

Abstract
Necdin is a multifunctional signaling protein that stabilizes terminal differentiation of postmitotic neurons. The human necdin gene in chromosome 15q11-q12 is maternally imprinted, paternally transcribed, and not expressed in Prader-Willi syndrome, a human genomic imprinting-associated neurodevelopmental disorder. Although necdin-deficient mice display several abnormal phenotypes reminiscent of this syndrome, little is known about molecular mechanisms that lead to the neurodevelopmental defects. Here, we demonstrate that paternally expressed necdin is required for physiological development of nerve growth factor (NGF)-dependent sensory neurons. Mouse embryos defective in the paternal necdin allele displayed absent necdin expression in the dorsal root ganglia, in which the tropomyosin-related kinase A (TrkA) receptor tyrosine kinase and the p75 neurotrophin receptor were expressed in a normal manner. Necdin interacted with both TrkA and p75 to facilitate the association between these receptors. NGF-induced phosphorylation of TrkA and mitogen-activated protein kinase was significantly diminished in the necdin-null sensory ganglia. Furthermore, the mice lacking the paternal necdin allele displayed augmented apoptosis in the sensory ganglia in vivo and had a reduced population of substance P-containing neurons. These mutant mice showed significantly high tolerance to thermal pain, which is often seen in individuals with Prader-Willi syndrome. These results suggest that paternally expressed necdin facilitates TrkA signaling to promote the survival of NGF-dependent nociceptive neurons.
Introduction
Necdin is a 325 aa residue protein expressed in terminally differentiated neurons (Maruyama et al., 1991; Aizawa et al., 1992; Uetsuki et al., 1996; Kuwajima et al., 2004). Necdin strongly suppresses the proliferation of several cell lines (Hayashi et al., 1995; Taniura et al., 1998) and interacts with cell cycle regulatory transcription factors E2F1 and E2F4 (Taniura et al., 1998; Kobayashi et al., 2002), which are also the targets of retinoblastoma tumor suppressor protein (Rb) (Nevins, 1992). Furthermore, necdin interacts with p53, a tumor suppressor protein with proapoptotic activity (Taniura et al., 1999). Thus, necdin may be involved in both the exit from the cell cycle and the inhibition of apoptosis to stabilize neuronal terminal differentiation (Yoshikawa, 2000).
Necdin is highly expressed in NGF-dependent neurons in the dorsal root ganglia (DRGs), and that suppression of endogenous necdin expression with antisense oligonucleotides induces apoptosis of primary NGF-dependent neurons (Takazaki et al., 2002). Necdin and its homologous melanoma antigen (MAGE) family proteins such as neurotrophin receptor-interacting MAGE homolog (NRAGE) (also termed MAGE-D1 or Dlxin-1), MAGE-H1, and MAGE-G1 (also termed necdin-like 2) bind to the p75 neurotrophin receptor (p75NTR) (Salehi et al., 2000; Tcherpakov et al., 2002; Kuwako et al., 2004), a receptor involved in the regulation of neuronal life and death (Dechant and Barde, 2002; Chao, 2003). These findings suggest that necdin modulates the survival signals in NGF-dependent sensory neurons. However, detailed mechanisms whereby necdin mediates NGF signal transduction remain to be elucidated.
The human necdin gene (NDN) is mapped to chromosome 15q11-q12, a region deleted in Prader-Willi syndrome (PWS) (Nakada et al., 1998). PWS is a typical human genomic imprinting-associated disease characterized by hyperphagia leading to severe obesity, short stature, hypogonadism, and behavioral problems (Holm et al., 1993), which are believed to be attributable to hypothalamic defects. This disorder is caused by the absence of paternally expressed genes located in the 15q11-q13 region. In fact, NDN is expressed from only the paternal allele and not expressed in individuals with PWS (Jay et al., 1997; MacDonald and Wevrick, 1997; Sutcliffe et al., 1997). The mouse necdin gene (Ndn) in chromosome 7C is also maternally imprinted and paternally expressed (MacDonald and Wevrick, 1997). Necdin is expressed at the highest level in the mouse hypothalamus, a region most affected by PWS (Aizawa et al., 1992; Uetsuki et al., 1996). Furthermore, necdin-null mice display phenotypes resembling PWS such as postnatal lethality, impaired neuronal development, and abnormal behaviors (Gerard et al., 1999; Muscatelli et al., 2000). These findings suggest that necdin deficiency is responsible for at least a subset of neuronal abnormalities in PWS.

