

Abstract
Functional interactions between dopamine D1-like receptors and NMDA subtype glutamate receptors have been implicated in the maintenance of normal brain activity and neurological dysfunction. Although modulation of NMDA receptor functions by D1 receptor activation has been the subject of extensive investigation, little is known as to how the activation of NMDA receptors alters D1 function. Here we report that NMDA receptors regulate D1 receptor function via a direct protein–protein interaction mediated by the carboxyl tail regions of both receptors. In both cotransfected cells and cultured hippocampal neurons the activation of NMDA receptors increases the number of D1 receptors on the plasma membrane surface and enhances D1 receptor-mediated cAMP accumulation via a SNARE-dependent mechanism. Furthermore, overexpression of mini-genes encoding either NR1 or D1 carboxyl tail fragments disrupts the D1–NR1 direct protein–protein interaction and abolishes NMDA-induced changes in both D1 cell surface expression and D1-mediated cAMP accumulation. Our results demonstrate that the D1–NR1 physical interaction enables NMDA receptors to increase plasma membrane insertion of D1 receptors and provides a novel mechanism by which the activation of NMDA receptors upregulates D1 receptor function. Understanding the molecular mechanisms by which D1 and NMDA receptors functionally interact may provide insight toward elucidating the molecular neurobiological mechanisms involved in many neuropsychiatric illnesses, such as schizophrenia.
- dopamine receptors
- NMDA receptors
- protein–protein interactions
- cAMP accumulation
- receptor trafficking
- G-proteins
Introduction
NMDA receptors and dopamine D1-like receptors represent two major structurally and functionally divergent families of neurotransmitter receptors in the CNS. The former is composed of a class of ligand-gated ion channels consisting of diverse subunits responsible for fast synaptic transmission. The latter belongs to the seven transmembrane domain receptor super-family exerting its biological effects primarily via the activation of adenylate cyclase by the G-protein signaling cascades. Numerous studies have shown that G-protein-coupled receptors regulate ligand-gated ion channel functions either by the activation of their intracellular signal transduction pathways (Greengard, 2001) or via direct protein–protein interactions (Liu et al., 2000; Lee et al., 2002). However, there have been few studies investigating the functional modulation of G-protein-coupled receptors by the activation of ligand-gated ion channels. Therefore, it is the subject of our present study to investigate whether the activation of NMDA receptors is able to regulate dopamine D1 receptor-mediated functions and the potential molecular mechanism underlying this process.
NMDA receptors, activated by the principal excitatory neurotransmitter glutamate, are important in activity-dependent synaptic plasticity and excitotoxicity that underlie learning, memory, neural development, and many neurological disorders (Michaelis, 1998). NMDA receptors, which mediate cation flux, exist as heteromeric assemblies of multiple subunits including NR1 and NR2 subunits (Michaelis, 1998). The NR1 subunit mRNA is spliced alternatively at three exons to form eight splice variants, which results in the presence/absence of C1, C2, or both cassettes at the C terminus (Ziff, 1997; Guilarte and McGlothan, 2003). NR2 subunits are encoded by four different gene products (NR2A, B, C, and D) (Durand et al., 1992; Sugihara et al., 1992; Hollmann et al., 1993; Hollmann and Heinemann, 1994).
Dopamine D1-like receptors, which include the D1 and D5 receptors, play a major role in regulating neuronal motor control, cognition, event prediction, and emotion (Missale et al., 1998; Goldman-Rakic, 1999). D1 and D5 receptors preferentially couple to Gs proteins, stimulating the activity of adenylate cyclase and protein kinase A-dependent (PKA) pathways. Previous studies have shown the functional interaction between D1-like receptors and NMDA receptors. Activation of D1 receptors enhances the NMDA currents via a PKA-dependent pathway that most likely involves the phosphorylation and activation of DARPP-32 (Greengard, 2001). Similar results are demonstrable on the EPSC of the pharmacologically isolated NMDA receptor-mediated component of synaptic transmission (Cepeda et al., 1992; Colwell and Levine, 1995). In the hippocampus, dopamine has been shown to produce a synapse-specific enhancement of long-term potentiation (LTP) via D1/D5 receptors and cAMP (Huang and Kandel, 1995). Furthermore, we have obtained evidence that D1 receptors can inhibit NMDA receptor currents and NMDA receptor-mediated excitotoxicity via direct protein–protein interactions (Lee et al., 2002). Similarly, a recent report has confirmed the direct interaction between D1 and NMDA receptors via the use of bioluminescence resonance energy transfer in cotransfected COS-7 cells (Fiorentini et al., 2003). In addition, several studies have suggested that NMDA receptors may modulate D1-mediated functions, because blockade of NMDA receptor activity led to attenuation of the ability of D1 in the modulation of neuronal activity (Huang et al., 1998) and in the induction of immediate early gene expression (Konradi et al., 1996; Keefe and Ganguly, 1998). A recent study also suggests that the activation of NMDA receptors recruits D1 receptors to the cell plasma membrane and enhances D1-mediated cAMP accumulation (Scott et al., 2002). Despite the above observation the mechanisms that enable NMDA receptors to modulate D1-mediated functions remain mostly unclear.
Materials and Methods
cAMP accumulation assay. COS-7 cells were transiently transfected with the indicated cDNA constructs by electroporation (40 μg of each indicated cDNA per 2.5 × 107 cells; 48 Ω, 135 mA, 500 mF), placed in 24-well plates, and grown for 4–5 d. Cells were washed with 0.5 ml of prewarmed Dulbecco's α-MEM containing 1-methyl-3-isobutylxanthine and 1 μm propranolol and then were incubated in the above medium in the presence or absence of antagonist/agonist for the indicated time period at 37°C and 5% CO2. The reaction was terminated by the addition of 0.5 ml of 0.2N HCl and incubation for 20 min at 4°C. Cellular debris was pelleted, and aliquots of the supernatant were used to determine the cAMP content via immunodetection (Amersham Biosciences, Oakville, Ontario, Canada) as described previously (Liu et al., 1995). To ensure equivalence of whole-cell cAMP assay comparisons, we monitored receptor densities for [3H] SCH-23390 binding (3.0 nm final concentration).
Primary cultures from hippocampus were prepared from fetal Wistar rats (embryonic day 17–19) and placed on 24-well plates for 10–14 d as previously described (Liu et al., 2000). cAMP assay with the use of primary culture neurons was performed identically as with COS-7 cells.
Ligand binding assay. COS-7 cells were transiently transfected as described above, placed into 150 mm plates, and cultured for 4–5 d. COS-7 cells were maintained in Dulbecco's α-MEM supplemented with 10% fetal calf serum at 37°C and 5% CO2. Next the cells were collected, and membranes were prepared for binding assays as described previously (Liu et al., 1995). For saturation experiments 0.5 ml aliquots of tissue homogenate (∼50–100 μg of membrane protein) were incubated in duplicate with increasing concentrations of [3H] SCH-23390 (85.5 Ci/mmol; 30–6800 pm final concentration) for 120 min at room temperature in a total volume of 1.5 ml. For competition binding studies 0.5 ml of membranes was incubated in duplicate with [3H] SCH-23390 (250–400 pm) and increasing concentrations of competing ligands (10–13 to 10–4 m) for 120 min. Experiments were terminated by rapid filtration, and filters were monitored for tritium. Nonspecific binding was defined in the presence of 10 μm (+)-butaclamol. Binding data were analyzed by the nonlinear least-square curve-fitting program KaleidaGraph (Abelbeck Software, Synergy, Reading, PA) as described previously (Liu et al., 1995).
