

Abstract
The presynaptic and postsynaptic properties of synapses change over the course of postnatal development. Therefore, synaptic plasticity mechanisms would be expected to adapt to these changes to facilitate alterations of synaptic strength throughout ontogeny. Here, we identified developmental changes in long-term depression (LTD) mediated by group 1 metabotropic glutamate receptors (mGluRs) and dendritic protein synthesis in hippocampal CA1 slices (mGluR-LTD). In slices prepared from adolescent rats [postnatal day 21 (P21) to P35], mGluR activation induces LTD and a long-term decrease in AMPA receptor (AMPAR) surface expression, both of which require protein synthesis. In neonatal animals (P8-P15), mGluR-LTD is independent of protein synthesis and is not associated with changes in the surface expression of AMPARs. Instead, mGluR-LTD at neonatal synapses results in large decreases in presynaptic function, measured by changes in paired-pulse facilitation and the rate of blockade by the use-dependent NMDA receptor blocker (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate. Conversely, mGluR-LTD at mature synapses results in little or no change in presynaptic function, suggesting a postsynaptic mechanism of expression. The developmental switch in the synaptic mechanisms of LTD would differentially affect synapse dynamics and perhaps information processing over the course of postnatal development.
- metabotropic glutamate receptor
- long-term depression
- protein synthesis
- AMPA receptor endocytosis
- hippocampus
- CA1
Introduction
There is growing evidence that both the presynaptic and postsynaptic properties of synapses change over the course of postnatal development. Nascent synapses are characterized by high presynaptic release probability and few postsynaptic AMPA receptors (AMPARs). Synapse maturation is accompanied by the acquisition of AMPARs and decreases in neurotransmitter release probability (Bolshakov and Siegelbaum, 1995; Pouzat and Hestrin, 1997; Liao et al., 1999; Petralia et al., 1999; Reyes and Sakmann, 1999; Pickard et al., 2000; Wasling et al., 2004) (but see Dumas and Foster, 1995; Hsia et al., 1998). For developing synapses to remain plastic over the course of synapse maturation, plasticity mechanisms must adapt with these changing synaptic properties. Indeed, there is evidence for changes in the mechanisms of longterm potentiation during synapse development (Yasuda et al., 2003). Here, we demonstrate that both the synaptic and molecular mechanisms of synaptic long-term depression (LTD) change over the course of synapse maturation in area CA1 of the rat hippocampus. Specifically, we examined the developmental changes of LTD, which rely on group 1 metabotropic glutamate receptors (mGluRs) and dendritic protein synthesis (mGluR-LTD) (Huber et al., 2000). This work was motivated by findings that group 1 mGluR function (measured as phosphoinositide turnover) and synaptic polyribosome number peak during the period of synapse formation and maturation [approximately postnatal day 7 (P7) to P15 in the hippocampus], suggesting that mGluR-LTD plays a significant role in the plasticity of developing synapses (Steward and Falk, 1985, 1991; Nicoletti et al., 1986; Dudek et al., 1989; Palmer et al., 1990; Casabona et al., 1997).
The developmental regulation and site of expression of mGluR-LTD in the hippocampus has been controversial. Some studies suggest that mGluR-LTD is only observed at early developmental time periods in area CA1 (P7-P11) (Bolshakov and Siegelbaum, 1994; Overstreet et al., 1997). However, others indicate that mGluR-LTD is restricted to adult animals (Kemp et al., 2000). There is general agreement that mGluR-LTD is induced postsynaptically (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Watabe et al., 2002). However, many studies of mGluR-LTD of immature neurons (P4-P11) conclude that mGluR-LTD is expressed presynaptically using both electrophysiological and optical methods (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Fitzjohn et al., 2001; Zakharenko et al., 2002; Feinmark et al., 2003; Rammes et al., 2003).
Evidence for a postsynaptic mechanism of mGluR-LTD expression comes from studies demonstrating that the selective group 1 mGluR agonist R,S-dihydroxyphenylglycine (DHPG), which induces LTD, results in endocytosis and a persistent decrease in postsynaptic AMPARs and NMDA receptors (NMDARs), which requires protein synthesis (Snyder et al., 2001; Xiao et al., 2001). DHPG-induced LTD is blocked by the postsynaptic injection of endocytosis and protein synthesis inhibitors (Snyder et al., 2001; Xiao et al., 2001), suggesting a postsynaptic mechanism of expression.
Here, we find a developmental switch in the protein synthesis dependence and synaptic locus of mGluR-LTD, which occurs between the second and fourth postnatal week. This switch may occur to accommodate the changing properties of synapses and may have consequences for information processing over the course of postnatal development.
Materials and Methods
Drugs. d,l-AP-5, (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten5,10-imine maleate (MK801) (Tocris Cookson, Ellisville, MO), anisomycin, and picrotoxin (Sigma, St. Louis, MO) were prepared fresh in artificial CSF (ACSF). The cannabinoid 1 receptor (CB1R) antagonist 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1 H-pyrazole-3-carboxamide (AM281) and R-(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-DE]-1,4-benzoxazin-6-yl]-(1-naphtmalenyl)methanone mesylate (WIN55,212-2) (Tocris Cookson) were prepared as a 1000× stocks in DMSO, used fresh or kept as stocks at -20°C, and diluted to final concentration in ACSF. Slices were preincubated in antagonists or inhibitors for 20-30 min before DHPG or paired-pulse low-frequency stimulation (PP-LFS). The effects of all of the pharmacological treatments on LTD were evaluated by comparing interleaved control and treated slices.
Electrophysiology. Long-Evans hooded rats were obtained from Charles River Laboratories (Wilmington, MA). Hippocampal slices (400 μm) were prepared from 8- to 35-d-old rats. Rats were anesthetized with the barbiturate pentobarbital (50 mg/kg) and decapitated soon after the disappearance of corneal reflexes. The brain was removed and dissected and then sliced using a vibratome (VT 1000S; Leica, Nussloch, Germany) in ice-cold dissection buffer containing the following (in mm): 2.6 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, 212 sucrose, and 10 dextrose. Area CA3 was surgically removed from each slice immediately after sectioning. The slices were transferred into a reservoir chamber filled with ACSF containing the following (in mm): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2 MgCl2, and 10 dextrose. Slices were allowed to recover for 2-5 h at 30°C. ACSF and dissection buffer were equilibrated with 95% O2 and 5% CO2.