Necdin expression is absent in DRGs of mutant mice lacking the paternal necdin gene. A, The restriction map of mouse Ndn locus. A 7 kb Ndn sequence containing coding (black) and 3′,5′ noncoding (hatched) regions was subcloned into the targeting vector. The 1.5 kb expression unit containing the pgk promoter with Neor gene (Pgk1/neor), polyadenylation signal [Poly(A) signal], was inserted to disrupt the Ndn coding sequence. The vector was introduced to TT2 cells for homologous recombination as described in Materials and Methods. B, Absence of necdin expression in the brain and spinal cord of mice deficient in the paternal necdin gene. Frozen sections of the hypothalamus and spinal cord from E13.5 wild-type (Ndn+m/+p) and necdin-deficient (Ndn+m/-p) littermates were immunostained for necdin. HT, Hypothalamus; SC, spinal cord; DBB, diagonal band of Broca; ML, mantle layer. Scale bars: HT, 50 μm; SC, 100 μm. C, Expression patterns of TrkA and p75NTR in necdin-deficient DRGs. Frozen sections of cervical DRGs from E12.5 wild-type (Ndn+m/+p) and necdin-deficient (Ndn+m/-p) littermates were immunostained for necdin, TrkA, and p75NTR. Scale bar, 50 μm. D, E, Western blot analysis of necdin, TrkA, p75NTR, and phosphorylated MAPK. Equal amounts of DRG lysates from E12.5 embryos were analyzed by Western blotting. Each lane represents pooled DRGs from a single embryo. Molecular sizes in kilodaltons are shown to the right. E, Signal intensities of TrkA, p75NTR, and pMAPK shown in D were normalized to those of tubulin (TB) and MAPK (mean ± SEM; n = 5 for each protein). *p < 0.05. NS, Not significant (p > 0.05).
Using mice lacking the paternal necdin gene, we demonstrate that paternally expressed necdin is required for the survival of NGF-dependent sensory neurons. We also find that necdin interacts with tropomyosin-related kinase A (TrkA) receptor tyrosine kinase and p75NTR, facilitates their association, and promotes NGF/TrkA signaling. Furthermore, we show that the necdin-deficient mice display impaired development of NGF-dependent nociceptive neurons and have a high tolerance to heat-induced pain, which is frequently observed in individuals with PWS.
Materials and Methods
Knock-out mice. Ndn in the mouse germ line was inactivated by conventional gene targeting (Nada et al., 1993). The mouse necdin chromosomal gene was isolated from TT2 embryonic stem (ES) cell genomic DNA by screening the library with mouse necdin cDNA as a hybridization probe. TT2 ES cells established from the first filial blastocyst between C57BL/6 and CBA/JNCrj mice were used to generate chimera mice. A linearized targeting vector was introduced by electroporation into TT2 cells, which were cultured with 150 μg/ml G418 (Sigma, St. Louis, MO) to select colonies carrying the integrated targeting sequence. The genotypes of selected colonies were analyzed by PCR. TT2 cells carrying disrupted necdin allele were injected into eight-cell stage ICR embryos, which were transplanted into the uterus of pseudopregnant females. The complete absence of necdin mRNA in the brain and DRGs of paternal Ndn-defective mice was confirmed by reverse transcription-PCR. Heterozygous male mice (Ndn+/-) were crossed with wild-type female mice (Ndn+/+) (ICR strain; SLC, Shizuoka, Japan) for >10 generations to obtain mutant mice for the following analyses. All Ndn+m/-p mice were fertile. Because the loci of wild-type p (pink-eyed dilution) and mutated Ndn in mouse chromosome 7C region are tightly linked, Ndn+m/-p mice were sorted out by their brown-eye phenotype from the wild-type (Ndn+/+) littermates that have mutated p in the ICR background. All of the mice with the brown-eyed phenotype were consistent with those with mutated Ndn genotype as analyzed by PCR. Experiments using gene-targeted mice were approved by the Recombinant DNA and Animal Experiment Committees of the Institute for Protein Research, Osaka University, and performed in accordance with institutional guidelines and regulations.
Antibodies. Primary antibodies used for immunohistochemistry are as follows: rabbit polyclonal antibodies against necdin (NC243; 1:1000) (Niinobe et al., 2000), TrkA (RTA; 1:1000; gift from Dr. L. F. Reichardt, Howard Hughes Medical Institute, University of California, San Francisco, CA), p75NTR (1:300; Promega, Madison, WI), activated caspase-3 fragment (1:2000) (Uetsuki et al., 1999), and substance P (1:3000; gift from Dr. J.-S. Hong, National Institute for Environmental Health Sciences, Research Triangle Park, NC). For the double labeling experiment, anti-necdin antibody GN1 was raised in guinea pig against mouse necdin protein (residues 83-325; 1:500). Secondary antibodies are rhodamine B (or fluorescein isothiocyanate)-conjugated anti-rabbit IgG (1:500; Cappel, Durham, NC) and cyanine 3-conjugated anti-guinea pig IgG (1:500; Jackson ImmunoResearch, West Grove, PA). The antibodies used for Western blotting are as follows: Necdin (NC243; 1:3000), TrkA (RTA; 1:1000), p75NTR (1:500; Promega), β-tubulin (1:1000; MP Biomedicals, Irvine, CA), phospho-p44/42 mitogen-activated protein kinase (MAPK) (E10; 1:1000; Cell Signaling Technology, Beverly, MA), MAPK [ERK1 (extracellular signal-regulated kinase 1)] (K-23; 1:5000; Santa Cruz Biotechnology, Santa Cruz, CA), amyloid protein precursor (APP) (AC-1; 1:3000) (Yoshikawa et al., 1992), FLAG (M2; 1:500; Sigma), phospho-TrkA (Tyr490; 1:1000; Cell Signaling Technology), and β-galactosidase (1:300; Chemicon, Temecula, CA). Antibodies used for coimmunoprecipitation assay are as follows: FLAG (M2; 1:50; Sigma), Necdin (NC243; 1:50), and p75NTR (MC192; 1:10; Alomone Labs, Jerusalem, Israel).