Confocal imaging. Human embryonic kidney-293 (HEK-293) cells were transiently transfected with the indicated cDNA constructs via the Lipofectamine method (6–10 μg of each indicated cDNA per 7.5 × 106 cells), placed in 35 mm plates, and grown for 2–4 d. For preblocking immunostaining the cells first were incubated with the monoclonal anti-hemagglutinin (anti-HA) antibody (10 μg/ml; Babco, Berkeley, CA) for 45 min and then a cold (nonconjugated) secondary antibody (10 μg/ml; Sigma, St. Louis, MO) for another 45 min at 4°C. After treatment with 500 μm NMDA/10 μm glycine or extracellular solution [ECS; (containing in mm): 25 HEPES, 140 NaCl, 1.3 CaCl2,33 d-glucose, 0.003 glycine, and 5.4 KCl, pH 7.35; osmolarity, 310–320 mOsm] for 30 sec at room temperature, the cells were fixed for 20 min at room temperature with 4% paraformaldehyde in PBS and stained with the same anti-HA primary antibody and Cy3-conjugated anti-mouse secondary antibody to detect the newly inserted HA-D1 receptors on the plasma membrane under nonpermeabilized conditions. The HLA (human leukocyte antigen) class I antigen then was stained with the FITC-conjugated monoclonal anti-HLA antibody (Sigma) under permeabilized conditions (cells were permeabilized by using PBS/0.1% Triton X-100 for 10 min). Optical images were collected by confocal scanning with dual channels for Cy3 and FITC fluorescence with a Zeiss 100 confocal microscope (Oberkochen, Germany) with a 100× oil objective lens. To ensure that the detection levels for the red (Cy3) and green (FITC) channels were consistent between experiments (control/treatment), we initially scanned randomly acquired fields (typically four to six) from each coverslip/experimental condition and then had the intensity levels adjusted to a minimum of 150% over background values for each color detector (“thresholding” pixel intensities 1.5 times above detected background values were considered to be representative of specific receptors). Under these conditions no detectable bleedthrough from one channel was observed. Detector and intensity levels were matched for each particular coverslip; settings were maintained throughout the coverslip. The same ratios determined between background/staining intensity were used to compare between control and NMDA-treated cells.
Cell-ELISA assays. Cell-ELISA assays (colorimetric assays) were done essentially as previously described (Lee et al., 2002). HEK-293 cells were transiently transfected with the indicated cDNA constructs by the Lipofectamine method (6–10 μg of each indicated cDNA per 7.5 × 106 cells), distributed equally to two six-well plates (35 mm/well), and grown for 2–4 d. The same density of cotransfected cells was treated with 500 μm NMDA/10 μm glycine or ECS before being fixed in 4% paraformaldehyde for 10 min in the absence (nonpermeabilized conditions) or the presence (permeabilized conditions) of 1% Triton X-100. Cells were incubated with a monoclonal antibody against the HA epitope (Babco, Richmond, CA; 1 μg/ml to detect the HA epitope inserted into the extracellular N terminus of D1 receptors) for the purpose of labeling the receptors on the cell surface under nonpermeabilized conditions or the entire receptor pool under permeabilized conditions. After an incubation with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma), the HRP substrate o-phenylenediamine (OPD; Sigma) was added to produce a color reaction that was stopped with 3N HCl. The cell surface expression of HA-D1 after pretreatment with NMDA was presented as the ratio of colorimetric readings under nonpermeabilized conditions to those under permeabilized conditions and then normalized to their respective control groups (pretreated with ECS). Analysis was done by using at least 12 separate dishes in each group. Cell-ELISA assays that used primary hippocampal neurons were performed identically with assays that used HEK-293 cells, with the exception that anti-D1 antibody (Chemicon, Temecula, CA) instead of anti-HA was used as the primary antibody.
Primary cultures, recombinant adenovirus construction, and infection. Primary cultures from hippocampus were prepared from fetal Wistar rats (embryonic day 17–19) on Cell+ (Sarstedt, N∫mbrecht, Germany) culture dishes for 14 d as previously described (Liu et al., 2000). Recombinant adenoviruses were formed by cotransfecting cDNAs encoding the D1-t2, D1-t3 in the shuttle vector pDC315 (Microbix Biosystems, Toronto, Ontario, Canada) with replication-deficient adenovirus type 5 DNA into HEK-293 cells. The recombinant adenoviruses containing the D1-t2, D1-t3 cDNAs were isolated, confirmed by restriction analysis, plaque-purified, expanded, and titered. For infection the primary hippocampal cultures were infected with 10 to ∼20 plaque-forming units per neuron [multiplicity of infection (moi)] of recombinant adenovirus in 500 μl of culture medium. Cultures were supplemented with 1.5 ml of fresh medium 1 hr after infection and were analyzed 12–24 hr after infection (Lee et al., 2002).
GST fusion proteins and mini-genes. Dopamine D1CT, D1-t1, D1-t2, D1-t3, NR1-1aCT, NR1-C0, NR1-C1, NR1-C2, and NR2ACT cDNA-encoding fragments were amplified by PCR from full-length cDNA clones. All 5′ and 3′ oligonucleotides incorporated BamHI and EcoRI sites, respectively, to facilitate subcloning into pcDNA3 or pGEX4T-2. Initiation methionine residues and stop codons also were incorporated where appropriate. GST-fusion proteins were prepared from bacterial lysates as described by the manufacturer (Amersham Biosciences). To confirm appropriate splice fusion and the absence of spurious PCR-generated nucleotide errors, we resequenced all constructs.
Coimmunoprecipitation, protein affinity purification (pull-down), and Western blotting. Coimmunoprecipitation, affinity pull-down, and Western blot analyses were performed as previously described (Liu et al., 2000; Lee et al., 2002). Rat brain hippocampus (100 mg) or transfected COS-7 cells (∼2 × 107) were homogenized in buffer containing (in mm) 50 Tris-Cl, pH 7.6, 150 NaCl, 2 EDTA, 1 PMSF plus 1% Igepal CA-630, 0.5 to ∼1% sodium deoxycholate, 1% Triton X-100, and protease inhibitor mixture (5 μl/100 mg of tissue; Sigma); after being centrifuged at 10,000 × g at 4°C for 20 min, the supernatant was extracted and protein concentrations were measured (Pierce, Rockford, IL). For coimmunoprecipitation experiments, solubilized hippocampal/cell extracts (500 to ∼700 μg of protein) were incubated in the presence of primary antibodies anti-NR1 (Pharmingen, San Diego, CA), anti-D1 (Chemicon), or mouse IgG (1 to ∼2 μg) for 4 hr at 4°C, followed by the addition of 20 μl of protein A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA) for 12 hr. Pellets were washed four times in the buffer described above, boiled for 5 min in SDS sample buffer, and subjected to SDS-PAGE. In each experiment 20 to ∼50 μg of tissue-extracted protein was used as control. For affinity purification experiments the solubilized hippocampal extracts (50–100 μg of protein) were incubated with glutathione-Sepharose beads (Pharmacia, Dorval, Québec, Canada) bound to the indicated GST-fusion proteins (50 to ∼100 μg) at room temperature for 1 hr. Beads were washed three times with 600 μl of PBS containing 0.1–0.5% Triton X-100 before the bound proteins were eluted with glutathione elution buffer. Elutes were incubated in sample buffer and subjected to 10% SDS-PAGE for Western blot analysis. Blots were blocked with 5% nonfat dried milk dissolved in TBST buffer (10 mm Tris, 150 mm NaCl, and 0.1% Tween-20) for 1 hr at room temperature, washed three times with TBST buffer, and then incubated with the appropriate primary antibody (anti-D1, anti-D5, or anti-NR1, diluted in 1% milk in TBST) overnight at 4°C and washed again with TBST buffer three times; the membrane was incubated with horseradish peroxidase-conjugated secondary antibody (diluted in 1% milk in TBST; Sigma) for 1.5 hr at room temperature. The proteins were visualized with enhanced chemiluminescence reagents as described (Amersham Biosciences).
