelerated from 4 to 40 rpm over 5 min and then remained at 40 rpm for an additional 5 min. Each trial lasted until the mouse fell from the rod or for a maximum of 10 min. Statistical significance was assessed by the Student's t test.
Results
Generation of a conditional model of SCA1
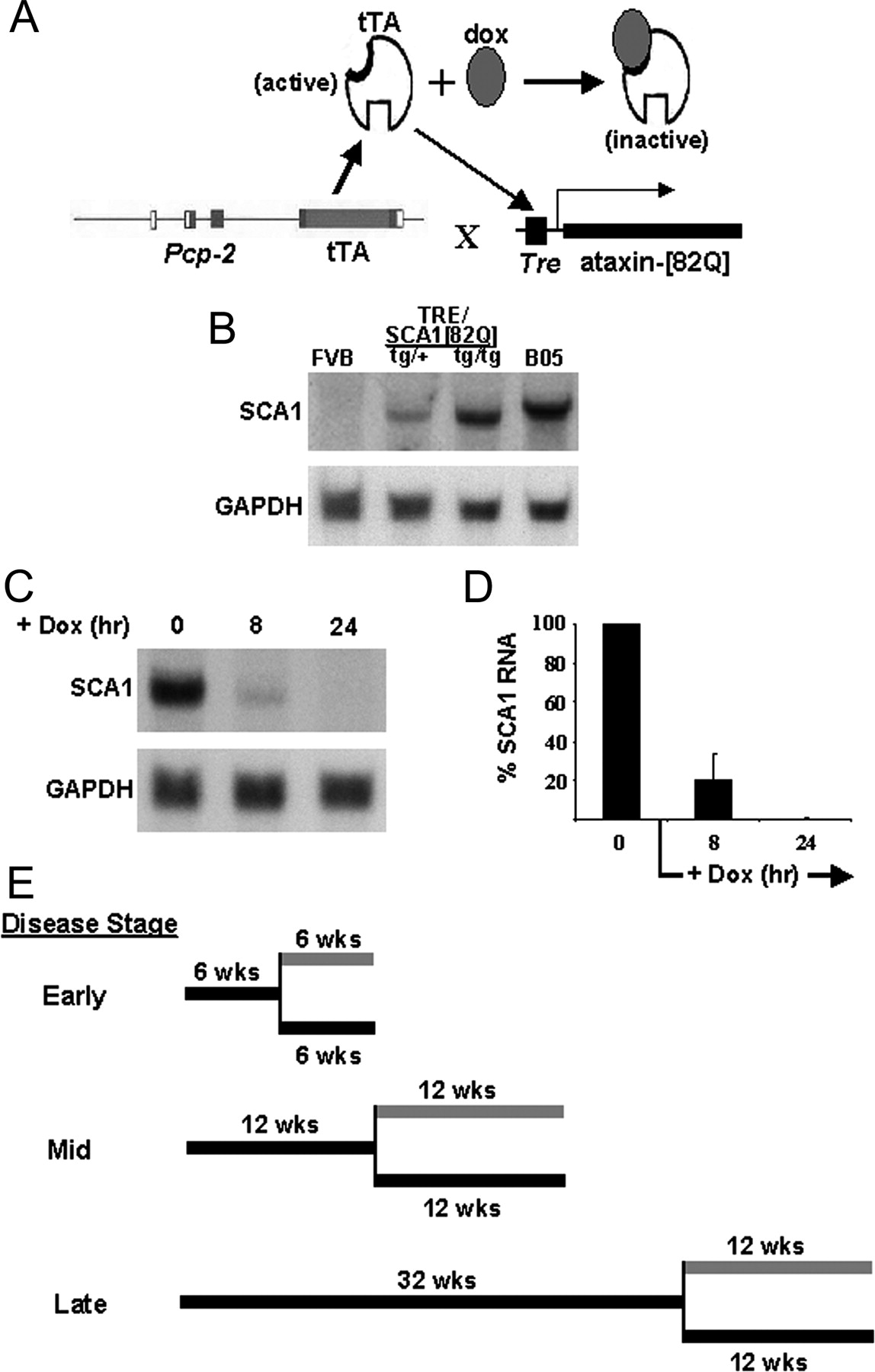
To create a conditional transgenic mouse model of SCA1, we used an adaptation of the tetracycline-regulated system (Gossen and Bujard, 1992). This system has been used to establish the inducible expression of individual genes in mammalian cells and transgenic animals (Yamamoto et al., 2001). In the absence of tetracycline, tTA binds to tetO elements, inducing expression of the respective downstream gene. In the presence of tetracycline or its derivative doxycycline, the ability of the tTA protein to bind tetO sequences is abolished because of a conformational change in tTA brought about by the interaction of tetracycline with the tTA. Thus, expression of the respective gene is suppressed. To generate a transgenic mouse expressing full-length ataxin-1[82Q] in a tet-regulated manner, two transgenic lines were required (Fig. 1A). The first line was constructed to express tTA in a Purkinje cell-specific manner using the Pcp2/L7regulatory region (Oberdick et al., 1990; Vandaele et al., 1991). The second line was constructed to contain the target gene, a SCA1 cDNA encoding a mutant polyglutamine tract with 82 repeats, joined to a TRE element. The two lines were crossed to place both genes in a single mouse.