Immunohistochemistry. Frozen 10-μm-thick tissue sections were prepared from mouse embryos at embryonic day 12.5 (E12.5), E13.5, and postnatal day 0 (P0), and immunostained as described previously (Kuwajima et al., 2004). The sections were incubated at 4°C with primary antibodies overnight and with fluorescence dye-conjugated secondary antibodies at room temperature for 90 min. The images were observed by fluorescence microscopy (BX50-34-FLAD1; Olympus, Tokyo, Japan) and taken by CCD camera system (M-3204C; Olympus). Nuclear DNA fragmentation was analyzed by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) and visualized with Texas Red as described previously (Uetsuki et al., 1999). TUNEL- and activated caspase-3-positive cells in cervical DRG sections from five and four embryos, respectively, in each group were counted and presented as the number per square millimeter. Substance P immunoreactivity was detected by the avidin-biotin peroxidase complex method using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA). The immunoreactive cells in cervical DRG sections from three embryos in each group were quantified as above. Antibodies used are described above. To determine the total neuron number in DRGs at P0, frozen sections were stained with 0.1% cresyl violet (Nissl staining).
Western blotting. DRG tissues of ICR mouse embryos were homogenized with a lysis buffer (TNE buffer) containing 10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, protease inhibitors (Complete; Roche, Basel, Switzerland), and 1 mm orthovanadate, and centrifuged at 12,000 × g for 30 min at 4°C to obtain the supernatant. The lysates (10-40 μg of protein per lane) were separated by 7.5 or 10% SDS-PAGE, blotted onto Immobilon membrane (Millipore, Billerica, MA), and incubated with the antibodies described above. After incubation with peroxidase-conjugated anti-rabbit (or anti-mouse) IgG (Cappel), the proteins were detected by chemiluminescence method (Chemiluminescence Reagent Plus; PerkinElmer, Boston, MA). Signal intensities were quantified with an image analyzer (LAS-1000 Plus; Fuji Film, Tokyo, Japan).
Immunoaffinity assay. Tissue extract from the DRG and spinal cord was applied to a HiTrap N-hydroxysuccinimide-activated affinity column (Amersham Biosciences, Uppsala, Sweden) coupled with IgG fractions of anti-necdin (NC243) antibody or preimmune antiserum (Kuwako et al., 2004). Bound proteins were eluted with 0.1 m glycine-HCl, pH 2.5. Fractions were precipitated with 10% trichloroacetic acid, rinsed with cold acetone, separated by 10% SDS-PAGE, and detected by Western blotting.
Coimmunoprecipitation assay. Human embryonic kidney 293A (HEK293A) cells were transfected with cDNAs as described previously (Kuwako et al., 2004). cDNAs encoding rat TrkA (a gift from Dr. E. M. Shooter, Stanford University School of Medicine, Stanford, CA), human p75NTR (a gift from Dr. T. Yamashita, Chiba University Medical School, Chiba, Japan), and human APP (Yoshikawa et al., 1992) were subcloned into p3xFLAG-CMV14 (Sigma) for FLAG tagging. FLAG-tagged rat TrkA mutants (Y499A, K547A, Y794A) were generated by PCR-based overlap mutagenesis. The cells were harvested 48 h after transfection and lysed in TNE buffer. The lysates (200 μg) were incubated at 4°C for 2 h with antibodies. The complexes were pelleted with protein A-Sepharose (Amersham Biosciences), separated by 10% SDS-PAGE, and detected by Western blotting (Kuwako et al., 2004).
Pheochromocytoma 12 cells. Pheochromocytoma 12 (PC12) cells were maintained in DMEM containing 10% fetal calf serum and 5% horse serum. PC12 cells plated on collagen-coated dishes were infected with Ad-Necdin and Ad-LacZ at multiplicity of infection of 40 and cultured in DMEM/F12 medium containing mouse 2.5S NGF (Invitrogen, Carlsbad, CA). Recombinant adenoviruses expressing mouse necdin (Ad-Necdin) and LacZ (Ad-LacZ) were constructed and prepared as described previously (Nishimura et al., 1998). The cells were harvested 38 h after NGF treatment and lysed in TNE buffer for the coimmunoprecipitation assay. Neurite outgrowth of NGF-treated PC12 cells was analyzed by counting cells with extended neurites (more than two times the cell-body diameter) among >200 infected cells after immunostaining for necdin and β-galactosidase as described previously (Kobayashi et al., 2002). Phosphorylation of signal proteins was analyzed by Western blotting using phosphoprotein-specific antibodies described above.

Necdin is coexpressed with TrkA or p75NTR in developing DRGs. A, B, Double immunostaining for necdin and TrkA or p75NTR. Frozen DRG sections of E13.5 mouse embryo were double immunostained for necdin with antibody GN1 (red) and TrkA (or p75NTR) (green), and two images were merged (yellow). Scale bars: A, 50 μm; B, 10 μm. C, Immunoaffinity assay for endogenous necdin-interacting proteins. Tissue lysate (1 mg) of the DRG and spinal cord from E13.5 embryos was applied to the immunoaffinity columns with anti-necdin IgG (NC243) (αNecdin IgG) and control preimmune IgG (Preimmune IgG). The eluates were analyzed by Western blotting for endogenous complexes of necdin with TrkA, p75NTR, and APP. Lysate, Tissue lysate (30 μg).