In vitro binding assays. Glutathione beads carrying GST fusion proteins (D1-t2, D1-t3) or GST (10 to ∼20 μg each) alone were incubated with [35S]-methionine-labeled NR1-C0, NR1-C1, or NR1-C2 probe, respectively. Then the beads were washed six times with PBS containing 0.5% Triton X-100 and eluted with 10 mm glutathione elution buffer. Eluates were separated by SDS-PAGE and visualized by autoradiography.
Results
Activation of NMDA receptor increases dopamine D1 receptor-mediated cAMP accumulation in cotransfected cells
As the first step toward investigating whether the activation of NMDA receptors will modulate D1 receptor function, we tested the effects of NMDA receptor activation on D1 receptor-mediated cAMP accumulation in COS-7 cells coexpressing D1 and NMDA receptors. As depicted in Figure 1A, left, the selective D1-like receptor agonist SKF-81297 (10 μm for 15 min at 37°C) stimulated D1-mediated cAMP accumulation, an effect that was blocked by the selective D1-like receptor antagonist SCH-23390 (1 μm for 15 min at 37°C). Interestingly, when the cotransfected cells were pretreated with 500 μM NMDA/10 μm glycine for 30 sec, the SKF-stimulated adenylate cyclase activity was increased by ∼45% (n = 12; p < 0.05). The fact that NMDA failed to modulate D1-mediated cAMP accumulation in cells expressing D1 receptor alone (data not shown) and that both AP-5 and MK801 could block the NMDA/glycine-induced effect on D1-mediated function addresses the requirement for the activation of NMDA receptors and calcium influx. To confirm that the observed cAMP activity enhancement actually is mediated by D1 receptors, we pretreated cotransfected cells with SCH-23390 and observed that the NMDA receptor activation-induced cAMP enhancement was abolished by the application of SCH-23390, strongly suggesting that the enhanced cAMP accumulation was mediated by D1 receptors. Furthermore, the activation of NMDA receptors showed no effects on D5-mediated cAMP accumulation in COS-7 cells expressing both D5 and NMDA receptors (Fig. 1A, right), although dopamine D1 and D5 receptors share similar pharmacological profiles and strong amino acid homology. We also observed that, in cells coexpressing D1 and NMDA receptors, the estimated EC50 for D1-stimulated cAMP production in NMDA-pretreated cells (312 ± 56 nm; n = 3) is virtually identical to that for untreated control cells (307 ± 71 nm; n = 3) despite the enhancement in apparent maximal accumulation of cAMP levels (Fig. 1B). Thus the enhancement of D1-mediated cAMP production cannot be attributed to NMDA-induced competitive increase of D1 receptor agonist efficacy. Furthermore, the affinity for SKF-81297 to compete with [3H] SCH-23390 binding at D1 receptors in cells coexpressing D1 and NMDA receptors after NMDA pretreatment (Ki = 33 ± 3 nm; n = 4) was not significantly different from that for untreated cells (Ki = 30 ± 1 nm; n = 4) or cells expressing D1 receptors alone (Ki = 35 ± 6 nm; n = 4) (Fig. 1C). NMDA pretreatment did not modify significantly either the estimated KD (untreated control, 360 ± 70 pm; NMDA-treated, 220 ± 40 pm; n = 3) or Bmax (untreated control, 0.22 ± 0.04 pmol/mg protein; NMDA-treated, 0.18 ± 0.03 pmol/mg protein; n = 3) for [3H] SCH-23390 (0.03–6.8 nm; 85 Ci/mmol) binding to D1 receptors in cells coexpressing D1 with NMDA receptors (Fig. 1D). Some of the data points in Figure 1D are compressed on the bottom of the curve; despite the scatter in the Scatchard plots (Fig. 1D, inset) the affinity is not significantly different between control and NMDA-treated samples (360 vs 220 pm). Analyses via both nonlinear regression and Scatchard plot transformation (data not shown) resulted in virtually identical outcomes, strongly indicating that these results are robust. In addition, the competition binding studies also strongly suggest that ligand-binding affinity is not altered with NMDA treatment. Taken together, these data suggest that the activation of NMDA receptors enhances D1 receptor-mediated cAMP accumulation without altering the pharmacological properties or the expression level of D1 receptors.
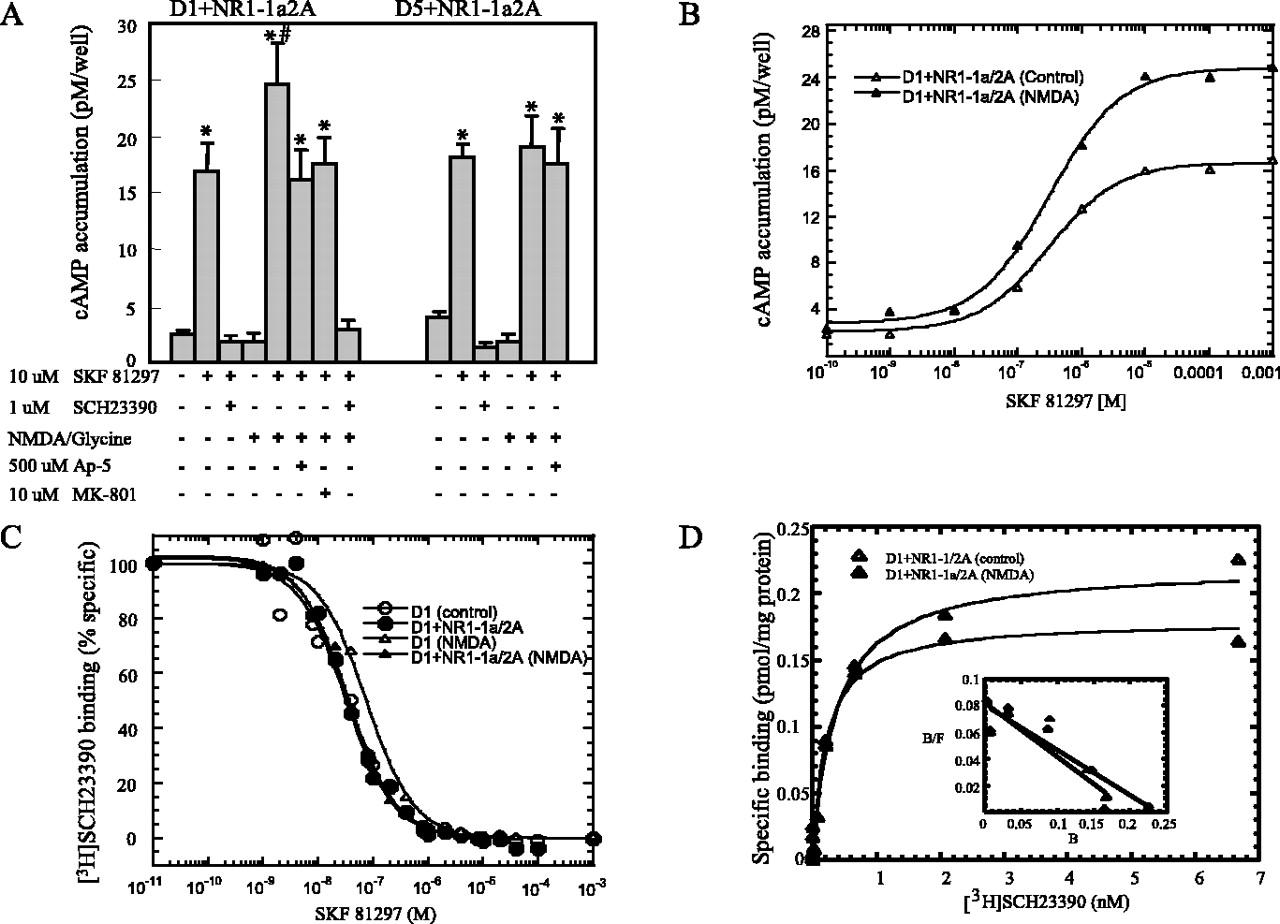
NMDA receptors modulate D1, but not D5, receptor-mediated cAMP accumulation in COS-7 cells coexpressing both receptors. A, NMDA receptor stimulation (500 μm NMDA/10 μm glycine; 30 sec at room temperature) enhanced D1-mediated, but not D5-mediated, cAMP accumulation by ∼45%, an effect that could be blocked by the selective D1-like receptor antagonist SCH-23390 or the NMDA receptor antagonist AP5 or MK-801, respectively. Data are representative as the means ± SEM of 12 independent experiments. Data were analyzed by ANOVA, followed by post hoc Student–Newman–Keuls test. *Significantly different from control group (p < 0.05); #significantly different from SKF treatment group (p < 0.05). B, NMDA receptor stimulation increases the efficacy, but not affinity, of D1 receptor-mediated cAMP accumulation. Estimated EC50 values are listed in Results. C, The affinity of SKF-81297 for D1 receptors is unchanged after NMDA treatment both in cells expressing D1 only and cells coexpressing D1 with NMDA receptors, as indexed by [3H] SCH-23390 binding. Ki values are listed in Results. The graph is a representative plot of four independent experiments. D, NMDA pretreatment did not alter the estimated KD or Bmax of [3H] SCH-23390 binding cells coexpressing D1 with NMDA receptors. The graph is a representative plot of three independent experiments.