For recording, slices were transferred to a submerged recording chamber, maintained at 30°C, and perfused continuously with ASCF at a rate of 2-3 ml/min. Field potentials (FPs) were recorded with extracellular recording electrodes (1 MΩ) filled with ACSF and placed in stratum radiatum of area CA1. FPs were evoked by monophasic stimulation (duration, 200 μs) of Schaffer collateral/commissural afferents with a concentric bipolar tungsten stimulating electrode (Frederick Haer Company, Bowdoinham, ME). NMDA receptor-mediated FPs were isolated in a modified ASCF containing the following (in mm): 3 CaCl2, 0.1 MgCl2, 2.5 KCl, 0.02 DNQX, 0.001 glycine, and 0.1 picrotoxin. Stable baseline responses were collected every 30 s using a stimulation intensity (10-30 μA), yielding 50-60% of the maximal response. FPs were filtered at 2 kHz, acquired, and digitized at 10 kHz on a personal computer using custom software (LabVIEW; National Instruments, Austin, TX). The initial slope of the FP was used to measure the stability of synaptic responses and quantify the magnitude of LTD. mGluR-LTD was induced by pairs of stimuli (interstimulus interval, 50 ms) delivered at 1 Hz for 15 min (1800 pulses; PP-LFS), by stimulation delivered at 5 Hz for 3 min (900 pulses; 5 Hz), or by application of 100 μm DHPG (Tocris Cookson) for 5 min. For paired-pulse facilitation (PPF) experiments, pairs of presynaptic stimulation (interpulse interval, 50 ms) were delivered every 30 s throughout the experiment.
The group data were analyzed as follows: (1) the initial slopes of the FP were expressed as percentages of the preconditioning or DHPG baseline average; (2) the time scale in each experiment was converted to time from the onset of conditioning or DHPG; and (3) the time-matched, normalized data were averaged across experiments and expressed as means ± SEM. The PPF ratio was obtained by dividing the initial field potential slope from the second pulse (FP2) by that of the first pulse (FP1) and then normalizing to the pre-DHPG baseline PPF value. For example, a 20% increase in PPF represents a change in the PPF or FP2/FP1 ratio from 1.6 to 1.92. Time constants (τ) of the rate of MK801 blockade of NMDAR FPs were determined by fitting the decay of the maximum amplitude of NMDAR FPs in MK801 with a double exponential using Origin analysis software (Microcal Software, Northampton, MA). Significant differences were determined by a Student's independent or paired t test (if indicated). p < 0.05 was considered to represent significant differences. For correlation analysis, correlation coefficients were determined, and z tests were performed to determine p values using StatView software (SAS Institute, Cary, NC).
Biochemical measurements of surface-expressed AMPA receptors. Biotinylation experiments were performed as described previously (Chung et al., 2000; Heynen et al., 2003). Hippocampal slices were prepared as described for electrophysiology experiments. After a 2-3 h recovery period in ACSF, slices were treated with DHPG (5 min), NMDA (3 min), or ACSF (control). DHPG experiments were performed in the presence of 100 μm d,l-AP-5. From each rat, two to three slices were pooled together for one condition. Slices were placed on ice to stop endocytosis and then were washed with ice-cold ACSF and incubated in ACSF containing 1 mg/ml sulfo-NHS-LC-biotin (Pierce, Rockford, IL) for 10 min on ice. To quench the biotin reaction, slices were washed three times with Trisbuffered saline and homogenized in a modified radioimmunoprecipitation assay (RIPA) buffer containing 50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 0.1% SDS, 0.5% Na-deoxycholate, 150 mm NaCl, 2 mm EDTA, 50 mm NaH2PO4, 50 mm NaF, 10 mm Na4P2O7, 1 mm Na3VO4, and Protease Inhibitor Cocktail III (Calbiochem, La Jolla, CA). The homogenates were centrifuged at 14,000 × g for 10 min at 4°C. Protein concentrations were measured with a BCA Protein Assay (Pierce). Protein (15 μg) was removed for total protein measurements. Protein (150 μg) was then mixed with 150 μl of UltraLink immobilized NeutrAvidin beads (Pierce) by rotating for 2 h at 4°C. The beads were washed with 10 vol of RIPA buffer and then eluted with SDS-PAGE sample buffer supplemented with 50 mm dithiothreitol for 20 min at 90°C. Both total and biotinylated proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and probed with anti-GluR1 C-terminal antibody (1:5000; Upstate Biotechnology, Lake Placid, NY), anti-GluR2/3 N-terminal antibody (1:1000; Chemicon, Temecula, CA) or anti-α-actin antibody (1:1000; Chemicon), or anti-GluR6/7 (1:5000; Upstate Biotechnology). Immunoreactive bands were visualized by enhanced chemiluminescence, captured on autoradiography film (Eastman Kodak, Rochester, NY). Digital images were produced by densitometric scans of autoradiographs on a ScanJet 4300C (Hewlett Packard, Palo Alto, CA) and quantified using Scion (Frederick, MD) Image software. The surface/total protein ratio was calculated for each condition. When duplicate conditions were performed within one animal, the ratio values were averaged to obtain an animal average for that condition. Therefore, the n values for the biotinylation experiments (see Figs. 4, 5, 6) represent the number of rats, as opposed to slices. Significant differences between surface/total ratios of treated slices and within-animal control slices were determined using a paired t test (for ratio and p values, see Table 1). Although the raw ratio values were used for statistical comparisons, the group data are presented in Figures 4, 5, 6 as a percentage of condition control to compare across different treatment conditions.