DRG explants. DRG explants were isolated from E13.5 embryos and cultured in a 24-well plate in Ham's F-14 medium (Imperial Laboratories, Andover, UK) supplemented with N-2 supplement (Invitrogen) in the presence of 10 ng/ml NGF (for neurite outgrowth assay) or 20 ng/ml NGF (for Western blotting) for 15 min or 16 h. The phase-contrast microscopic images were captured to measure the length of eight randomly selected neurites of each DRG explant. For Western blot analysis, the explants were homogenized with TNE buffer and centrifuged at 12,000 × g for 30 min at 4°C to obtain the supernatant. The lysates (10-20 μg/lane) were separated by 10% SDS-PAGE, blotted onto Immobilon membrane, and incubated with the above antibodies.

Necdin interacts with both TrkA and p75NTR. A, B, Immunoprecipitation assay for interactions of necdin with TrkA and p75NTR. Lysates of HEK293A cells transfected with expression vectors for FLAG-TrkA, FLAG-p75NTR, FLAG-APP, and necdin were immunoprecipitated (IP) with anti-FLAG antibody M2 (αFLAG) and immunoblotted (IB) with anti-necdin antibody NC243 (αNecdin) (A, top panel). Conversely, the lysates were immunoprecipitated with antibody NC243 and immunoblotted with antibody M2 (B, top panel). Expressed proteins in cell lysates are shown in the bottom panels. C, Necdin-enhanced association between TrkA and p75NTR. Lysates of cells transfected with expression vectors for HA-p75NTR (4 μg), FLAG-TrkA (4.5 μg), and necdin (0.1 and 0.5 μg) were immunoprecipitated with antibody M2 and immunoblotted with anti-p75NTR antibody (top panel). D, Interactions of necdin with TrkA mutants. Lysates of cDNA-transfected cells expressing necdin, FLAG-tagged wild-type (WT) TrkA, and the mutants (K547A, Y499A, Y794A) were immunoprecipitated as in A. Phosphorylated Tyr499 in rat TrkA was analyzed using the antibody against phospho-human TrkA (Tyr490) (αpTrkA) (D, second panel from the top).
Behavioral analysis. Hotplate and tail-flick tests were performed as described previously (Crowley et al., 1994). Mice were placed on a 55°C hotplate, and the latency to foreleg lick, hindleg shaking, or jumping was measured. The distal centimeter of the mouse tail was immersed in a 50°C water bath, and the latency to tail flick was recorded.
Statistical tests. Statistical significance was tested using an unpaired Student's t test or one-way ANOVA followed by Tukey's post hoc test. A significance of p < 0.05 was required for rejection of the null hypothesis.
Results
Necdin expression is absent in mice lacking the paternal Ndn
To study the role of endogenous necdin in NGF signaling, we produced mutant mice defective in the necdin gene Ndn by conventional targeting in the mouse germ line (Fig. 1A). Because mice defective in the paternal Ndn (Ndn+m/-p) are expected to show absent necdin expression, heterozygous male mice (Ndn+m/-p) were crossed with wild-type female mice (Ndn+m/+p) (both in the ICR background) to obtain Ndn+m/-p and Ndn+m/+p littermates. Unlike the previous Ndn mutant C57BL/6 mice (Gerard et al., 1999; Muscatelli et al., 2000), the present necdin-deficient ICR mice showed no postnatal lethality. The ratio of Ndn+m/-p to Ndn+m/+p littermates was consistently 1:1 from E12.5 to P200 (data not shown). This is consistent with the previous findings that mutant mice in the genetic backgrounds other than C57BL/6 show a striking reduction in postnatal lethality (Gerard et al., 1999; Muscatelli et al., 2000).
We performed immunohistochemical analysis for necdin expression in Ndn+m/-p mice. Little or no necdin immunoreactivity was detected in the nervous tissues such as in the hypothalamus, spinal cord, and DRG (Fig. 1B). Dissection of Ndn+m/-p and Ndn+m/+p mice revealed no gross morphological abnormalities in the nervous system (data not shown). Necdin was undetected in DRGs of paternal Ndn-deficient embryos at E12.5, whereas the expression patterns and staining intensities of TrkA and p75NTR in necdin-deficient DRGs were similar to those of the wild-type littermates (Fig. 1C). The expression levels of necdin, TrkA, and p75NTR in DRGs were examined by Western blot analysis (Fig. 1D). The levels of TrkA and p75NTR had no significant differences between Ndn+m/-p and Ndn+m/+p littermates (Fig. 1E). These data suggest that nascent neurons expressing TrkA and p75NTR are differentiated in a normal manner even in the absence of necdin at this stage of DRG development.
NGF induces autophosphorylation of TrkA followed by phosphorylation of signaling molecules such as MAPK (Huang and Reichardt, 2003). To examine the state of NGF signaling in DRGs in vivo, we analyzed phosphorylation levels of TrkA and MAPK by Western blotting using antibodies specific to the phosphoproteins. The signal intensity of phosphorylated forms of TrkA was below the detection limits of the Western blot analysis (data not shown), whereas the phosphorylated MAPK levels in DRGs were barely detectable (Fig. 1D, second panel from the bottom). The level of phosphorylated MAPK in necdin-deficient DRGs in vivo was significantly reduced to ∼30% of the wild-type control (Fig. 1D,E), suggesting that TrkA signaling is reduced in necdin-null DRG neurons, which express the NGF receptor system in a normal manner.