NMDA receptor activation increases D1 receptor-mediated cAMP accumulation via an interaction with the carboxyl tail of the D1 receptor
Given that (1) NMDA receptor activation increased D1, but not D5-mediated cAMP accumulation; (2) D1 and D5 receptors display particular sequence divergence within the C terminus (CT), a region that might confer subtype-selective functional attributes and coupling to distinct effectors independent of those classically associated with G-protein activation; and (3) that NMDA receptors can couple directly to the CT of the D1, but not D5, receptor (Lee et al., 2002), we investigated whether the D1 receptor CT is responsible for the observed NMDA receptor modulation of D1-mediated cAMP accumulation by using chimeric D1 and D5 receptors (D1/D5CT, D5/D1CT) in which the CTs were swapped (Demchyshyn et al., 2000). As illustrated in Figure 2A, D5/D1CT fully reconstituted the ability of NMDA receptor activation to enhance D5/D1CT-mediated cAMP accumulation by ∼48% (n = 6; p < 0.05), whereas D1/D5CT failed to establish functional interactions with NMDA receptors. These data suggest that the CT of the D1 receptor is required for the expression of the functional modulation of D1 by NMDA receptors.

NMDA receptor modulation of D1 cAMP production is dependent on D1-CT and NR1-1a-CT sequences in cells cotransfected with D1/NMDA receptors. A, Chimeric D1 receptors in which the carboxyl tail was swapped with sequences encoded by D5-CT (D1/D5-CT360–477) are unresponsive to NMDA receptor stimulation, whereas D5/D1-CT332–446 receptor mutants fully reconstitute NMDA receptor-mediated increases in cAMP activity. B, The ability of the NMDA receptors to enhance D1 receptor-mediated cAMP accumulation functionally was abolished by coexpressing with a mini-gene encoding the D1-t2 domain, but not the D1-t1 or D1-t3 domains. C, The ability of the NMDA receptors to enhance D1 receptor-mediated cAMP accumulation functionally was abolished by coexpressing with a mini-gene encoding the NR1-1a-CT domain, but not the NR2A-CT domain. Data are representative as the means ± SEM of 6–10 independent experiments. Data were analyzed by ANOVA, followed by post hoc Student–Newman–Keuls test. *Significantly different from control group (p < 0.05); #significantly different from SKF treatment group (p < 0.05).
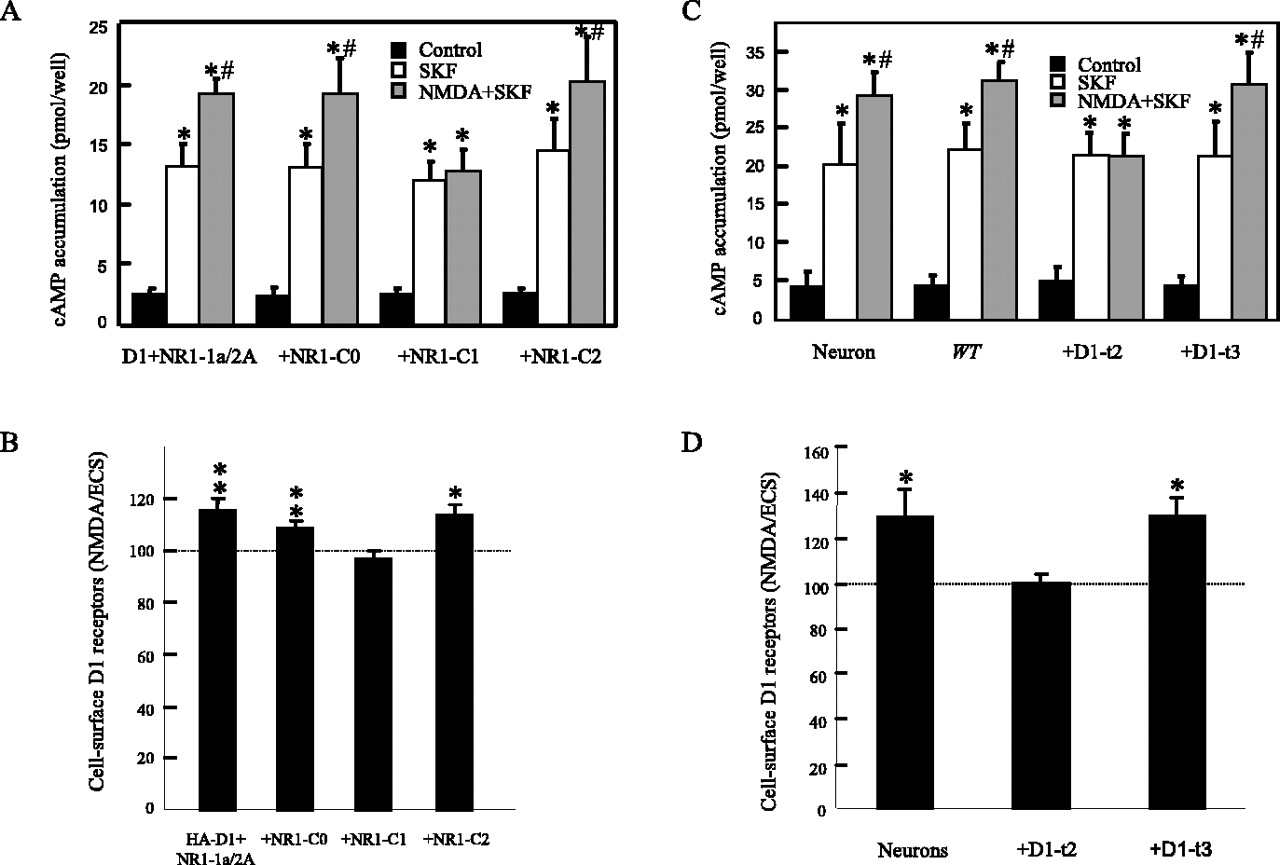
To confirm these results and to delineate the region(s) of the D1 receptor CT involved in the functional interaction with the NMDA receptors, we used three D1-CT mini-genes [A357-N386 (D1-t1), L387-L416 (D1-t2), S417-T446 (D1-t3)], of which D1-t2 and D1-t3 have been shown previously to be critical for direct binding to NR1-1a and NR2A subunits, respectively (Lee et al., 2002). As shown in Figure 2B, cAMP assays revealed that overexpression of the mini-gene encoding D1-t2, but not D1-t1 or D1-t3, was able to abolish completely the ability of NMDA receptor to modulate D1 receptor-mediated cAMP accumulation functionally. In addition, overexpression of a mini-gene encoding the NR1-1a CT (NR1-1aCT), but not NR2A CT (NR2ACT), significantly blocked the NMDA-induced D1-mediated cAMP enhancement (Fig. 2C). These data indicate the absolute requirements of the D1-t2 motif and the possible involvement of the D1-t2: NR1-1a direct interaction in this process.