Conditional SCA1 mouse design and expression. A, tTA expression was driven by the Purkinje cell regulatory region from the murine Pcp2/L7 gene. Active tTA binds to the TetO sequence to induce expression of the SCA1[82Q] transgene. The presence of doxycycline inhibits expression of SCA1[82Q] by binding to tTA, making it inactive. B, Cerebellar mRNA levels in conditional SCA1 mice compared with those in B05 SCA1[82Q] animals. To reach levels comparable to those in B05 mice, the TetO-SCA1[82Q] transgene had to be bred to homozygosity. C, A Northern blot of cerebellar mRNA showing the loss of SCA1[82Q] mRNA after administering dox to 12-week-old conditional SCA1[82Q] mice for 8 and 24 hr. D, Quantitative analysis of SCA1[82Q] mRNA levels in 12-week-old conditional SCA1[82Q] mice given dox for 8 and 24 hr. Error bars indicate SEM. E, A scheme depicting the experimental strategy used to assess the ability of Purkinje cells to recover from the effects of mutant ataxin-1. The experimental design is shown for assessing recovery at three stages of disease: early, middle, and late. Black bars depict periods of SCA1[82Q] gene-on, and gray bars depict periods of SCA1[82Q] gene-off. The vertical black line denotes the age at which dox was administered to the experimental group of mice for each disease stage.
Nine Pcp2-tTA founder animals were identified by Southern blot analysis, and lines were established from each. To determine the level of transgene expression in these Pcp2-tTA transgenic mice, Northern blotting on cerebellar extracts was performed. The line with the highest level of tTA RNA was selected and bred to each of the seven TRE-SCA1[82Q] founders generated. To obtain SCA1 mRNA levels comparable to those seen in the well characterized SCA1 B05 line (Burright et al., 1995), mice were bred to homozygosity for the TRE-SCA1[82Q] transgene (Fig. 1B).
To assess the ability of dox to silence SCA1 expression, 12-week-old Pcp2-tTA(tg/+)/TRE-SCA1[82Q](tg/tg) mice were treated with 2 mg/ml dox in their drinking water. Total RNA was isolated from cerebella after 8 and 24 hr of dox administration. Northern blot analyses revealed that by 8 hr of dox treatment the amount of SCA1 RNA had decreased by 80%. By 24 hr of dox treatment SCA1 mRNA was essentially undetectable (Fig. 1C,D). Thus, in these mice dox administration was very effective in rapidly silencing cerebellar transcription of the TRE-SCA1[82Q] transgene.
Reversal of disease in conditional SCA1 mice: experimental design
The ability of the conditional SCA1[82Q] mice to recover from disease after stopping transgene expression was examined at an early, middle, and late stages of disease (Fig. 1E). For the early stage of disease Pcp2-tTA(tg/+)/TRE-SCA1[82Q](tg/tg) mice, designated as the conditional SCA1[82Q] mice, were aged for 6 weeks with the SCA1[82Q] gene on, at which time the gene was then turned off. This age was selected because at 6 weeks of age, conditional SCA1[82Q] mice showed a moderate deficiency on the accelerating Rotarod and no signs of neurological abnormality by home cage behavior. These mice also had a mild Purkinje cell pathology that included cytoplasmic vacuoles and some dendritic pruning, heterotopic Purkinje, and nuclear inclusions. The ability of animals to recover from mid-stage disease was assessed by allowing conditional expression of the SCA1[82Q] transgene up to 12 weeks of age before shutting it off. At 12 weeks, conditional SCA1[82Q] animals began to show signs of ataxia by home cage behavior and had a significantly impaired Rotarod performance. In addition, Purkinje cell pathology in 12-week-old conditional SCA1[82Q] mice was quite extensive. To assess recovery potential at a late stage of disease, conditional SCA1[82Q] mice were allowed to age until 32 weeks with the SCA1[82Q] gene on. These animals had overt ataxia by home cage behavior, were unable to stay on the accelerating Rotarod, and had dramatic Purkinje cell pathology, but no noticeable Purkinje cell loss.
At each disease stage, conditional SCA1[82Q] mice were divided into three groups. In the first group disease status was assessed at the age at which the SCA1[82Q] transgene was turned off: 6 weeks for the early stage, 12 weeks for the middle, and 32 weeks for the late stage. In the other two groups, disease status was monitored as the mice aged with the SCA1[82Q] transgene either remaining on or shut off (Fig. 1E). The gene-off group was administered 2 mg/ml dox plus 5% sucrose in their drinking water. The length of this period of gene-off was either 6 weeks for animals at the early disease stage or 12 weeks for the animals at the middle and late stages. At each stage, the disease control group was given 5% sucrose in their drinking water and designated the gene-on mice. In addition, a group of nontransgenic mice were administered dox in their drinking water for the full extent of the gene-off period.
Disease reversal at an early stage of disease
Six-week-old mice allowed to age for an additional 6 weeks with the SCA1[82Q] gene on demonstrated a progressive deterioration in their ability to perform on the accelerating Rotarod (Fig. 2). In contrast, within 2 weeks of administering dox to 6-week-old conditional SCA1[82Q] mice, this progressive loss of performance on the Rotarod had stopped and returned to that of age-matched controls (Fig. 2A,C). Moreover, the dox-treated conditional SCA1[82Q] animals showed no signs of ataxia by home cage behavior.