Necdin and TrkA are coexpressed in DRG cells in vivo
We next examined whether necdin is coexpressed with TrkA and p75NTR, two types of receptors involved in NGF signaling, in developing DRGs. Immunohistochemistry revealed that necdin, TrkA, and p75NTR were distributed predominantly in the cytoplasm of DRG cells at E13.5 (Fig. 2A,B). At this stage of development, ∼80% of neuron-like cells in DRGs were immunopositive for necdin or TrkA, whereas ∼40% were for p75NTR. Double-labeling analysis revealed that virtually all of the TrkA- and p75NTR-immunopositive cells overlap with necdin-immunopositive cells. These results suggest that necdin coexists with TrkA, p75NTR, or both in DRG cells in vivo. Necdin was present not only in small TrkA-positive neurons but also in large cells, which presumably are TrkC-positive neurotrophin-3 (NT-3)-dependent sensory neurons (Mu et al., 1993). TrkA was undetected in some of these large cells. Likewise, there was another large cell population containing both necdin and p75NTR. Thus, it is likely that some large sensory neurons also express necdin at this stage of DRG development.
Previous studies have shown that necdin forms stable complexes with p75NTR (Tcherpakov et al., 2002; Kuwako et al., 2004). In addition, TrkA interacts with p75NTR when these two receptors are coexpressed (Chao, 2003). Therefore, we performed the immunoaffinity assay to detect the endogenous complexes of necdin with TrkA and p75NTR (Fig. 2C). The extracts of the DRG and spinal cord, which comprise a large number of necdin-expressing neurons, were applied to the immunoaffinity column carrying anti-necdin IgG. Both TrkA and p75NTR bound to necdin, whereas APP, a transmembrane protein expressed abundantly during neuronal differentiation (Yoshikawa et al., 1992), failed to bind. This indicates that necdin forms stable complexes with TrkA, p75NTR, or both under physiological conditions.
Necdin interacts with TrkA
To further study physical interactions of necdin with TrkA, we performed the coimmunoprecipitation assay using cDNA-transfected HEK293A cells. For comparable immunoprecipitation conditions, TrkA, p75NTR, and APP, all of which are single-span transmembrane proteins, were tagged with FLAG epitope at their C-terminal ends. Necdin was coprecipitated with TrkA and p75NTR, but not with APP (Fig. 3A). Conversely, TrkA and p75NTR, but not APP, were coprecipitated with necdin (Fig. 3B). These data confirmed that necdin forms stable complexes with TrkA and p75NTR. We then examined the effect of necdin on the association between TrkA and p75NTR (Fig. 3C). When necdin was coexpressed with TrkA and p75NTR, it increased the amount of p75NTR coprecipitated with TrkA to two to four times the control level. These results indicate that necdin interacts with these two NGF receptors to facilitate their association.

Necdin enhances NGF/TrkA signaling in PC12 cells. A, Coimmunoprecipitation assay for the association between TrkA and p75NTR. PC12 cells infected with recombinant adenoviruses expressing necdin (Ad-Necdin) and β-galactosidase (Ad-LacZ) were treated with NGF (50 ng/ml) for 38 h. Cell lysates (400 μg) were immunoprecipitated (IP) with mouse anti-p75NTR antibody MC192 (αp75NTRm) and immunoblotted (IB) with anti-TrkA antibody (top panel). Expressed proteins in cell lysates are shown in the bottom panels. B, Neurite outgrowth assay. Ad-Necdin- and Ad-LacZ-infected PC12 cells were treated with NGF for 38 h, and cells bearing extended neurites were counted (mean ± SEM; n = 4; p < 0.005). C, D, Western blot analysis of phosphorylated forms of TrkA and MAPK. PC12 cells were infected with Ad-Necdin and Ad-LacZ, incubated for 36 h, and treated with NGF (10 ng/ml) for the indicated durations. C, Phosphorylated proteins (pTrkA, pMAPK) were analyzed by Western blotting using phosphoprotein-specific antibodies. Similar data were obtained from two more sets of Western blot analysis. D, Signal intensities of pTrkA and pMAPK were normalized to those of respective total proteins including unphosphorylated forms. The values relative to those of the maximum intensities (=1) at 5 min are presented (mean ± SEM; n = 3). *p < 0.005 (compared with the controls treated with Ad-LacZ).
After NGF binding, activated TrkA recruits signaling molecules such as the adaptor protein Shc and the effector phospholipase C-γ1 (PLC-γ1), both of which recognize specific phosphotyrosine residues in the intracellular domain of TrkA (Huang and Reichardt, 2003). Thus, we investigated whether necdin, like these TrkA-interacting proteins, binds to phosphorylated TrkA. Necdin failed to bind to the kinase-dead mutant TrkA K547A in which Lys547 (position numbers based on the rat TrkA sequence) was converted to Ala (Fig. 3D). Autophosphorylation of the specific sequence Tyr499 in the juxtamembrane region was confirmed by Western blotting using an antibody against human Tyr490 (phospho-TrkA). These data indicate that necdin recognizes only the kinase-active form of TrkA. Phosphotyrosine residues at positions 499 and 794 of rat TrkA are recognized by Shc and PCL-γ1, respectively. We produced the TrkA mutants Y499A and Y794A by converting Tyr to Ala. Necdin interacted with both Y499A and Y794A, suggesting that necdin interacts with TrkA in a manner distinct from those of Shc and PLC-γ1. Furthermore, it is unlikely that necdin binds indirectly to TrkA via these signaling proteins.