Activation of NMDA receptor enhances D1 receptor membrane expression
Changes in G-protein-coupled receptor distribution by altering receptor trafficking/internalization/desensitization long have been identified as a means by which receptor functions are modulated (Claing et al., 2002). Therefore, we subsequently examined whether the observed enhancement of D1 receptor-mediated cAMP accumulation by the activation of NMDA receptors is a consequence of increased D1 receptor cell surface expression. Because increased cell surface receptor expression might arise from enhanced membrane insertion or, alternatively, by affecting the rate of receptor internalization, we used an immunocytochemistry-based method that allows for specific visualization of newly inserted receptors (Lu et al., 2001). Thus in HEK-293 cells coexpressing NMDA and HA-D1 receptors (HA epitope is inserted into the extracellular N terminus of the D1 receptor), the preexisting cell surface HA-D1 receptors first were blocked with anti-HA primary antibody and a nonfluorescence-conjugated secondary antibody. After treatment for 30 sec with 500 μm NMDA/10 μm glycine or ECS, the cells then were labeled with the same primary antibody and a Cy3-conjugated secondary antibody to label the newly inserted membrane HA-D1 receptors. To determine whether the insertion of HA-D1 receptors occurred at the plasma membrane, we subsequently stained cells, under cell-permeabilized conditions, with anti-HLA that predominantly will recognize a monomorphic epitope on the α-chain polypeptide of human class 1 HLA molecules present at the cell surface (Schreiber et al., 1984). The lack of Cy3 immunofluorescence in control cells confirmed the complete blockade of preexisting HA-D1 receptors at the plasma membrane by the unconjugated secondary antibody. However, NMDA pretreatment significantly increased the intensity and number of HA-D1 receptor clusters at the cell surface, as shown in Figure 3A, left panels. Consistent with the cAMP assay data presented in Figure 2B, NMDA-induced enhancement of HA-D1 receptor membrane insertion also could be blocked by the overexpression of the D1-t2 mini-gene (Fig. 3A, middle panels), but not with the D1-t3 mini-gene (Fig. 3A, right panels). We further quantified this NMDA-induced increase in HA-D1 receptor membrane insertion by using a cell-ELISA assay (Lee et al., 2002). Analogous to the immunofluorescent staining results, the ELISA assay revealed a ∼25% increase in HA-D1 receptor on the cell surface by NMDA receptor activation (n = 12; p < 0.01). Furthermore, this process also could be blocked by pretreatment with MK-801 or by the overexpression of the D1-t2, but not D1-t3, mini-genes, indicating the absolute requirement of the Ca2+ influx and that the enhanced D1-mediated cAMP accumulation may be a result of the increased D1 receptor on the cell surface. Moreover, NMDA-induced enhancement of D1 receptor on the cell surface was blocked at 4°C (Fig. 3B), which is consistent with previous reports that endocytosis/exocytosis is blocked at 4°C (Man et al., 2000).
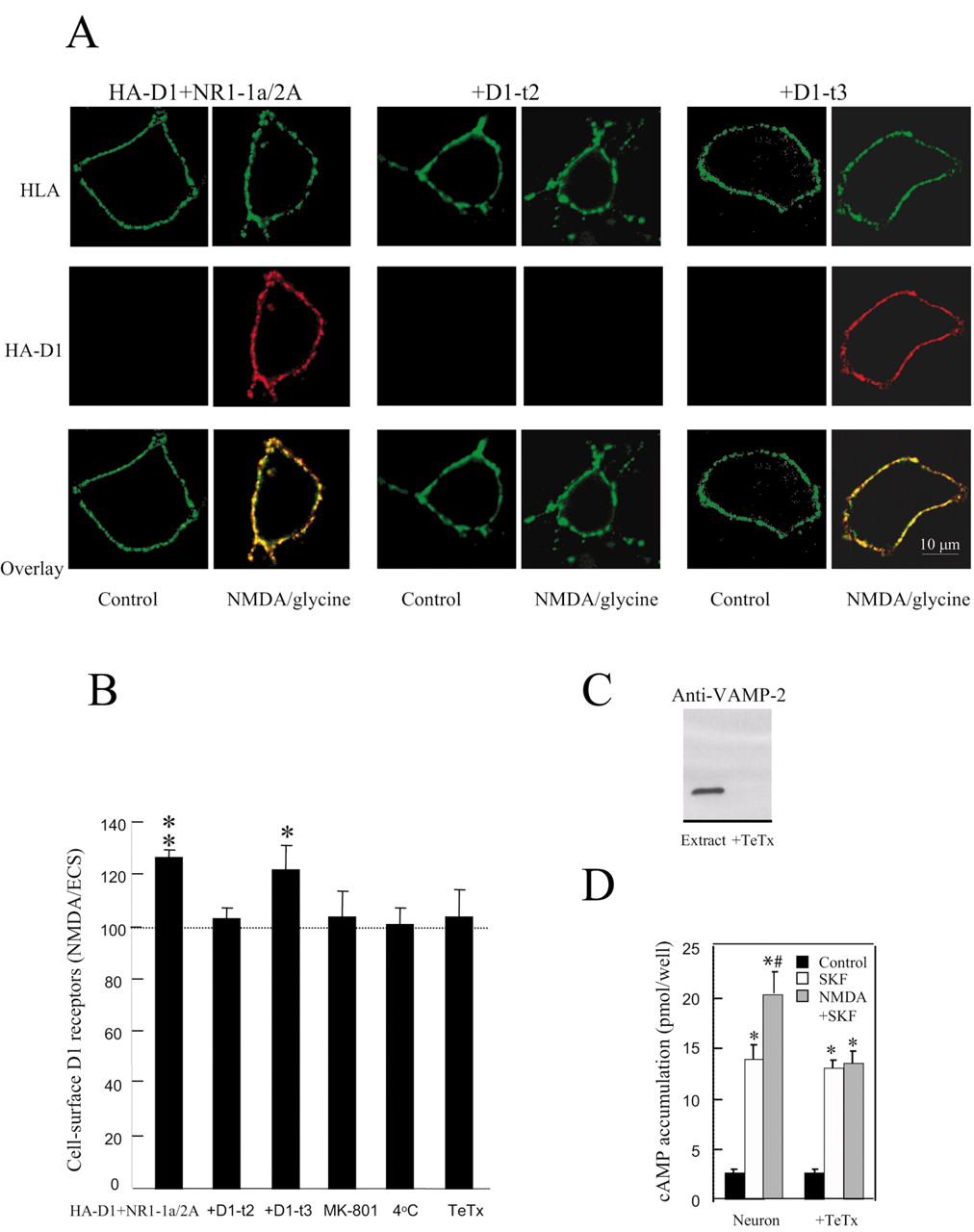
NMDA facilitates membrane expression of HA-D1 receptors in cells coexpressing D1 and NMDA receptors. A, After preblocking existing cell surface HA-D1 receptors with anti-HA and nonfluorescent secondary antibody, we treated the cells with 500 μm NMDA/10 μm glycine or extracellular solution (ECS) for 30 sec. Control and NMDA/glycine-treated cotransfected HEK-293 cells were stained sequentially for HA-D1 (red) and HLA (green) under nonpermeabilized and permeabilized conditions, respectively. Individual (HLA, green; HA-D1, red) and superimposed (overlay) confocal images show that activation of NMDA receptors modulates HA-D1 receptor membrane expression in cells coexpressing D1 and NR1-1a/2A subunits (left panels), an effect that is blocked with overexpression of the D1-t2 mini-gene (middle panels), but not the D1-t3 mini-gene (right panels). B, Summarized data indicating effects of NMDA stimulation on HA-D1 membrane expression. Columns show the means ± SEM of the ratios of colorimetric readings under nonpermeabilized conditions versus those under permeabilized conditions from cells treated with NMDA/glycine, normalized to their respective control groups (dashed line) treated with ECS. *p < 0.05; **p < 0.01 (Student's t test). C, Western blot of VAMP-2 from solubilized hippocampal culture neurons with or without the pretreatment of TeTx (150 ng/ml) for 48 hr. D, TeTx blocks NMDA receptor modulation of D1-mediated cAMP accumulation in cultured rat hippocampal neurons. Data are representative as the means ± SEM of six independent experiments. Data were analyzed by ANOVA, followed by post hoc Student–Newman–Keuls test. *Significantly different from control group (p < 0.05); #significantly different from SKF treatment group (p < 0.05).