Restoration of a normal performance level on the Rotarod in conditional SCA1[82Q] mice treated with dox beginning at 6 weeks of age. A, Performance on the accelerating Rotarod of 6 week gene-on—2 week gene-off mice compared with that of 8 week gene-on mice. Error bars indicate SEM. B, Performance on the accelerating Rotarod of 6 week gene-on—4 week gene-off mice compared with that of 10 week gene-on mice. Error bars indicate SEM. C, Performance on the accelerating Rotarod of 6 week gene-on—6 week gene-off mice compared with that of 12 week gene-on mice. Error bars indicate SEM.
Restoration of a normal neurological phenotype in the 6-week-old mice from the conditional SCA1[82Q] line was accompanied by a similar reversal of Purkinje cell pathology (Fig. 3). Purkinje cells in mice in which the SCA1[82Q] transgene was expressed for 6 weeks showed signs of mild pathology (Fig. 3A). These included some dendritic atrophy, as assessed by both calbindin immunostaining (Fig. 3B) and measurement of the thickness of the molecular layer (Fig. 3F), and heterotopic Purkinje cell bodies that became considerably worse by 12 weeks of expression. In contrast, morphology of the Purkinje cells in the 6 week gene-on—6 week gene-off mice was dramatically improved (Fig. 3C), approaching that seen in the 12-week-old nontransgenic mice (Fig. 3D). In the dox-treated conditional SCA1[82Q] animals, the molecular layer was not noticeably reduced in thickness, and the dendritic arborization, although not as extensive as seen in the 12 week nontransgenic mice, was more elaborate than in the 6-week-old SCA1[82Q] mice (Fig. 3, compare C,A). Interestingly, one morphological feature that was not corrected was the presence of heterotopic Purkinje cell bodies in the molecular layer that remained in the 6 week gene-on—6 week gene-off mice. Overall, stopping SCA1[82Q] expression at this early stage of disease resulted in an essentially complete recovery of a normal phenotype by both neurological and morphological measures.

Restoration of normal Purkinje cell morphology in 6 week gene-on—6 week gene-off conditional SCA1[82Q] mice. A, In 6 week gene-on mice, calbindin immunostaining revealed a detectable degree of Purkinje cell dendritic atrophy. B, Calbindin immunostaining showing the severe level Purkinje cell atrophy in 12 week gene-on mice. C, In 6 week gene-on—6 week gene-off animals, calbindin immunostaining showed an essentially normal Purkinje cell morphology with the typical robust dendritic trees. D, Calbindin immunostaining showing Purkinje cell morphology in 12-week-old wild-type mice. E, A diagram of a mid-sagittal cerebellar section depicting the posterior location of the folia presented in A-D. F, Thickness of the cerebellar molecular layer in conditional SCA1 mice. Thickness of the molecular layer was determined by measuring the distance between the Purkinje cell body layers and dividing in half (n = number animals measured).
Disease reversal at a middle stage of disease
To determine if the recovery capability of mice decreased with disease progression, we next examined the effect of halting ataxin-1[82Q] expression at a middle stage of disease.
Twelve-week-old conditional SCA1[82Q] mice were divided into a dox-treated group and an untreated disease control group (Fig. 1E). The disease status of both groups was assessed at intervals of 4 weeks. The period on dox was limited to 12 weeks because of the development of diabetes and polyuria in treated animals, typically induced in rodents on a sucrose-rich diet (Brenner et al., 2003).
Whereas 12-week-old conditional SCA1[82Q] mice administered dox for 4 weeks (i.e., 12 week gene-on—4 week gene-off mice), showed signs of neurological improvement by home cage behavior, these mice showed no improvement in their performance on the accelerating Rotarod. Mice in which SCA1[82Q] was on for 12 weeks and off for 4 weeks had a deficit on the Rotarod that matched that of 12 week and 16 week gene-on conditional SCA1[82Q] mice (Fig. 4A). After 8 weeks of treatment (12 week gene-on—8 week gene-off mice), Rotarod performance improved (Fig. 4B). However, the level of performance did not equal that of the nontransgenic mice at 19 weeks of age. Moreover, an additional 4 weeks of dox administration did not result in any further improvement in Rotarod performance. The 12 week gene-on—12 week gene-off mice had the same level of performance on the Rotarod as did the 12 week gene-on—8 week gene-off mice (Fig. 4B). Thus, based on their Rotarod performance, mice in which mutant ataxin-1 expression was halted at 12 weeks of age exhibited only a partial recovery from disease.

A, In 12 week gene-on—4 week gene-off mice, Rotarod performance was not improved compared with the performance level of 12 week gene-on and 16 week gene-on mice. Error bars indicate SEM. B, In 12 week gene-on—8 week gene-off and 12 week gene-on-12 week gene-off animals, their performance on the Rotarod improved above the performance level of 20 week gene-on and 24 week gene-on mice, but did not reach the performance level of 19-week-old wild-type nontransgenic FVB/N animals.