DHPG treatment of acute hippocampal slices from adolescent rats results in a protein synthesis-dependent decrease in AMPAR surface expression. A-F, Sample Western blots of total (T) and surface (S) GluR2/3 or GluR1 subunits of the AMPA receptor. Quantitative data of the ratio of surface to total GluR2/3 or GluR1 in hippocampal slices taken either 15 or 60 min after DHPG application (100 μm; 5 min). The number of experiments per group is indicated on each bar. *p < 0.05. A, B, DHPG treatment of hippocampal slices prepared from adolescent rats results in a long-term decrease of GluR2/3 and GluR1 surface expression using receptor biotinylation. C, D, Preincubation in the mGluR antagonist LY341495 (100 μm) blocks DHPG-induced decreases in GluR2/3 and GluR1 surface expression. Quantitative data of the ratio of surface to total GluR2/3 or GluR1 in hippocampal slices taken 15 min after DHPG application (100 μm; 5 min) in the presence of LY341495. E, F, Preincubation of slices in the protein synthesis inhibitor anisomycin (20 μm) specifically blocks the late (60 min) decrease in AMPAR surface expression induced by DHPG. Error bars represent SEM.

DHPG does not affect the surface expression of kainate receptors in adolescent rats. A, Sample Western blot of total (T) and surface (S) GluR6/7. Quantitative data of the ratio of surface to total GluR6/7 in hippocampal slices taken either 15 or 60 min after DHPG application (100 μm; 5 min). The number of experiments per group is indicated on each bar. B, Sample Western blot of total actin and actin “pulled down” (P) with avidin beads, demonstrating that intracellular proteins are not biotinylated in this assay. Error bars represent SEM.

NMDAR activation, but not mGluR activation, reduces AMPAR surface expression in neonatal rat slices. A, Sample Western blot of total (T) and surface (S) GluR2/3. Quantitative data of the ratio of surface to total GluR2/3 in hippocampal slices taken either 15 or 60 min after DHPG application (100 μm; 5 min). The number of experiments per group is indicated on each bar. *p < 0.05. B, Sample Western blot of GluR1 and quantitative data of the same samples as those used in A. C, Sample Western blot and quantitative data of the ratio of surface to total GluR2/3 in hippocampal slices taken either 10 or 60 min after NMDA application (20 μm; 3 min). D, Sample Western blot of GluR1 and quantitative data of the same samples as those used in C. Error bars represent SEM.
Raw ratio values from surface biotinylation experiments
Results
mGluR-LTD can be induced at immature synapses
We first determined whether mGluR-LTD can be induced pharmacologically and synaptically at immature synapses. Hippocampal slices were prepared from neonatal (P8-P15) or adolescent (P21-P35) rats. Extracellular field potential recordings were obtained in area CA1 and were elicited by Schaffer collateral stimulation. LTD was induced using either the group 1 mGluR agonist DHPG (100 μm; 5 min) or synaptic stimulation (900 pairs of stimulation pulses with a 50 ms interval delivered at 1 Hz; PP-LFS). Because the NMDAR-dependent form of LTD is robust at this developmental age (Dudek and Bear, 1993), all LTD experiments presented in this study were performed in the presence of the NMDAR antagonist d,l-AP-5 (100 μm). We find that LTD can be induced in slices of neonatal rats with DHPG or PP-LFS in the presence of AP-5 (Fig. 1). Although group 1 mGluR function peaks during the early developmental period, DHPG-induced LTD is not enhanced at immature synapses (83 ± 3% of pre-DHPG baseline; n = 10) compared with LTD in slices from adolescent rats (84 ± 4%; n = 7) (Fig. 1A). In contrast, LTD induced with PP-LFS is enhanced in slices from P8-P15 rats (63 ± 4%; n = 6) compared with P21-P35 rats (84 ± 3%; n = 7; p < 0.02) (Fig. 1B). Pretreatment of neonatal slices for 5 min in the broad mGluR antagonist (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495) (100 μm) (Fitzjohn et al., 1998) abolished DHPG-induced LTD (97 ± 2%; n = 6). In a subset of experiments, DHPG was applied after LY341495 washout and induced LTD (79 ± 4%; n = 3) (Fig. 1C). Preincubation (20-30 min) in LY341495 significantly reduced PP-LFS-induced LTD (92 ± 5%; n = 7) compared with interleaved control slices (68 ± 6%; n = 11; p = 0.004) (Fig. 1C). Although there was a slight residual depression observed with PP-LFS in LY341495, it was not significant compared with baseline values (p = 0.14). Our results indicate that an mGluR-dependent LTD can be induced in immature synapses by synaptic stimulation (PP-LFS) or DHPG.

Chemically and synaptically induced mGluR-dependent LTD in neonatal hippocampus. All of the experiments shown in the figures were performed in the NMDA receptor antagonist d,l-AP-5 (100 μm). A-D, Plotted are FP slopes (mean ± SEM) as a function of time from onset of DHPG or conditioning stimulation. A, DHPG (100 μm; 5 min; arrow) induces LTD of FP slope values, which is similar in magnitude in neonatal (P8-P15) and adolescent (P21-P35) rats. B, LTD induced with PP-LFS is enhanced in slices from neonatal rats compared with adolescent rats. C, DHPG-LTD in neonatal rats is blocked by the broad mGluR antagonist LY341495 (100 μm). D, PP-LFS-induced LTD in neonatal rats is inhibited by LY341495 (100 μm). Representative FPs (average, 1 min) are shown for each experiment at the times indicated by the numbers on the graph. Calibration: 0.5 mV, 5 ms.