The enhancement of D1-mediated cAMP accumulation and membrane expression depends on membrane fusion exocytosis
Previous studies have shown that Clostridium tetanus toxin (TeTx) selectively cleaves vesicle-associated membrane protein (VAMP) and prevents exocytosis (Maletic-Savatic and Malinow, 1998; Hua and Charlton, 1999). To test whether the D1 receptor enhancement of the response to NMDA could be produced by insertion of new receptors into the plasma membrane from the intracellular vesicular pool, we repeated the experiments after impairing the fusion of intracellular vesicles with the plasma membrane by using TeTx. We first confirmed that pretreatment with TeTx (100 ng/ml) for 48 hr cleaved the v-SNARE synaptobrevin/VAMP2 in neurons. In cultured neurons significant VAMP2 cleavage occurred on preincubation with TeTx, as illustrated by the lack of an anti-VAMP2 immunoreactive band comparable to a band seen in nontreated neurons, as shown in Figure 3C. Under the same TeTx pretreatment condition the enhancement of D1 receptor membrane expression by the activation of NMDA receptors was prevented completely (Fig. 3B). Similarly, the elimination of VAMP2 also blocked the observed enhancement of D1-mediated cAMP accumulation with NMDA stimulation (Fig. 3D). These data suggest that activation of NMDA receptors modulates D1 receptor function via a membrane fusion-dependent exocytotic process.
NMDA receptor NR1 subunit directly couples to dopamine D1 receptors through the C1 region of the carboxyl tail
We have shown previously that two regions (D1-t2, D1-t3) in the D1 receptor carboxyl tail can couple directly and selectively to the NMDA glutamate receptor subunits NR1-1a and NR2A (Lee et al., 2002). The observed D1–NMDA protein–protein interactions enable dopamine regulation of NMDA receptor-mediated functions. The fact that NMDA receptor modulation of D1-mediated cAMP accumulation and D1 receptor membrane expression could be abolished by the overexpression of D1-t2 mini gene, as presented in Figures 2B and 3B, suggests that the D1–NMDA protein–protein interaction also may be responsible for the observed NMDA receptor functional modulation of D1 receptors. However, it is unknown which specific region in the carboxyl tail of the NR1-1a subunit is essential for the direct coupling to D1 receptors. We first confirmed the existence of D1–NMDA receptor complex in rat hippocampal extract. As depicted in Figure 4A, NMDA receptor NR1 subunit antibody can coimmunoprecipitate D1, but not D5, receptors, confirming the specific physical interaction between D1 and NMDA receptors. Given that NMDA receptor modulation of D1-mediated cAMP accumulation and D1 receptor membrane expression could be abolished by the overexpression of D1-t2 mini gene as presented in Figures 2 B and 3B, we examined whether the coimmunoprecipitation of D1 receptors by the NR1 antibody can be blocked by the overexpression of D1-t2 mini-gene. As shown in Figure 4 B, overexpression of D1-t2 mini-gene blocked the ability of NR1 antibody to coimmunoprecipitate D1 receptors in the cotransfected D1 and NR1-1a subunit of the cells, but not in cotransfected D1 and NR1-1a/2A subunits of the cells because of the coexistence of D1-t3–NR2A interaction (Lee et al., 2002). To determine the CT regions of NR1-1a subunit involved in the formation of D1–NMDA receptor complex, we constructed various glutathione S-transferase (GST) fusion proteins encoding the NR1-C0, E834-D864; NR1-C1, D864-T900; and NR1-C2, T900-S938 (Fig. 4C). As shown in affinity purification assays GST-NR1-C1, but not GST-NR1-C0, GST-NR1-C2, or GST alone, precipitated solubilized D1 receptor, indicating that the NMDA receptor NR1-1a subunit can interact with D1 receptors through its NR1-C1 region of the carboxyl tail (Fig. 4 D). In addition, we confirmed that the D1–NR1-1a complex is formed via a direct interaction between D1-t2 and NR1-C1 via in vitro binding assay. GST-D1-t2 and GST-D1-t3 were preincubated with in vitro-translated [35S]-methionine-labeled peptides encoding NR1-C0 ([35S]-NR1-C0), NR1-C1 ([35S]-NR1-C1), or NR1-C2 ([35S]-NR1-C2) sequences. As shown in Figure 4 E, the [35S]-NR1-C1 probe bound with GST-D1-t2, but not with GST-D1-t3 or GST. The binding of [35S]-NR1-C1 with GST-D1-t2 was specific, because GST-D1-t2 did not bind with [35S]-NR1-C0 or [35S]-NR1-C2. Therefore, it appears that the D1-t2 region of the D1 receptor carboxyl tail and C1 region of the NMDA receptor NR1-1a subunit are responsible for mediating the direct interaction between D1 receptor and NR1-1a subunit of NMDA receptors. Given the fact that the observed D1-t2: NR1-C1 may be responsible for the NMDA modulation of D1 receptor function, we tested whether the activation of NMDA receptors affects the observed protein–protein interactions. As shown in Figure 4F, D1 receptor antibody failed to coimmunoprecipitate NMDA receptors in cotransfected cells pretreated with NMDA-specific antagonist AP-5, but not with D1 receptor-selective antagonist SCH-23390, suggesting that NMDA receptor stimulation, but not D1 receptor activation, is essential to form D1 and NMDA receptor complexes. However, in rat tissue and cotransfected cells, D1 and NMDA receptors could associate without exogenous NMDA receptor agonist stimulation. It is possible that constitutive NMDA receptor activation may occur because of glutamate found in the growth media (Garcia-Gallo et al., 1999).