When SCA1[82Q] remained on from 12 to 24 weeks of age there was progressive atrophy of Purkinje cell bodies and dendrites (Fig. 5A-D). In contrast, halting SCA1[82Q] expression for 4 weeks (12 week gene-on—4 week gene-off) halted the progressive Purkinje cell atrophy (Fig. 5E). After halting SCA1[82Q] expression further, for 8 weeks (Fig. 5F) and 12 weeks (Fig. 5G), the cerebellar molecular layer showed an increase in thickness (Fig. 3F) and arborization of the Purkinje cell dendritic tree. These data clearly demonstrate that cessation of SCA1[82Q] expression at this middle stage of disease halted the further progression of Purkinje cell disease. Moreover, within the 12 week time frame in which SCA1[82Q] remained off in this experiment, there was a partial restoration of the normal phenotype based on Rotarod performance and Purkinje cell morphology.
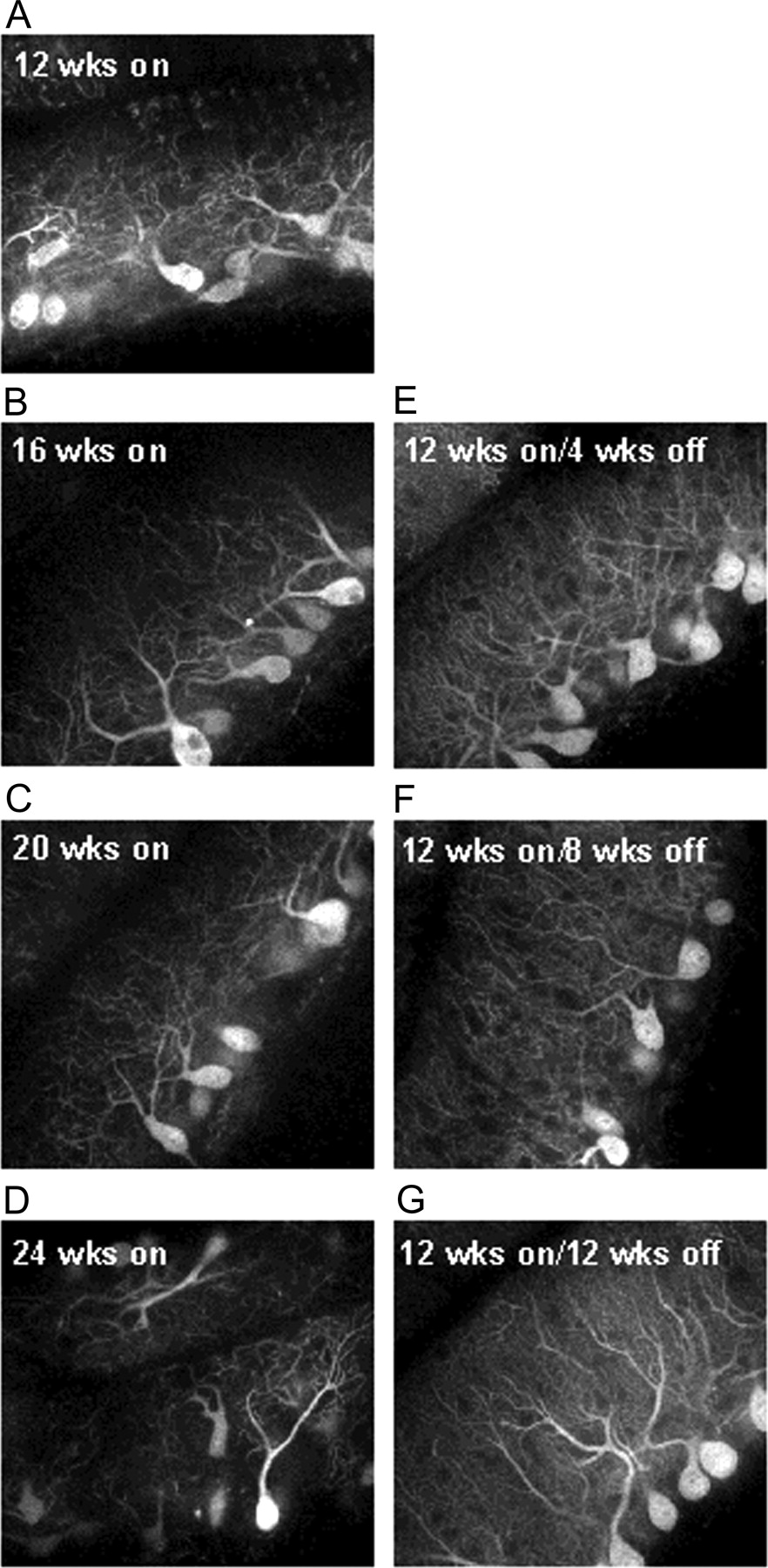
Halting SCA1[82Q] expression at 12 weeks of age stopped progression of pathology, and Purkinje cells showed signs of morphological recovery. A—D depicts the progression in Purkinje cell pathology, as revealed by calbindin immunostaining, that occurred in SCA1[82Q] gene-on mice that were aged from 12 to 24 weeks of age. E—G shows that when the in SCA1[82Q] gene was shut-off in 12-week-old mice for 4 weeks (E), 8 weeks (F), and 12 weeks (G), not only was the progression of Purkinje cell pathology halted, but with longer gene-off periods the dendritic morphology of Purkinje cells improved considerably.