Developmental switch in the protein synthesis dependence of mGluR-LTD
mGluR-LTD in adolescent rats (P21-P35) is dependent on dendritic protein synthesis and is independent of transcription (Huber et al., 2000). Because numerous synaptic polyribosomes are observed at developing synapses (Steward and Falk, 1985, 1991), we hypothesized that mGluR-LTD at neonatal synapses would also be protein synthesis dependent. Surprisingly, we find that mGluR-LTD in neonatal rats is not sensitive to the protein synthesis inhibitors (Fig. 2). In neonatal rat slices, DHPG induced significant LTD in slices pretreated with anisomycin (88 ± 2%; n = 10) (Fig. 2A), which was not different from LTD in interleaved control slices (83 ± 3%; n = 10; p = 0.21). The control DHPG-LTD values are the same data as those presented in Figure 1A and were replotted in Figure 2A for comparison with anisomycin-treated slices. In contrast, as reported previously, DHPG-induced LTD in adolescent rats was completely inhibited by anisomycin (100 ± 5%, n = 6; interleaved control slices, 84 ± 3%, n = 7; p = 0.01) (Fig. 2C). Similar results were observed with another protein synthesis inhibitor, cycloheximide (60 μm). DHPG-LTD was completely blocked by cycloheximide in adolescent rats (103 ± 2%, n = 4; control slices, 77 ± 5%, n = 4; p = 0.01) (Fig. 2D) and was unaffected in neonatal rats (80 ± 3%, n = 6; control slices, 81 ± 3%, n = 5; p = 0.79) (Fig. 2C). Likewise, LTD induced with synaptic stimulation (PP-LFS) only requires protein synthesis at mature synapses (P8-P15: control, 63 ± 4%, n = 6; anisomycin, 60 ± 5%, n = 4; P21-P35: control, 77 ± 3%, n = 18; anisomycin, 99 ± 4%, n = 16; p = 0.0001) (Fig. 3A,B). PP-LFS-induced LTD in neonatal rats was also insensitive to cycloheximide (62 ± 6%, n = 8; control slices, 64 ± 2%, n = 7; p = 0.01) (Fig. 3C) in contrast to adolescent rats (Huber et al., 2000).

The protein synthesis dependence of chemically induced mGluR-LTD is developmentally regulated. A, C, DHPG-induced LTD in slices from neonatal rats is insensitive to the protein synthesis inhibitors anisomycin (20 μm; A) or cycloheximide (60 μm; C). B, D, In contrast, anisomycin (B) and cycloheximide (D) block DHPG-LTD in adolescent rats.
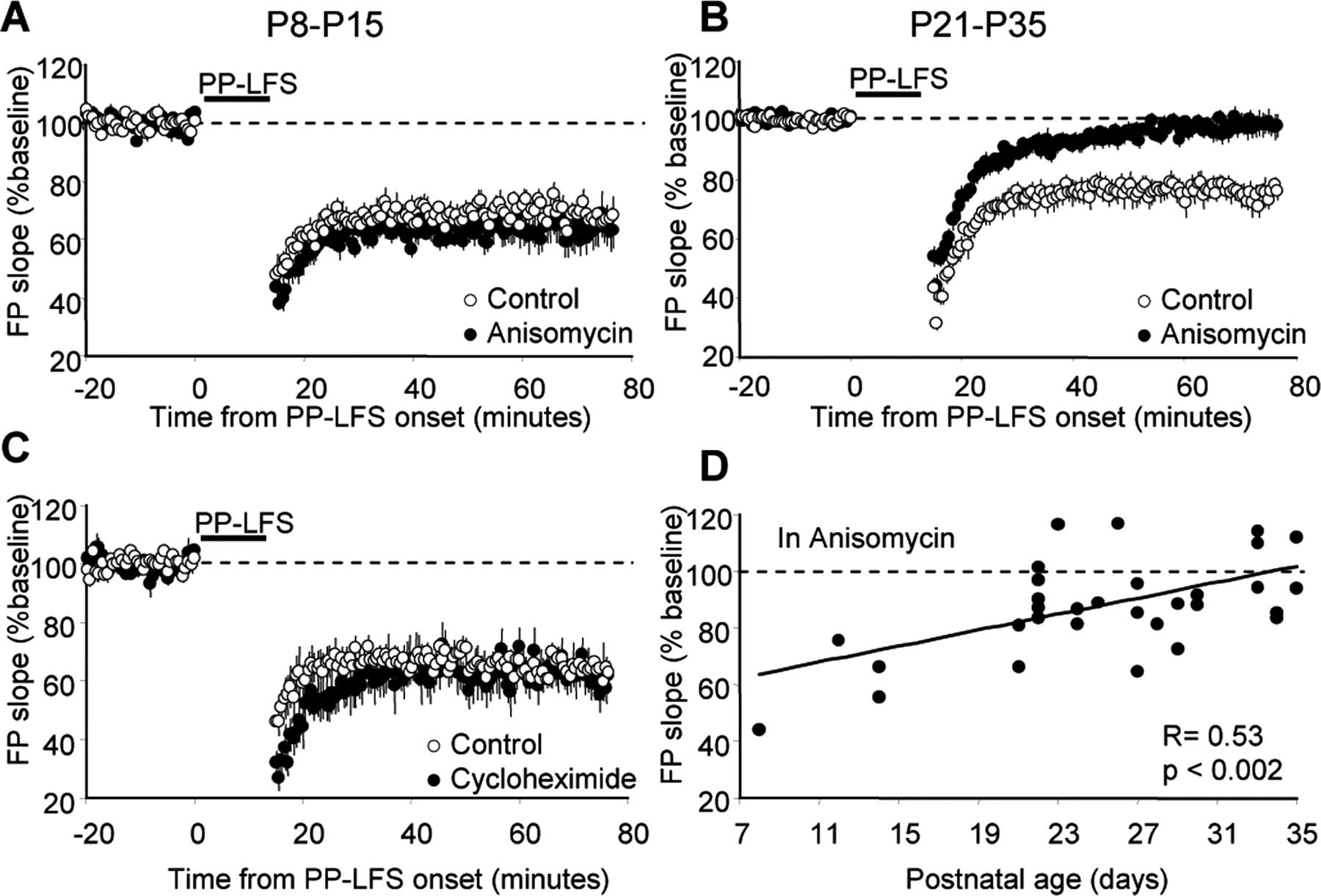
The protein synthesis dependence of synaptically induced mGluR-LTD is developmentally regulated. A, C, LTD induced with synaptic stimulation (PP-LFS) in neonatal rats is not affected by anisomycin (A) or cycloheximide (C). B, PP-LFS-induced LTD in slices from adolescent rats is blocked by anisomycin. D, The ability of anisomycin to block PP-LFS-induced LTD is significantly correlated with the postnatal age of the rats.