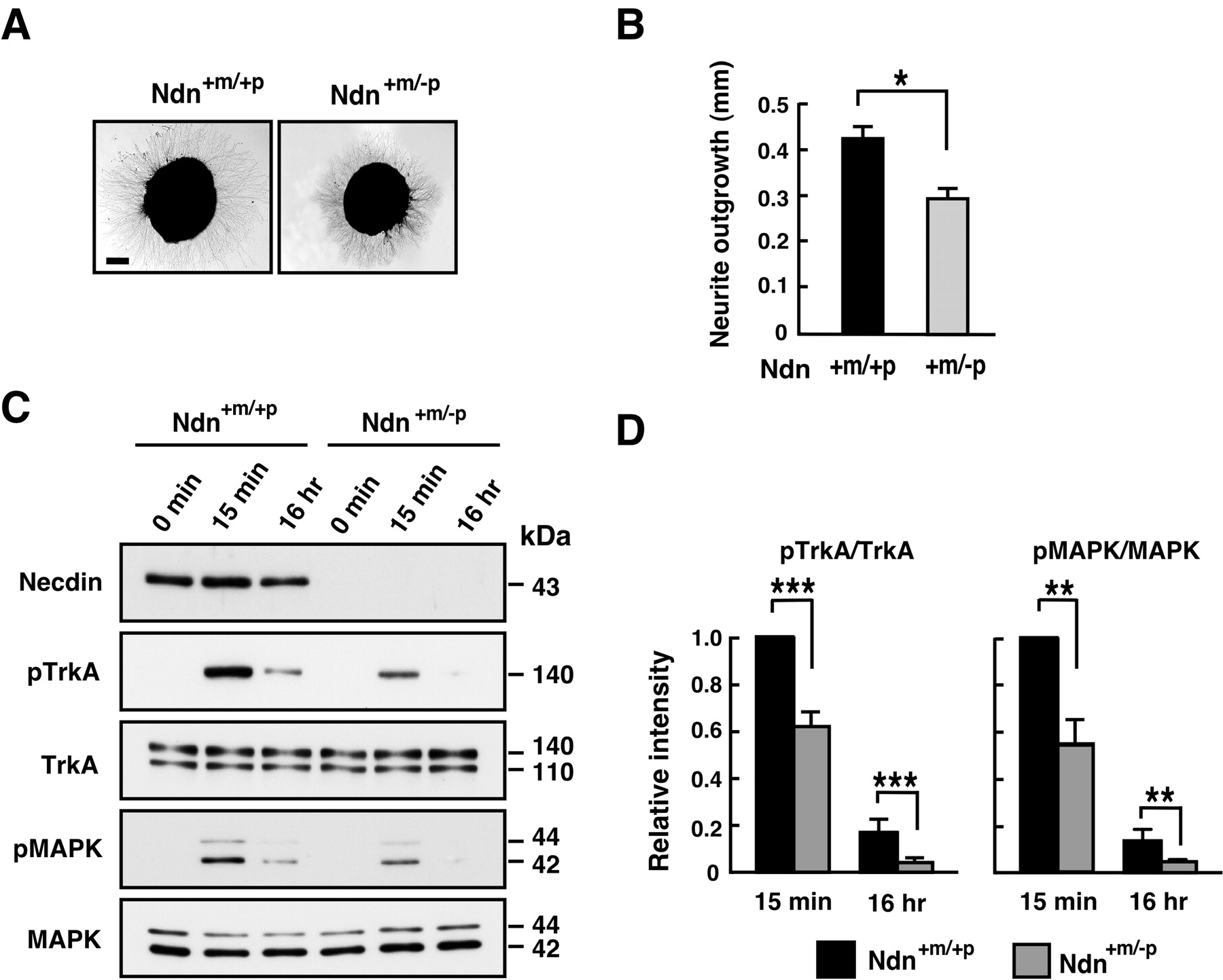
NGF/TrkA signaling is diminished in necdin-null DRG explants. A, B, Neurite outgrowth assay. DRG explants were prepared from E13.5 embryos, treated with NGF for 16 h, and analyzed by phase-contrast microscopy. A, Typical images of NGF-treated DRG explants. B, The neurite length of the explants was measured after NGF treatment for 16 h (explants examined: +m/+p, n = 105; +m/-p, n = 102). Scale bar: A, 200 μm. C, D, Western blot analysis of phosphorylated forms of TrkA and MAPK. DRG explants from E13.5 littermates were cultured in the presence of NGF (20 ng/ml) for the indicated durations. Phosphorylated proteins (pTrkA, pMAPK) were analyzed by Western blotting using phosphoprotein-specific antibodies. The values relative to those of the wild-type controls (=1) at 15 min were presented (mean ± SEM; n = 4). B, D, Graphs show mean ± SEM. *p < 0.01; **p < 0.005; ***p < 0.001.