Motor recovery is associated with appearance of mGluR1 at the Purkinje cell-parallel fiber synapse
To better correlate changes in Purkinje cell morphology with restoration of motor coordination, we examined the 12 week gene-on-4 week gene-off and 8 week gene-off mice in more detail. Based on previous work, we focused on two aspects of Purkinje cell dendritic morphology, spine density, and localization of mGluR1 at the Purkinje cell-parallel fiber synapse. mGluR1 is expressed at a high level in Purkinje cell dendritic spines that are innervated by parallel fibers (Takacs et al., 1997). Importantly, the presence of mGluR1 in Purkinje cells is essential for proper motor coordination and synaptic plasticity at the Purkinje cell-parallel fiber synapse (Aiba et al., 1994; Ichise et al., 2000).
Figure 6A shows the spine density along Purkinje cell dendrites in a 12-week-old wild-type FVB mouse that are richly decorated with mGluR1, as assessed by immunostaining for mGluR1α (Fig. 6B). In mice, n = 4 examined, in which SCA1[82Q] was expressed for 12 weeks, there was a detectable decrease in dendritic arborization and distribution of spines along the remaining dendrites (Fig. 6C). However, mGluR1α was detected at the Purkinje cell-parallel fiber synapses remaining (Fig. 6D). Despite not being able to perform the accelerating Rotarod task (Fig. 4), Purkinje cell dendritic arborization and spine density in 12 week gene-on-4 week gene-off (Fig. 6E) appeared to be identical to both 12-week-old control mice (Fig. 6A) and 12 week gene-on-8 week gene-off mice (Fig. 6G). Thus, an increase in dendritic arborization and spine density alone was not sufficient to restore performance on the accelerating Rotarod. Interestingly, mGluR1α immunostaining of the molecular layer of 12 week gene-on-4 week gene-off mice was similar to that of 12 week gene-on mice (Fig. 6F). In contrast, in 12 week gene-on-8 week gene-off mice, mGluR1α was found to be present at Purkinje cell-parallel fiber synapses throughout the molecular layer (Fig. 6H). Consistent with the immunostaining, Western blot analysis showed that the amount of mGluR1α was reduced, compared with wild-type, in the cerebellum of 12 week gene-on and 12 week gene-on-4 week gene-off mice. In contrast, the level of mGluR1α expression of 12 week gene-on-8 week gene-off mice was identical to that in 12 week wild-type mice (Fig. 6I,J). These data show that, in addition to dendritic arborization and spine density, localization of mGluR1α to the Purkinje cell-parallel fiber synapse has a role in restoration of a complex motor task, i.e., performance on the accelerating Rotarod.

Recovery on the Rotarod correlates with appearance of mGluR1α at the Purkinje cell-parallel fiber synapse. A, Calbindin immunostaining showing Purkinje cell dendritic arborization and spine density in 12-week-old wild-type mice. B, Calbindin immunostaining (green) and mGluR1α staining (red) showing the presence of mGluR1α at Purkinje cell-parallel fiber synapses in 12-week-old wild-type mice. C, Calbindin immunostaining showing the decreased amount Purkinje cell dendritic arborization and spine density in 12 week gene-on mice. D, Calbindin immunostaining (green) and mGluR1α staining (red) showing the presence of mGlur1α at the remaining Purkinje cell-parallel fiber synapses in 12 week gene-on mice. E, Calbindin immunostaining showing restoration of Purkinje cell dendritic arborization and spine density in 12 week gene-on—4 week gene-off mice. F, Calbindin immunostaining (green) and mGluR1α staining (red) showing the lack of mGluR1α at Purkinje cell-parallel fiber synapses in 12 week gene-on—4 week gene-off mice; n = 4 mice. G, Calbindin immunostaining showing restoration of Purkinje cell dendritic arborization and spine density in 12 week gene-on—8 week gene-off mice; n = 4 mice. H, Calbindin immunostaining (green) and mGlur1α staining (red) showing the presence of mGlur1α at the remaining Purkinje cell-parallel fiber synapses in 12 week gene-on—8 week gene-off mice. I, Western blot showing mGluR1α expression in cerebella of 12 week wild-type, 12 week gene-on, 12 week gene-on-4 week gene-off, and 12 week gene-on-8 week gene-off mice.α-Tubulin blots shown as controls of total protein loading. J, Quantification of mGluR1α expression in cerebella of 12 week wild-type, 12 week gene-on, 12 week gene-on-4 week gene-off, and 12 week gene-on-8 week gene-off mice. The amount of mGluR1α, both monomer and dimer together, is expressed as the percentage of the mGluR1α in wild-type mice. Error bars indicate SEM (n = 3).