Previous studies of mGluR-LTD in neonatal hippocampus have used 5 Hz synaptic stimulation (3 min) (Bolshakov and Siegelbaum, 1994; Bolshakov et al., 2000; Zakharenko et al., 2002). Therefore, we also examined the protein synthesis dependence of mGluR-LTD using this protocol. As in LTD induced with DHPG and PP-LFS at this age, 5 Hz-induced LTD was also insensitive to anisomycin (P8-P15: interleaved controls, 81 ± 5%, n = 7; anisomycin, 78 ± 4%, n = 7; p = 0.89).
These results indicate that, as synapses mature, mGluR-LTD becomes increasingly dependent on protein synthesis. In support of this idea, the ability of anisomycin to block PP-LFS-induced LTD is significantly correlated with postnatal age (r = 0.53; p < 0.002) (Fig. 3D).
Developmental changes in mGluR-induced decreases in AMPAR surface expression
In dissociated cultured neurons, activation of group 1 mGluRs results in a long-term decrease in the surface expression of AMPARs and NMDARs, which requires protein synthesis (Snyder et al., 2001; Xiao et al., 2001). These results indicate that the new proteins required for mGluR-LTD most likely function to control AMPAR endocytosis or trafficking. Therefore, mGluR-LTD at immature synapses may not be mediated by AMPAR internalization or there may be sufficient levels of protein(s) to maintain internalization in the absence of new synthesis.
To determine whether mGluR-dependent AMPAR endocytosis was developmentally regulated, we measured DHPG-induced changes in surface expression of AMPAR subunits GluR1 and GluR2/3. Previous studies of DHPG-induced AMPAR endocytosis have been performed in dissociated cultured hippocampal neurons (Snyder et al., 2001; Xiao et al., 2001). To correlate the developmental changes in mGluR-LTD with DHPG-induced AMPAR endocytosis in the same preparation, we performed these experiments in acute hippocampal slices using receptor biotinylation. We first verified that we could observe decreases in AMPAR surface expression in acute slices from adolescent rats. Slices were incubated in control ACSF or treated with DHPG (100 μm; 5 min). Surface receptors were labeled with biotin either 15 or 60 min after application of DHPG. Biotinylated receptors were precipitated, and the ratio of surface to total GluRs was determined by quantitative Western blotting. The biochemical analysis confirmed that DHPG treatment of hippocampal slices (P21-P35) results in a long-term (60 min) decrease in biotinylated (surface)/total ratio of GluR2/3 (15 min after DHPG, 81 ± 6% of control slices, p = 0.03, n = 7; 60 min after DHPG, 74 ± 6% of control slices, n = 6, p = 0.004) (Fig. 4A) and GluR1 (15 min after DHPG, 81 ± 4%, n = 9, p = 0.001; 60 min after DHPG, 80 ± 5%, n = 6, p = 0.018) (Fig. 4B; for raw ratio values, see Table 1). DHPG did not affect the total levels of GluR2/3 (15 min, 99 ± 3% of control, p = 0.87; 60 min, 103 ± 12%, p = 0.96) or GluR1 (15 min, 106 ± 13%, p = 0.4; 60 min, 101 ± 22%, p = 0.78). As in mGluR-LTD, the DHPG-induced decreases in surface expression were blocked by the broad-range mGluR antagonist LY341495 (100 μm; GluR1, 15 min after DHPG, 113 ± 12%, n = 6, p = 0.39; GluR2/3, 15 min after DHPG, 113 ± 3%, n = 5, p = 0.02) (Fig. 4C,D).
As observed previously in neuronal culture, DHPG-induced decreases in AMPAR surface expression in hippocampal slices were sensitive to the protein synthesis inhibitor anisomycin (20 μm). Slices preincubated in anisomycin displayed significantly reduced AMPAR surface expression at 15 min after DHPG treatment but not at 60 min after DHPG treatment (GluR2/3, 15 min after DHPG, 84 ± 6%, n = 8, p = 0.036; 60 min after DHPG, 102 ± 5%, n = 7, p = 0.97; GluR1, 15 min after DHPG, 84 ± 1%, n = 7, p = 0.0003; 60 min after DHPG, 94 ± 10%, p = 0.55, n = 9) (Fig. 4E,F).
To determine whether the effects of DHPG are specific to the AMPA subtype of receptors, we measured changes in surface expression of kainate receptor subunits GluR6 and GluR7. In area CA1, kainate receptors are expressed primarily presynaptically on both excitatory and inhibitory synapses and postsynaptically on interneurons (Huettner, 2003). DHPG did not affect the surface expression of GluR6/7 (15 min after DHPG, 100 ± 10%, p = 0.77, n = 6; 60 min after DHPG, 93 ± 5%, p = 0.58, n = 7) (Fig. 5A), indicating that DHPG causes a specific reduction in AMPAR surface expression. Additional control experiments confirmed that intracellular proteins, such as actin, were not biotinylated in this assay (n = 3) (Fig. 5B). Together, these experiments confirm that DHPG causes an mGluR- and protein synthesis-dependent long-term decrease in AMPAR surface expression in acute slices from adolescent rats.
In contrast to adolescent rats, DHPG treatment of slices from neonatal animals did not decrease the surface expression of GluR2/3 and GluR1 subunits (GluR2/3: 15 min after DHPG, 98 ± 5%, n = 10, p = 0.8; 60 min after DHPG, 104 ± 5%, n = 12, p = 0.69; GluR1: 15 min DHPG, 100 ± 4%, n = 9; 60 min after DHPG, 111 ± 4%, p = 0.003, n = 10) (Fig. 6A,B).
In addition to mGluRs, chemical activation of NMDARs induces LTD and internalization of AMPARs (Lee et al., 1998; Colledge et al., 2003). We performed additional experiments, treating neonatal slices with NMDA to determine whether there is a general deficit in activity-induced AMPAR internalization in neonatal slices or whether the deficit is specific for the mGluR pathway. We first confirmed that NMDA (20 μm; 3 min) induced LTD in CA1 of neonatal hippocampal slices (80 ± 3% of baseline at 55-60 min after NMDA application; n = 5). Unlike DHPG treatment of neonatal slices, we observed decreases in surface expression of GluR1 and GluR2/3 at 10 min (GluR1, 82 ± 2% of control slices; n = 6, p < 0.001; GluR2/3, 74 ± 6%, n = 5, p = 0.03) and 60 min (GluR1, 75 ± 5%, n = 5, p = 0.008; GluR2/3, 89 ± 2%, n = 4, p = 0.02) after NMDA (20 μm; 3 min) application (Fig. 6C,D; Table 1). These data demonstrate that mGluR-induced, but not NMDA-induced, decreases in AMPAR surface expression are developmentally regulated and do not occur at immature synapses. This finding also suggests that mGluR-LTD at this age is not mediated by a decrease in postsynaptic receptor number.