Association of dopamine D1 and NMDA receptors. A, Coimmunoprecipitation of D1 receptors, but not D5 receptors, by NR1 antibody in rat hippocampal tissue. B, Blockade of the coimmunoprecipitation of D1 receptors by NR1 antibody with the overexpression of the D1-t2 mini-gene, but not the D1-t3 mini-gene, in COS-7 cells coexpressing D1 and NR1-1a subunit, but not in cells coexpressing D1 and NR1-1a/2A subunits. C, Schematic representation of the generated GST-fusion proteins encoding NR1-C0, NR1-C1, and NR1-C2. D, Western blots of D1 receptors after affinity precipitation by GST-NR1-C1, but not by GST-NR1-C0, GST-NR1-C2, or GST alone. E, In vitro binding assay depicting the [35S]-NR-C1 binding with GST-D1-t2, but not with GST or GST-D1-t3. GST fusion proteins (D1-t2, D1-t3) or GST (10 to ∼20 μg each) alone was incubated with [35S]-methionine-labeled NR1-C0, NR1-C1, or NR1-C2 in vitro-translated peptide, respectively. Then the beads were washed six times with PBS containing 0.5% Triton X-100 and eluted with 10 mm glutathione elution buffer. Eluates were separated by SDS-PAGE and visualized by autoradiography. Data are representative of three independent experiments. F, Blockade of the coimmunoprecipitation of NR1 by D1 antibody in cotransfected COS-7 cells pretreated with AP-5, but not with SCH-23390.
NMDA modulates D1-mediated cAMP accumulation and D1 receptor membrane expression via D1-t2: NR1-C1 interaction in both cotransfected cells and neurons
Although we have shown a direct interaction between NR1-C1 of the NR1-1a subunit and the D1-t2 of the D1 receptors, there was no direct evidence that NR1-C1 is responsible for the observed NMDA receptor modulation of D1-mediated functions. To identify whether NR1-C1 is involved directly in the functional modulation of D1 receptors, we used NR1-C0, NR1-C1, and NR1-C2 mini-genes in coexpression experiments. As shown in Figure 5A, the ability of the NMDA receptor to enhance D1-mediated cAMP accumulation could be blocked by the coexpression of NR1-C1, but not NR1-C0 or NR1-C2, mini-gene in COS-7 cells cotransfected with D1 and NR1-1a/2A subunits. Similarly, NMDA receptors failed to modulate D1 receptor membrane expression in HEK-293 cells overexpressing the NR1-C1 mini-gene with D1 and NMDA receptors (Fig. 5B). Together with the data showing that D1-t2 mini-gene was able to block the NMDA receptor modulation of D1-mediated cAMP accumulation and D1 receptor membrane expression presented in Figures 2B and 3B, we conclude that direct coupling between D1-t2 and NR1-C1 enables NMDA receptors to modulate functionally the D1-mediated cAMP accumulation and HA-D1 receptor membrane expression. In addition, we used primary rat hippocampal neurons to confirm that the modulation of D1 receptor-mediated function by the NMDA receptor occurs in a more physiologically relevant system. In cultured hippocampal neurons NMDA pretreatment significantly increased D1-like receptor-mediated cAMP accumulation (Fig. 5C) and D1 receptor membrane expression (Fig. 5D). Although SKF-81297 activates both D1 and D5 receptors, it is likely that the increased cAMP accumulation by NMDA is mediated by the D1 receptor because NMDA failed to modulate D5-mediated cAMP accumulation (Fig. 1 A). Furthermore, adenoviral-mediated expression of D1-t2 mini gene, but not D1-t3 mini gene, was able to block effectively the NMDA receptor-mediated enhancement of D1 receptor cAMP responsiveness and D1 receptor membrane expression in cultured hippocampal neurons (Fig. 5C,D).

A, In COS-7 cells expressing both D1 and NMDA receptors, the ability of the NMDA receptors to enhance D1 receptor-mediated cAMP accumulation functionally was abolished by coexpression with a mini-gene encoding the NR1-C1 domain, but not the NR1-C0 or NR1-C2 domains. Statistics analysis was performed by ANOVA, followed by post hoc Student–Newman–Keuls test. *Significantly different from control group (p < 0.05; n = 6); #significantly different from SKF treatment group (p < 0.05; n = 6). B, Summarized data indicating effects of NMDA stimulation on HA-D1 membrane expression in D1/NMDA receptor cotransfected HEK-293 cells with the overexpression of NR1-C0, NR1-C1, or NR1-C2 mini-gene, respectively. Columns show the means ± SEM of the ratios of colorimetric readings under nonpermeabilized conditions versus permeabilized conditions from cells treated with 500 μm NMDA/10μm glycine, normalized to their respective control groups (dashed line) treated with extracellular solution (ECS). *p < 0.05; **p < 0.01 (Student's t test). C, In rat primary cultured neurons the ability of the NMDA receptors to enhance D1 receptor-mediated cAMP accumulation functionally was abolished after infection with the recombinant D1-t2, but not D1-t3, adenovirus. Data were analyzed by ANOVA, followed by post hoc Student–Newman–Keuls test. *Significantly different from control group (p < 0.05; n = 6); #significantly different from SKF treatment group (p < 0.05; n = 6). D, Summarized data indicating the effects of NMDA stimulation on D1 membrane expression by infecting the cultured hippocampal neurons with the recombinant D1-t2, D1-t3 adenovirus. Columns show the means ± SEM of the ratios of colorimetric readings under nonpermeabilized conditions versus those under permeabilized conditions from neurons treated with 500 μm NMDA/10 μm glycine, normalized to their respective control groups (dashed line) treated with ECS. *p < 0.05 (Student's t test).
Discussion
In summary, we provide evidence in both cotransfected cells and cultured hippocampal neurons that activation of NMDA receptors enhances dopamine D1 receptor-mediated cAMP accumulation by recruiting more D1 receptors to the plasma membrane. Furthermore, the observed NMDA modulation of D1-mediated function is regulated by the direct protein–protein interaction between the D1-t2 region of the carboxyl tail of the D1 receptors and the C1 region of the NR1-1a subunit, because overexpression of D1-t2 or NR1-C1 mini-gene significantly blocked NMDA receptor enhancement of D1 receptor-mediated function and D1 receptor membrane expression. Several proteins have been shown to bind the C1 cassette of the NR1 receptor, including calmodulin, actinin (Ehlers et al., 1996; Wyszynski et al., 1997, 1998), yotiao (Lin et al., 1998), and neurofilament-L (Ehlers et al., 1998); these remain as candidates for playing a role in the regulation of NMDA receptor function. In addition, the C1 cassette of the NR1 subunit also contains an endoplasmic reticulum (ER) retention motif (Standley et al., 2000), and the masking/unmasking of this motif may be critical in the subcellular localization of the NMDA receptor.
NMDA receptor stimulation enhances dopamine D1 receptor-mediated cAMP accumulation in both cotransfected cells and primary hippocampal cultures. We conclude that NMDA-mediated functional modulation of D1 receptor-dependent cAMP accumulation is not a product of altered pharmacological properties, based on the observations that, in cotransfected COS-7 cells, activation of NMDA receptors did not alter (1) the estimated EC50 for SKF-stimulated D1-mediated cAMP accumulation (Fig. 1B), (2) the estimated inhibitory constant (Ki) for SKF from [3H] SCH-23390 competition binding assays (Fig. 1C), or (3) the estimated dissociation constant (KD) and the maximal [3H] SCH-23390 binding (Bmax) to the D1 receptor (Fig. 1D). Previous studies have shown that both dopamine D1 and D5 receptors specifically couple to Gs protein to stimulate cAMP accumulation and that they share a high degree of amino acid homology (Sunahara et al., 1990, 1991). It is difficult to differentiate D1 and D5 receptors functionally because they exhibit almost identical pharmacological profiles. However, dopamine D5, but not D1, receptors exhibit functional cross-talk with GABAA receptors via a direct protein–protein interaction between the carboxyl tail of D5 receptor and the second intracellular loop of the γ2 subunit of the GABAA receptors (Liu et al., 2000). In addition, we have reported recently that dopamine D1 receptors, but not D5 receptors, can modulate NMDA receptor currents via direct coupling between the D1-t3 region (S417-T446) of the carboxyl tail of the D1 receptor and NR2A subunit. In each case the receptor cross-talk is independent of classical G-protein signaling and is facilitated by interactions with specific sequence motifs that do not share homology between D1 and D5 receptors. The direct protein–protein interactions enable functional differentiation of dopamine D1 and D5 receptors and may provide an additional signal transduction pathway for neurotransmitter receptors to exert their physiological functions.