Disease reversal at a late stage of disease
In another series of experiments the ability of Purkinje cells to recover from a late stage of disease was examined by turning off SCA1[82Q] in 32-week-old mice (Fig. 1E). Thirty-two-week-old conditional SCA1[82Q] animals in which the transgene was conditionally expressed developed a very severe ataxia based on home cage behavior and performance on the accelerating Rotarod. Mice in which SCA1[82Q] expression was halted by dox at 32 weeks of age showed some signs of an improved neurological phenotype only after an extended gene-off period. By home cage behavior and Rotarod assessments (Fig. 7A), 32 week gene-on-4 week gene-off mice showed no signs of improvement in their neurological status, remaining severely ataxic. After extension of the gene-off period to 8 and 12 weeks, the conditional SCA1[82Q] mice showed a progressing improvement in their home cage behavior such that by 12 weeks with the SCA1[82Q] gene off, the animals had a mild form of ataxia by home cage behavior similar to that manifested by 12 week gene-on mice. On the Rotarod 32 week gene-on—8 week gene-off and 32 week gene-on—12 week gene-off mice showed consistent signs of improved performance (Fig. 7B,C, respectively). However, these changes failed to reach statistical significance.

Conditional SCA1[82Q] mice treated with dox beginning at 32 weeks of age failed to show evidence of significant improvement on the Rotarod. A, The Rotarod performance of 32 week gene-on—4 week gene-off and the performance of 36-week-old gene-on mice were equally compromised. Error bars indicate SEM. B, Although the Rotarod performance of 32 week gene-on—8 week gene-off mice was slightly better than the performance of the 40 week gene-on mice on trial days 3 and 4, it did not reach statistical significance. Error bars indicate SEM. C, The Rotarod performance of 32 week gene-on—12 week gene-off mice was better than the performance of 44 week gene-on mice on all four trial days, yet, it failed to reach statistical significance. Error bars indicate SEM.
Pathologically, 32-week-old conditional SCA1[82Q] mice have very severe Purkinje cell atrophy (Fig. 8A). In contrast, mice in which SCA1[82Q] expression was halted for 12 weeks beginning at 32 weeks of age demonstrated a considerable restoration of Purkinje cell morphology, as determined by calbindin immunostaining (Fig. 8C). The intensity of staining increased noticeably. In many instances, the thickness of the molecular layer and arborization of Purkinje cell dendritic tree increased. There was a corresponding decrease in the number of Purkinje cells with cytoplasmic vacuoles and nuclear inclusions. Overall, the status of the cerebellar cortex in the 32 week gene-on—12 week gene-off mice had an appearance similar to the cerebellum of 12 week gene-on animals (Fig. 8D), indicating that even at this late stage of disease Purkinje cells have at least some ability to recover from the effects of ataxin-1[82Q]. Intriguingly, as seen in the reversal at an early and middle stage of disease, the presence of heterotopic Purkinje cell bodies in the molecular layer remained in the 32 weeks gene-on—12 weeks gene-off mice (Fig. 8C).

Halting SCA1[82Q] expression at 32 weeks of age stopped progression of pathology, and Purkinje cells showed some signs of morphological recovery. A, B, The degree of Purkinje cell pathology as revealed by calbindin immunostaining in a posterior folium from three conditional gene-on SCA1[82Q] mice at ages of 32 and 44 weeks, respectively. C, The calbindin immunostaining of Purkinje cells in a posterior folium (Fig. 3E) from five 32 week gene-on—12 week gene-off mice showed a considerable restoration in the intensity of calbindin staining and arborization of Purkinje cell dendritic trees. D, The calbindin immunostaining of Purkinje cells in a posterior folium (Fig. 3E) from four 12 week gene-on.
Clearance of ataxin-1[82Q] in response to dox administration
Evidence indicates that glutamine tract expansion compromises the ability of a cell to degrade the polyglutamine protein and perhaps other proteins (Kaytor and Warren, 1999; Bence et al., 2001). Thus, it was of interest to assess the clearance of mutant ataxin-1 in the conditional SCA1[82Q] mice with disease progression. However, the ability to assess clearance of mutant ataxin-1 by Purkinje cells was complicated by the fact that mutant ataxin-1 cannot be quantitatively extracted for Western blot analysis (Burright et al., 1995). Thus, ataxin-1 expression after dox administration was examined by immunostaining. As shown in Figure 9A (column 1), mutant ataxin-1 localized to the nuclei of Purkinje cells in 8-, 16-, and 30-week-old conditional SCA1[82Q] mice. At all ages, the amount of diffuse nuclear ataxin-1[82Q] detectable in Purkinje cells by Ab11750 immunofluorescence decreased to levels seen in nontransgenic FVB/N mice within 2 d after administering dox (Fig. 9A, columns 2, 3). These results suggest that Purkinje cells can readily clear mutant ataxin-1 after cessation of SCA1 expression.

Ataxin-1 response to dox administration in Pcp2-tTA(tg/+)/TRE-SCA1[82Q](tg/tg) mice. A, Ataxin-1 Ab 11750 immunostaining revealed a decrease in ataxin-1[82Q] after a 2 d treatment with dox. The first column shows the 11750 immunostaining in Purkinje cells in an anterior folium from 8-, 16-, and 30-week-old conditional SCA1[82Q] mice. After 2 d of dox administration the level of 11750 staining (second column) had decreased to that in nontransgenic wild-type FVB/N mice (third column). B, The time course for the loss of ataxin-1[82Q] intranuclear inclusions from Purkinje cells in 16-week-old conditional SCA1[82Q] mice treated with dox for 2, 6, and 10 d. Error bars indicate SEM.