Presynaptic changes accompany mGluR-LTD at neonatal synapses
Previous studies of mGluR-LTD in slices from young (P4-P18) rats show strong evidence that LTD expression is mediated by a long-term decrease in presynaptic function (Bolshakov and Siegelbaum, 1994; Fitzjohn et al., 2001; Zakharenko et al., 2002; Rammes et al., 2003). Therefore, based on these studies and our findings that mGluR-dependent decreases in AMPAR surface expression are only observed in adolescent rats, we propose that there is a developmental change in the synaptic mechanisms of mGluR-LTD expression. To test this idea, we used two parameters to measure changes in presynaptic release probability during LTD, PPF, and the rate of blockade of NMDA receptor-mediated synaptic responses by MK801. These parameters were measured after DHPG-induced LTD in slices from both neonatal (P8-P15) and adolescent (P21-P35) rats. DHPG-induced LTD was examined, as opposed to synaptically induced LTD, because a similar magnitude of DHPG-induced LTD is observed across these developmental ages (Figs. 1A, 7B) and would allow comparison of the degree of PPF changes or rate of MK801 blockade across development.

mGluR-LTD in neonatal rats is associated with changes in presynaptic function. A, Representative FPs elicited by paired-pulse stimulation in P8-P15 and P21-P35 hippocampal slices during baseline (1) and after 50 min of DHPG onset (2). Calibration: 0.5 mV, 20 ms. DHPG trace (2) was scaled to baseline FP1 amplitude for comparison of PPF changes within a single experiment. Note that, in the P8 rat, the second response is facilitated but is unchanged at P29. B, Group data of PPF (FP2 slope/FP1 slope as a percentage of pre-DHPG baseline) change during LTD in neonatal and adolescent animals. The asterisk indicates that PPF changes are greater in neonatal than adolescent rats (p < 0.02). C, DHPG-induced LTD of FP1 was not different among age groups. D, Representative NMDAR FPs taken at the times indicated in E and F from a P13 or P33 rat. Calibration: 0.2 or 0.1 mV (as indicated), 10 ms. E, F, Group average of decay of NMDAR FP amplitude in the presence of MK801 (10 μm) in control or after DHPG (100 μm; 5 min) treatment of neonatal (E) or adolescent (F) rat slices. Fast component (τ1) is slower in DHPG-treated neonatal slices.
Manipulations that alter presynaptic release probability, such as decreases in the Ca2+/Mg2+ ratio or adenosine, increase paired-pulse facilitation. Generally, the magnitude of PPF is inversely related to presynaptic release probability (Creager et al., 1980; Manabe et al., 1993; Debanne et al., 1996). However, preferential postsynaptic “silencing” of high- or low-release probability synapses also result in PPF changes (Poncer and Malinow, 2001). Pairs of stimulation (interstimulus interval, 50 ms) were delivered during baseline stimulation, during DHPG application, and for 1 h after DHPG. In slices from neonatal rats, PPF ratios increased during DHPG application and persisted for at least 60 min after DHPG (116 ± 2% of pre-DHPG baseline PPF values; n = 14; p < 0.0001), as reported previously (Fitzjohn et al., 2001). PPF values also increased during LTD in slices from adolescent rats (109 ± 2% of pre-DHPG baseline; n = 24; p = 0.008). However, the PPF increases in neonatal animals were significantly greater than those observed in adolescent rats (p < 0.02) (Fig. 7A). There was no difference in the magnitude of DHPG-induced LTD (P8-P15, 72 ± 2%, n = 14; P21-P35, 69 ± 2%, n = 24; p = 0.4) (Fig. 7C) or the absolute levels of PPF [P8-P15, 1.63 ± 0.05 (FP2/FP1 slope); P21-P35, 1.59 ± 0.05; p = 0.63] between these developmental ages. To further support a developmental change in the presynaptic contribution to mGluR-LTD, there is an inverse correlation of PPF changes during LTD and postnatal age (r = 0.4; p < 0.01).
As another means to measure developmental changes in presynaptic function associated with LTD, we used the irreversible, use-dependent NMDAR antagonist MK801. During repetitive presynaptic stimulation, the rate of blockade of NMDAR responses by MK801 has been used to determine presynaptic release probability (Pr) and detect changes in Pr during long-term plasticity, such as long-term potentiation and LTD (Hessler et al., 1993; Rosenmund et al., 1993; Weisskopf and Nicoll, 1995; Kullmann et al., 1996; Xiao et al., 1997; Kaneko and Takahashi, 2004). If Pr is relatively high, there is a greater number of NMDAR channels opened and a more rapid blockade of the response by MK801. We compared the rate of MK801 blockade of NMDAR-mediated FPs in control slices and those in which LTD had been induced using DHPG from both neonatal and adolescent rats. DHPG was used to induce LTD in normal ACSF. After LTD (of the AMPAR response) was established (30 min after DHPG application), a modified ACSF (see Materials and Methods) was applied to the slice to isolate the NMDAR-mediated FP, and this response was allowed to stabilize (∼20-25 min). Synaptic stimulation was stopped for 10 min, and MK801 (10 μm) was applied to the slice. Synaptic stimulation (200 pulses) was resumed at 0.5 Hz in the presence of MK801. The rate of decay of the NMDAR FP amplitude by MK801 was fit by a double exponential, and time constants for the fast (τ1) and slow (τ2) components were obtained (Fig. 7E,F) (Rosenmund et al., 1993). These two components are thought to represent two populations of synaptic terminals with a high and low Pr (Rosenmund et al., 1993). In neonatal slices, DHPG caused an increase in τ1 (DHPG, τ1, 49 ± 8 s; control, τ1, 26 ± 3 s; n = 6; p = 0.02), consistent with a lower Pr after DHPG treatment. This change was evident by the slower decay curves in DHPG-treated slices (Fig. 7E). In contrast, DHPG treatment of adolescent slices did not significantly affect τ1, and the decay curves overlapped (DHPG, τ1, 49 ± 8 s; control, τ1, 33 ± 8 s; n = 6; p = 0.19) (Fig. 7F). There was no change in τ2 at either age (neonatal: DHPG, τ2, 222 ± 22 s; control, τ2, 178 ± 21 s; n = 6; p = 0.15; adolescent: DHPG, τ2, 188 ± 24 s; control, τ2, 254 ± 35 s; n = 6; p = 0.11). There were no differences in τ1 or τ2 between the neonatal and adolescent groups (p = 0.45 and 0.1, respectively). These results, together with the observed PPF changes during LTD, confirm that mGluR-LTD at neonatal synapses is accompanied by decreases in presynaptic release probability (Bolshakov and Siegelbaum, 1994; Fitzjohn et al., 2001; Zakharenko et al., 2002). Our results also indicate that the magnitude of these presynaptic changes diminish as synapses mature and suggest that other, perhaps postsynaptic, mechanisms contribute to mGluR-LTD in mature synapses.