Many receptor/ion channels are trafficked between the plasma membrane and the intracellular compartments via vesicle-mediated membrane insertion and internalization. Regulation of neurotransmitter receptor membrane insertion/internalization has been proven to be an important means of controlling the functions of many receptors, such as opioid receptors (Chu et al., 1997), the β-adrenergic receptors (Karoor et al., 1998), and AMPA receptors (Man et al., 2000). Generally, agonist stimulation of G-protein-coupled receptors leads to the internalization of the activated receptor. To date, there has been little evidence showing the recruitment of G-protein-coupled receptors to the cell surface with agonist stimulation. Recently, Dr. Aperia's group has shown that in primary cultures of rat neostriatal neurons the activation of NMDA receptors recruits D1 receptors from the interior of the cell to the plasma membrane and thus increases D1-like receptor-mediated cAMP accumulation (Scott et al., 2002). However, the molecular mechanism through which activation of NMDA regulates D1 receptor trafficking and D1-mediated cAMP accumulation is not identified. In contrast, Fiorentini et al. (2003) shows that coexpression of D1 and NR1/2B subunits abolished the agonist-induced D1 receptor intracellular sequestration, suggesting that NMDA receptors may modulate D1 receptor trafficking through multiple regulatory pathways dependent on the NMDA receptor subunit composition, which is consistent with previous reports that clearly have demonstrated functional differences with distinct NMDA receptor subunit compositions (Brimecombe et al., 1997; Vicini et al., 1998; Barria and Malinow, 2002). Although the study by Fiorentini et al. (2003) has similarities to our study reported here, there are still some clear discrepancies. As opposed to what Fiorentini and colleagues have reported, we have shown that NMDA activation not only promotes a recruitment of D1 receptors to the cell surface but that NMDA activation appears to increase the ability of these two receptors to form a complex (Fig. 4F). More importantly, our study examined the functional consequence of the recruitment of D1 receptors to the cell surface that are mediated by D1–NMDA direct protein–protein interaction. We speculate that this discrepancy in agonist dependence may be attributable to subunit differences. Whereas Fiorentini and colleagues use NR2B in their studies, we have used NR2A subunits in our experiments. Several studies have demonstrated differential functional NMDA receptor properties dependent on NR2 subunit subtype composition (Krupp et al., 1996; Brimecombe et al., 1997; Sprengel et al., 1998).
In our study the activation of NMDA receptors, for as short as 30 sec, can promote membrane expression of dopamine D1 receptors, thereby increasing the availability of D1 receptors for agonist-mediated activation of adenylate cyclase. Furthermore, because the D1-mediated increase in cAMP levels with activation of NMDA receptors is a result of a 30 sec pretreatment of NMDA, the effect is unlikely to be the consequence of increased D1 receptor synthesis. In addition, [3H] SCH-23390 saturation ligand-binding analysis on COS-7 homogenates, as opposed to intact whole-cell preparations, exhibits equivalent D1 receptor densities, as shown in Figure 1 D, irrespective of NMDA receptor activation, indicating that the total number of D1 receptors, localized intracellularly and on the cell surface, is not affected with the activation of NMDA receptors. Taken together, we speculate that the increased D1 receptor membrane expression may be attributable to increased membrane insertion from the available intracellular pool of receptors, as indicated in Figure 3. We also have shown that the increased D1 receptor membrane expression is dependent on the direct protein–protein interaction between D1-t2 and NR1-C1. The NMDA receptor activation may promote the D1–NMDA receptor interaction and thereby recruit D1 receptors to the cell surface. In addition, given that there are many reports of the functional modulation of NMDA receptors by D1 receptors through a PKA-dependent pathway (Greengard, 2001), NMDA-mediated increases in D1 receptors at the cell surface via the direct protein–protein interaction may provide a mechanism for the NMDA receptor to modulate/amplify its own signaling.
Many studies have suggested the involvement of dopamine D1-like receptors in the process of working and memory (Sawaguchi and Goldman-Rakic, 1991; Williams and Goldman-Rakic, 1995). The fact that D1-like receptor stimulation can alleviate some of the “negative” symptomology of schizophrenia (Davidson and Harvey, 1990; Lidow et al., 1998) and reverse antipsychotic-induced working memory deficits (Castner et al., 2000) indicated the potential role of D1-like receptors in the maintenance and expression of schizophrenia. Furthermore, cortical glutamatergic activity also has been postulated to play a key role in the pathophysiology of schizophrenia (Iversen, 1995; Olney and Farber, 1995; Thornberg and Saklad, 1996; Tamminga, 1998; Dean et al., 1999; Mohn et al., 1999). Reductions of NMDA receptor neurotransmission in the prefrontal cortex mimic most of the behavioral symptomology associated with cognitive deficits in schizophrenia (Jentsch et al., 1997). Postmortem studies have identified a relative decrease of the NR1 subunit in the hippocampus of schizophrenia brains (Gao et al., 2000). In therapeutic trials the agents that enhance NMDA receptor activity have improved selectively the persistent negative symptoms in schizophrenia patients (Goff and Coyle, 2001; Javitt, 2001). Furthermore, NMDA blocking agents such as phencyclidine induce a cluster of symptoms that is often indistinguishable from schizophrenia (Carlsson et al., 2001). These data strongly suggest that both D1-like and NMDA receptor-mediated neurotransmissions are perturbed in a number of cortical areas of the schizophrenia brain. Our data have shown that activation of NMDA receptors not only enables NMDA receptors to form a complex with D1 receptors but also enhances D1-mediated functions via the D1-t2: NR1-C1 direct interaction. Therefore, the hypodopaminergic states may explain why blocking NMDA activity by phencyclidine is able to induce schizophrenia-like symptoms and the reduced NR1 subunit expression is observed in postmortem schizophrenia brain, which is consistent with the fact that clinical agents enhancing NMDA receptor activity will lead to the improvement of negative symptoms of schizophrenia. Thus further research into D1–NMDA interactions may help us in the identification of novel targets for development of new therapeutic agents for schizophrenia.
Footnotes
This work was supported by the Canadian Institutes of Health Research (F.L.) and Canadian Psychiatric Research Foundation (F.L.). F.L. is a National Alliance for Research on Schizophrenia and Depression Young Investigator. We gratefully acknowledge Dr. Y.-T. Wang and Dr. M. P. Charlton for critical discussion and comments. We are thankful for the generous gift of the HA-D1 cDNA from Dr. M. Caron.
Correspondence should be addressed to Dr. Fang Liu, Department of Neuroscience, Centre for Addiction and Mental Health, Clarke Division, 250 College Street, Toronto, Ontario M5T 1R8 Canada. E-mail: f.liu.a{at}utoronto.ca or fang_liu{at}camh.net.
F. J. S. Lee's present address: Department of Neurology, Harvard Medical School, Children's Hospital, Boston, MA 02115.
DOI:10.1523/JNEUROSCI.3922-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/241149-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}