As is the case for most other mutant polyglutamine proteins, ataxin-1[82Q] accumulates into a single large nuclear inclusion in Purkinje cells of transgenic mice (Skinner et al., 1997). Evidence indicates that these inclusions form to some extent by the relative inability of the cell to degrade the polyglutamine protein (Cummings et al., 1998). Clearance of nuclear inclusions of ataxin-1[82Q] was examined in dox treated Pcp2-tTA(tg/+)/TRE-SCA1[82Q](tg/tg) mice by immunostaining using the anti-ataxin-1 antibody 11NQ, which preferentially stains ataxin-1 in nuclear inclusions (Skinner et al., 1997). In conditional SCA1[82Q] mice treated with dox beginning at 16 weeks of age, the frequency of Purkinje cells with detectable nuclear inclusions of ataxin-1[82Q] decreased from 50 to 20% after 2 d on dox (Fig. 9B). Furthermore, the size of nuclear inclusions was considerably smaller in Purkinje cells with detectable inclusions after 2 d on dox (data not presented). After 10 d of dox treatment, the frequency of Purkinje cells with detectable nuclear inclusions of any size was <1% (Fig. 9B). If SCA1[82Q]-induced disease was allowed to progress to a much more severe stage, out to 32 weeks of age, the frequency of Purkinje cells with detectable nuclear inclusions decreased from 63 to 8% after a 12 week exposure to dox. Thus, at all stages of disease examined, formation of nuclear inclusions of ataxin-1[82Q] was reversible.
Discussion
We developed a conditional mouse model of the polyglutamine neurodegenerative disease SCA1 using the regulatory region from the Pcp2 (L7) gene (Oberdick et al., 1990; Vandaele et al., 1991) to direct expression of the tetracycline-responsive activator, tTA, to cerebellar Purkinje cells, a prominent site of pathology in SCA1 (Koeppen, 1998). Similar to results reported using a conditional mouse model of HD expressing an N-terminal fragment of mutant huntingtin (Yamamoto et al., 2000), we found that in conditional SCA1 mice the pathogenic effects of a full-length mutant polyglutamine protein could be reversed after cessation of ataxin-1 expression. In addition, the ability of Purkinje neurons to recover from polyglutamine-induced disease decreased with age and progression of disease. Moreover, the age-related decrease in ability to recover was not associated with a reduced ability of older animals to clear mutant ataxin-1.
Oral administration of dox was proficient in terminating expression of the SCA1[82Q] transgene, resulting in a fast, within hours, decrease in the level of cerebellar SCA1[82Q] RNA (Fig. 1C,D). Clearance of diffuse nuclear mutant ataxin-1 from Purkinje cells was rapid after SCA1 expression was halted. After 2 d on dox, ataxin-1 detectable by Ab11750-immunostaining in Purkinje cells dropped to background levels (Fig. 9). In addition, the ability of Purkinje cells to clear mutant ataxin-1 did not seem to vary with disease progression from 8 to 30 weeks, a span of time when there was a dramatic decrease in neurological function and worsening of pathology. Mutant ataxin-1 localized to the nuclear inclusions was also readily cleared from Purkinje cells, having an apparent half-life of 2 d at 16 weeks of age. This result is consistent with an earlier observation using a conditional model of HD in which inclusions formed by a fragment of mutant huntingtin were cleared from the brain (Yamamoto et al., 2000), supporting the concept that formation of polyglutamine nuclear inclusions is not irreversible. Considerable evidence links the formation of nuclear inclusions to an inability of the protein to be properly degraded. In tissue culture cells inhibition of the proteasome increases formation of nuclear inclusions containing mutant polyglutamine protein. It was suggested that inclusions may directly block proteasome function (Bence et al., 2001). The conditional SCA1 mice revealed that Purkinje cells are capable of removing inclusions at a relatively rapid rate. Even after an extended period of ataxin-1[82Q] expression, despite having undergone considerable atrophy, Purkinje cells still were able to clear mutant ataxin-1 nuclear inclusions after cessation of SCA1[82Q] expression. Thus, if mutant ataxin-1 has a negative effect on the degradative machinery of a neuron, it is not irreversible, even at a late stage of disease.
The capacity of Purkinje neurons to recover from the adverse effects of ataxin-1[82Q] was examined at three stages of disease; at an early stage after 6 weeks of SCA1[82Q] expression, at a middle stage after 12 weeks of SCA1[82Q] expression, and at a late stage after 32 weeks of SCA1[82Q] expression. The extent to which Purkinje cells recovered decreased as the length of their exposure to ataxin-1[82Q] increased and pathology became progressively worse. When expression of ataxin-1[82Q] was stopped early in disease, after 6 weeks of SCA1[82Q] expression, disease progression was quickly halted, and neurological status improved such that the mice recovered to an essentially normal neurological phenotype and Purkinje cell morphology. One abnormal morphological feature that failed to show signs of improvement was the presence of heterotopic Purkinje cells in the molecular layer. We suggest that the failure of the mice to correct this feature is consistent with an earlier suggestion that the heterotopic Purkinje cells are a compensatory response to the dendritic pruning induced by mutant ataxin-1 (Clark et al., 1997).