CB1 receptor activation is not required for mGluR-LTD in neonatal rats
Previous work has demonstrated that the induction of mGluR-LTD at neonatal synapses requires postsynaptic Ca2+ influx and depolarization (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997). Because of the presynaptic changes associated with LTD, the existence of a retrograde messenger has been postulated. Here, we tested the involvement of potential retrograde messengers in mGluR-LTD at neonatal synapses: endocannabinoids.
Endocannabinoids are released from CA1 pyramidal neurons in response to group 1 mGluR activation and depress both excitatory and inhibitory synaptic transmission via a presynaptic CB1R (Misner and Sullivan, 1999; Varma et al., 2001; Ohno-Shosaku et al., 2002). Endocannabinoids have also been implicated in LTD at inhibitory synapses in area CA1 and at excitatory synapses in the basal ganglia and neocortex (Gerdeman et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003; Sjostrom et al., 2003). A previous study failed to find a role for CB1R in DHPG-induced LTD in area CA1 of adolescent mice (Rouach and Nicoll, 2003). We obtained similar results in neonatal rat hippocampal slices and found no role for CB1R in mGluR-LTD (Fig. 8B,C). The effects of the CB1R antagonist AM281 (1 μm) were tested on mGluR-LTD in neonatal slices, which blocks mGluR and depolarization-induced endocannabinoid release (Gifford et al., 1997; Maejima et al., 2001; Ohno-Shosaku et al., 2002; Melis et al., 2004). AM281 (1 μm) had no effect on either DHPG-induced LTD (AM281, 84 ± 6%, n = 7; control, 84 ± 4%, n = 7; p = 0.94) or LTD induced with PP-LFS (AM281, 73 ± 5%, n = 8; control, 67 ± 3%, n = 10; p = 0.26). In contrast, preapplication of AM281 greatly reduced the synaptic depression induced by the CB1R agonist WIN55,212-2 (2 μm; 10 min) (D'Ambra et al., 1992; Al-Hayani and Davies, 2000), indicating that the antagonist is effective in our slice preparation (WIN55,212-2, 47 ± 9% of baseline, n = 4; WIN55,212-2 plus AM281, 87 ± 2%, n = 4; p = 0.03) (Fig. 8A). These results confirm that CB1R activation depresses excitatory synaptic transmission in neonatal hippocampal area CA1 (Al-Hayani and Davies, 2000) and demonstrate that CB1Rs are not required for mGluR-induced LTD at this age.

CB1R activation is not required for mGluR-LTD in neonatal slices. A, CB1R antagonist AM281 (1 μm) is effective in the hippocampal slice preparation and inhibits synaptic depression induced by a brief application of the CB1R agonist WIN55,212-2 (WIN 55; 2 μm; 10 min). Solid bars indicate the time of drug application. Representative FPs are taken at the times indicated by the numbers on the graph. Calibration: 0.5 mV, 5 ms. B, C, Preincubation in AM281 does not affect LTD induced with DHPG (B) or PP-LFS (C) in neonatal rat slices.
Discussion
Here, we show, using both pharmacological and synaptic stimulation of group 1 mGluRs, that the synaptic mechanisms and protein synthesis dependence of mGluR-LTD change with developmental age. In neonatal synapses, mGluR-LTD does not rely on protein synthesis and is associated with large changes in PPF and the rate of MK801 blockade, suggesting a presynaptic site of LTD expression. Consistent with this idea, mGluR activation of neonatal slices does not cause a decrease in AMPAR surface expression. As synapses mature, mGluR-LTD and the associated decrease in AMPAR surface expression require new protein synthesis and result in a small or no change in presynaptic function.
Previous studies on the synaptic mechanisms of mGluR-LTD have yielded conflicting results. Many of the studies, which concluded there is a presynaptic site of mGluR-LTD expression, were performed in hippocampal slices from neonatal animals (P4-P18) (Bolshakov and Siegelbaum, 1994; Fitzjohn et al., 2001; Zakharenko et al., 2002; Rammes et al., 2003) (but see Watabe et al., 2002). However, the studies demonstrating a role for postsynaptic protein synthesis were performed at more mature synapses (14-21 d in vitro cultures or slices from P10-P30 rats) (Huber et al., 2000; Snyder et al., 2001; Xiao et al., 2001). Our results offer an explanation for these disparate results and indicate that the developmental state of the synapse determines its response to postsynaptic mGluR activation.