Necdin enhances NGF/TrkA signaling
To elucidate the effects of the interaction between necdin and TrkA, we attempted to set up a gain-of-function study using necdin-null DRG cells in primary cultures. However, biochemical analyses of the primary DRG cells were impossible because of their limited number and heterogeneity. Alternatively, we used NGF-treated PC12 cells in which TrkA and p75NTR, but not necdin, were endogenously expressed. Necdin was ectopically expressed in PC12 cells by infection with recombinant adenovirus carrying necdin cDNA. Necdin increased the amount of TrkA coprecipitated with p75NTR (Fig. 4A), indicating that necdin facilitates the association between endogenous TrkA and p75NTR in these cells. Furthermore, necdin increased the number of NGF-treated PC12 cells bearing extended neurites (Fig. 4B), consistent with the previous data (Tcherpakov et al., 2002). This suggests that ectopically expressed necdin enhances NGF signaling in PC12 cells.
We then investigated whether necdin affects the phosphorylation of TrkA and MAPK in NGF-treated PC12 cells (Fig. 4C). Phosphorylation of TrkA and MAPK in uninfected and Ad-LacZ-infected PC12 cells increased strikingly at 5 min, declined at 2 h, and remained low 24-72 h after NGF stimulation. In contrast, high phosphorylation levels of TrkA and MAPK were sustained for 24-72 h in necdin-expressing PC12 cells. The levels of phosphorylated TrkA and MAPK in necdin-expressing cells at 72 h were 4.2 and 4.6 times, respectively, those in LacZ-expressing controls (Fig. 4D). These results suggest that, in PC12 cells, ectopic necdin sustains the activation of the TrkA signaling pathway through its interaction with TrkA.
NGF/TrkA signaling is diminished in necdin-deficient DRGs
We examined whether the NGFs/TrkA signal transduction is reduced in necdin-deficient DRGs isolated from mutant mice. We analyzed the neurite outgrowth of DRG explants as an indication of NGF signaling activity. When DRG explants from E13.5 embryos were cultured in the presence of NGF, the length of neurites in Ndn+m/-p DRG neurons was significantly reduced to 70% of the Ndn+m/+p control (Fig. 5A,B), suggesting that necdin deficiency diminishes NGF signaling in DRG neurons.
Because necdin enhances TrkA signaling through sustained phosphorylation of TrkA and MAPK as shown in PC12 cells (Fig. 4C,D), the levels of the phosphorylated forms of these proteins were examined using NGF-treated DRG explants. Phosphorylation levels of TrkA in Ndn+m/-p DRGs were significantly reduced to 62 and 30% of the Ndn+m/+p DRG levels 15 min and 16 h, respectively, after NGF stimulation. The phosphorylation levels of MAPK in Ndn+m/-p DRGs were also significantly reduced to 56 and 45% of the Ndn+m/+p DRG levels at 15 min and 16 h, respectively, after NGF stimulation. These results suggest that NGF-induced TrkA signaling is significantly reduced in the absence of endogenous necdin in DRGs.
Necdin deficiency augments apoptosis in vivo
TrkA signal transduction is the major pathway for the survival of NGF-dependent neurons. Programmed cell death of NGF-dependent neurons is a classic example of target-deprived cell death (Oppenheim, 1991). We thus investigated whether apoptosis in necdin-deficient DRG is augmented in vivo in Ndn+m/-p mice at early stages of development. We used immunohistochemical TUNEL analysis for apoptotic cells in vivo (Fig. 6A). The TUNEL-positive cells in Ndn+m/-p cervical DRGs (C1-C8) at E12.5 were 1.8 times more than those in the Ndn+m/+p DRGs (mean number of TUNEL-positive cells per square millimeter: Ndn+m/+p, 66.5 ± 7.0, n = 138; Ndn+m/-p, 118.9 ± 13.6, n = 136; p < 0.005). We also examined the cell population containing activated caspase-3, a principal apoptosis-inducing protease, by immunohistochemistry using a cleaved end-specific antibody (Fig. 6B) (Uetsuki et al., 1999). Activated caspase-3-positive cells in Ndn+m/-p DRGs were 1.6 times more than those in the Ndn+m/+p DRGs (mean number of activated capase-3-positive cells per square millimeter: Ndn+m/+p, 27.3 ± 3.7, n = 129; Ndn+m/-p, 42.2 ± 4.7, n = 122; p < 0.05). These results suggest that necdin deficiency augments apoptosis in developing mouse DRGs in vivo and that endogenous necdin is required for the survival of embryonic DRG neurons under physiological conditions.

Apoptosis is augmented in necdin-deficient embryonic DRGs in vivo. A, TUNEL assay. Frozen sections of cervical DRGs from wild-type (Ndn+m/+p) and necdin-deficient (Ndn+m/-p) littermates at E12.5 were double stained for TUNEL and necdin with antibody NC243. B, Immunohistochemistry for activated capase-3. Frozen sections of cervical DRGs from wild-type and necdin-deficient littermates at E12.5 were double immunostained for activated caspase-3 (Casp3act) with rabbit cleaved caspase-3 end-specific antibody and for necdin with guinea pig anti-necdin antibody GN1. Scale bars, 50 μm. Quantitative data for A and B are given in Results.