In contrast to the full recovery from an early stage of disease, at later stages, we found a more protracted Purkinje cell recovery. Conditional SCA1[82Q] mice in which gene expression was ceased at 12 weeks of age, a middle stage, after ataxia was evident by both Rotarod and home cage assessments, demonstrated a partial recovery within the 12 week gene-off period. Mice in which the gene remained on until a much later stage of disease, 32 weeks when the animals had severe ataxia by Rotarod and cage behavior in addition to very extensive Purkinje cell pathology, showed the least amount of recovery during the 12 weeks of gene-off. Thus, the ability of the conditional SCA1 mice to recover decreased with age and disease progression. The extent to which the decreased ability of the older mice to recover was attributable to effects of disease progression versus effects of age remains to be determined. Nevertheless, it is important to note that within the confines of the experimental design, the disease process never became irreversible. Even at a very late stage, after Purkinje cells have undergone a substantial degree of atrophy, disease progression still required continuous expression of mutant ataxin-1.
Not only was disease progression halted after stopping SCA1[82Q] expression late in disease, neurological function and pathology also showed signs of improvement. First, an improvement in their cage behavior was seen. By 12 weeks of gene-off, their cage behavior had progressively improved from a very severe ataxia to a mild ataxia similar to that seen at 12 weeks gene-on. This improvement in cage behavior was accompanied by noticeable and steady improvement in their performance on the accelerating Rotarod (Fig. 7). Although their improvement on the Rotarod did not reach statistical significance, by 12 weeks of gene off performance of dox-treated mice was better than untreated SCA1[82Q] animals on all four of the trial days. Moreover, their performance on the Rotarod after 12 weeks of gene-off was identical to that of 12-week gene-on mice.
The ability of the 32 week gene-on—12 week gene-off mice to improve their neurological performance was accompanied by improvement in Purkinje cell morphology (Fig. 7). Intensity of calbindin immunostaining increased dramatically in all of the dox-treated mice examined. In some, the level of calbindin staining in 32 week gene-on—12 week gene-off animals increased beyond that seen in 12 week gene-on mice. In addition, there was a substantial decrease in the number of Purkinje cells with nuclear inclusions as well as cytoplasmic vacuoles. These changes were accompanied by a noticeable increase in dendritic arborization and thickness of the molecular layer. Thus, by both neurological and morphological assessments, disease status in the 32 week gene-on—12 week gene-off conditional SCA1[82Q] mice improved from a severe to a moderate form of disease.
By following recovery in the conditional SCA1[82Q] mice, a sequence of events leading to repair and restoration of neurological function can be assembled. In particular, after recovery after stopping SCA1[82Q] expression at a middle stage of disease (12 weeks) is especially informative. Within days of stopping SCA1[82Q] expression, mutant ataxin-1 was cleared by Purkinje neurons. After 4 weeks of the gene off, mice improved their neurological function in that by home cage behavior ataxia was no longer evident. Yet, their ability to perform the more challenging accelerating Rotarod task showed no signs of improvement. During the initial 4 week gene-off period Purkinje cell dendritic arborization and spine density improved considerably. However, compared with wild-type mice, many of the Purkinje dendritic spines in the 4 week gene-off mice were not positive for postsynaptic mGluR1. With an additional 4 weeks of the SCA1[82Q] gene being off, improvement on the Rotarod was seen. Improvement on the Rotarod at 8 weeks of gene-off correlated with a consistent appearance of the mGluR1 receptor at the Purkinje neuron-parallel fiber synapse in the molecular layer.
The importance of mGluR1 for proper motor function is consistent with previous data from mice with a targeted deletion of mGluR1 (Aiba et al., 1994; Conquet et al., 1994). Mice lacking mGluR1 were unable to perform on the Rotarod, whereas introduction of mGlur1α expression into Purkinje cells of mGluR1-/- mice restored normal Rotarod performance (Ichise et al., 2000). The conditional SCA1[82Q] mice revealed that, in addition to being key for synapse formation and plasticity during development, dendritic mGluR1 also has a role in Purkinje cell plasticity associated with repair from neurodegeneration in the adult.
Overall, the results on the conditional SCA1[82Q] mice support the concept that most if not all of pathological changes seen in polyglutamine disease, excluding cell loss, are reversible (Yamamoto et al., 2000). Our results also show that the earlier an intervention is administered, the more complete will be the restoration of normal function. Yet, even at a very late stage of disease the pathologic effects of mutant ataxin-1 were partially reversible. This demonstrates that Purkinje cells have a sustained ability to recover from long-term effects of mutant ataxin-1. A therapeutic intervention targeted at the reducing the expression of mutant ataxin-1 showed that SCA1 RNAi was effective in suppressing Purkinje cell pathology and neurological signs when SCA1 mice were injected at 7 weeks of age (Xia et al., 2004). Because our results on the conditional SCA1 mice indicate that at a late stage of disease Purkinje cells retain some ability to recover, it will be interesting to examine whether an RNAi strategy will be effective at a late stage.
Footnotes
This work was supported by National Institutes of Health-National Institute of Neurological Disorders and Stroke Grant NS22920 to H.T.O. We thank R. Ehlenfeldt for maintaining the mouse colony.
Correspondence should be addressed to Dr. Harry T. Orr, Institute of Human Genetics, University of Minnesota, Mayo Mail Code 206, Minneapolis, MN 55455. E-mail: orrxx002{at}umn.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248853-09$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}