To quantitate and compare the developmental changes in LTD, we grouped our data into two age groups: neonatal (P8-P15) and adolescent (P21-P35). However, the significant correlations of postnatal age with the effects of anisomycin on LTD (Fig. 3D) and the magnitude of PPF changes associated with LTD suggest that synaptic changes in mGluR-LTD do not occur abruptly but gradually as the synapses mature. Furthermore, the fact that there are small changes in PPF (Fig. 7B) and a nonsignificant reduction in τ1 of MK801 blockade, which occur during LTD in the adolescent group, suggest that a presynaptic expression mechanism, albeit reduced, may persist at mature synapses. However, because mGluR-LTD in adolescents is blocked by protein synthesis inhibitors, this suggests that any presynaptic contribution must also require protein synthesis. Alternatively, or in addition, PPF changes observed in the adolescent group may have a contribution from AMPAR removal at high release probability synapses (Poncer and Malinow, 2001).
There are alternative explanations for the greater PPF changes we observe during LTD at neonatal synapses. A recent study demonstrated that the baseline PPF magnitude is inversely correlated with the PPF changes observed during LTD (Santschi and Stanton, 2003). Therefore, a higher Pr at neonatal synapses would be reflected by a lower PPF and could explain why there are greater increases in PPF during LTD. In our study, we find no correlation with baseline PPF and the change in PPF with LTD (r = 0.05; p = 0.7). The difference in our findings may be attributable to a difference in the route of LTD induction (Santschi and Stanton, 2003). Furthermore, we do not find a correlation with developmental age and the baseline PPF (r = 0.13; p = 0.4) or differences in the average baseline PPF values or the τ1 of MK801 blockade between neonatal and adolescent groups (see Results). A recent study described developmental decreases in Pr at hippocampal synapses, as measured by PPF and the rate of MK801 blockade, but these changes occurred during an earlier developmental window (from P6 to P12) than our developmental switch (Wasling et al., 2004). Other studies of Pr later in hippocampal development (P15-P35) report an increase in Pr or no change (Dumas and Foster, 1995; Hsia et al., 1998). We think that the greater DHPG-induced changes in PPF and rate of MK801 blockade at neonatal synapses represent a greater presynaptic contribution to the LTD compared with more mature synapses.
Although we find that mGluR-LTD is independent of protein synthesis at developing synapses, our results do not address the question of whether mGluRs activate synaptic protein synthesis at this age. Instead, our data indicate that there are developmental changes in mGluR regulation of AMPAR trafficking. New proteins are required for the persistent decrease in surface AMPAR expression at mature synapses (Fig. 4E,F) (Snyder et al., 2001). Therefore, it is likely that LTD is protein synthesis independent in the neonatal slices because mGluR activation does not alter AMPAR surface expression (Fig. 6). Because NMDAR-dependent decreases in AMPAR surface expression are intact in neonatal synapses, we conclude that components of the mGluR-mediated AMPAR endocytosis process are developmentally regulated, as opposed to general AMPAR endocytosis machinery.
We measured changes in GluR surface expression and not endocytosis per se. Therefore, mGluR activation of neonatal synapses may induce endocytosis of AMPARs but also increase insertion rates so there is no net change in surface expression. In addition, with receptor biotinylation, we cannot determine whether we are measuring surface expression of synaptic or extrasynaptic receptors or both. Therefore, the developmental differences that we observe in DHPG-induced decreases in AMPAR surface expression could be attributable to a difference in the ability to detect surface changes in synaptic receptors with biotinylation. Using immunocytochemistry in dissociated neuronal culture, DHPG has been shown to reduce the number of synaptic AMPARs associated with presynaptic markers, and we would predict that similar changes are occurring in the slice (Snyder et al., 2001). However, it remains to be determined whether there is a developmental change in the effects of DHPG specifically on synaptic AMPARs. Recent work demonstrated that postsynaptic Ca2+ increases alone, independent of NMDARs or mGluRs, are sufficient to induce “silent” synapses (presumably by removing postsynaptic AMPARs) at immature (P3-P12), but not mature (P29-P32), synapses (Xiao et al., 2004). This previous work and our current results suggest that the mechanisms that induce AMPAR removal, such as those that regulate insertion, change with synapse development (Esteban et al., 2003; Yasuda et al., 2003).
The CB1R agonist WIN55,212-2 induced a small depression in the presence of 1 μm AM281 (Fig. 8A), suggesting an incomplete blockade of CB1Rs. This leaves open the possibility that a minority of CB1R activation can lead to full mGluR-LTD. However, we find this unlikely and think that our results are consistent with those of Rouach and Nicoll (2003) (from mature rats), who found no role for endocannabinoids in mGluR-LTD of excitatory synaptic transmission. Because endocannabinoids have been implicated in mGluR-dependent LTD of inhibitory synaptic transmission (Chevaleyre and Castillo, 2003), this suggests that mGluRs use distinct mechanisms to induce LTD of inhibitory and excitatory synaptic transmission. Another potential retrograde messenger for mGluR-LTD at immature synapses is the arachidonic acid metabolite 12-(S)hydroperoxyeicosatetraenoic acid, which is required for mGluR-LTD induced with 5 Hz stimulation (Feinmark et al., 2003). Additional experiments are required to confirm the role of arachidonic acid metabolism in DHPG and PP-LFS-induced LTD.
The developmental switch in the synaptic mechanisms of LTD may occur to accommodate the changing properties of synapses over the course of maturation. Because there are fewer surface AMPARs at immature synapses (Liao et al., 1999; Petralia et al., 1999; Pickard et al., 2000), it may be more efficacious to depress synaptic transmission at developing synapses by reducing presynaptic release probability, as opposed to endocytosis of AMPARs. The consequences of the developmental switch of LTD on hippocampal function are unclear. Because mGluR-LTD affects the short-term dynamics of nascent synapses (Fig. 7), it may differentially impact information processing and plasticity of developing and mature CA1 synapses (Fuhrmann et al., 2002).
Footnotes
This work was supported by National Institutes of Health Grant R01NS045711, the McKnight Foundation, and the FRAXA Research Foundation. K.M.H. is a Southwestern Medical Foundation Endowed Scholar in Biomedical Research. We thank Christine Daly and Lenora Volk for technical assistance and Ege Kavalali and Jay Gibson for helpful discussions.
Correspondence should be addressed to Kimberly Huber, Center for Basic Neuroscience, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390-9111. E-mail: kimberly.huber{at}utsouthwestern.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252992-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}