Necdin deficiency causes impaired development of nociceptive neurons
We investigated whether development of NGF-dependent neurons is impaired in necdin-null DRGs in which NGF/TrkA signaling is diminished. Development and survival of substance P-containing DRG neurons, which sense temperature and noxious stimuli, are dependent on NGF (Otten et al., 1980). Thus, we analyzed the population of substance P-immunoreactive neurons in necdin-deficient DRGs. The density of substance P-containing cells in Ndn+m/-p DRGs in vivo was significantly reduced to 59% of the Ndn+m/+p control at P0 (Fig. 7A,B). There was no significant difference in size between wild-type and necdin-deficient DRGs (mean area of randomly selected DRG slices examined: Ndn+m/+p, 0.11 ± 0.01 mm2; Ndn+m/-p, 0.12 ± 0.01 mm2; p > 0.05).
We also determined the number of total neurons in DRGs at P0 by counting them in Nissl-stained DRG sections and found that the total neuron number of Ndn+m/-p DRGs was significantly reduced to 88% of the Ndn+m/+p control, although the extent of the reduction was smaller than that of substance P-containing neurons (mean number of randomly selected DRG slices examined: Ndn+m/+p, 188 ± 3.6 per slice, n = 103; Ndn+m/-p, 165 ± 2.9 per slice, n = 104; p < 0.001). To evaluate the pain sensation of Ndn+m/-p mice, their reactions to thermal pain were quantified by hotplate and tail-flick tests (Fig. 7C,D). The latencies of Ndn+m/-p mice at P14 and P28 were approximately two times those of Ndn+m/+p mice, indicating a high tolerance to heat-induced pain in these necdin-null mice.
Discussion
The present study has shown that mice lacking the paternal necdin gene display reduced TrkA signaling and augmented apoptosis in embryonic sensory ganglia. In addition, the necdin-null mice have a reduced number of nociceptive neurons and show impaired pain sensation at postnatal stages. Similar reduction in sensory neuron number and impaired sensory dysfunction have been found in the mutant mice lacking NGF (Crowley et al., 1994) and TrkA (Smeyne et al., 1994). The nociceptive abnormality seen in the necdin-null mice is reminiscent of a surprisingly high tolerance to pain in PWS (Holm et al., 1993). Electrophysiological studies have shown that patients with PWS have abnormally low sensory nerve action potential amplitudes, which indicate reduced sensory neuron numbers (Brandt and Rosen, 1998). These observations suggest that necdin deficiency is responsible for the impaired development of sensory neurons in PWS as a result of reduced TrkA signaling.
Because several phenotypes of the necdin-null mice reported in the present and previous studies resemble several clinical symptoms of PWS, these mutant mice are useful for the analyses of PWS pathogenesis at molecular and cellular levels. A group of mutant mice defective in a single necdin gene show respiratory distress leading to postnatal lethality, which is also seen in PWS cases (Gerard et al., 1999). It has been demonstrated that the respiratory distress can be attributed to abnormal development of central respiratory rhythm-generating center in the brainstem (Ren et al., 2003). It has been reported most recently that these necdin-null mice display morphological abnormalities in axonal outgrowth and fasciculation in several brain regions, presumably for lack of the interactions of necdin with the proteins related to axonal fasciculation and elongation such as Fez1 and BBS4 (Lee et al., 2005). Another major PWS symptom is the hypogonadism leading to the failure of sexual maturation and infertility. This symptom has been suggested to be caused by abnormal development of hypothalamic neurons containing luteinizing hormone-releasing hormone (LHRH) [also termed GnRH (gonadotropin-releasing hormone)]. A group of necdin-null mice display a reduction in hypothalamic LHRH-containing neurons (Muscatelli et al., 2000). Intriguingly, a similar abnormality of hypothalamic LHRH neurons has been reported in mice deficient in the NSCL (neuronal stem cell leukemia) transcription factors, which upregulate necdin gene expression (Kruger et al., 2004). This is consistent with the idea that necdin plays a pivotal role in the development of LHRH neurons. Together, it is conceivable that necdin is required for normal development of specific neurons and that the absence of its expression causes certain types of neurodevelopmental abnormalities such as those seen in PWS.

Development of NGF-dependent DRG neurons is impaired in necdin-deficient mice. A, B, Immunohistochemistry for substance P neurons in DRGs. A, Frozen sections of mouse DRGs from wild-type (Ndn+m/+p) and necdin-deficient (Ndn+m/-p) littermates at P0 were immunostained for substance P (SP). Scale bar: A, 50 μm. B, Substance P-positive (SP+) cells (A, arrowheads) in each DRG section were counted (DRG slices examined: +m/+p, n = 108; +m/-p, n = 98). C, D, Behavioral analyses of heat-induced pain. Wild-type and necdin-deficient littermates at P14 and P28 were subjected to hotplate (C) and tail-flick (D) tests to measure the latencies (animals examined: +m/+p, n = 7; +m/-p, n = 6). Similar data were obtained from four more groups of littermates. B-D, Graphs show mean ± SEM. *p < 0.05; **p < 0.002; ***p < 0.001.
In the peripheral nervous system and CNS in vivo, necdin is expressed exclusively in postmitotic neurons (Uetsuki et al., 1996; Andrieu et al., 2003). In embryonic DRGs, necdin is expressed in postmitotic neurons (Takazaki et al., 2002). This is supported by the present finding that a large population of DRG cells express both necdin and TrkA (Fig. 2A,B), because TrkA is expressed in postmitotic neurons differentiating at a later stage of DRG neurogenesis (Ma et al., 1999). However, the principal growth suppressor Rb is expressed in proliferative neuronal progenitors and involved in their terminal mitosis during neurogenesis (Yoshikawa, 2000). The absence of Rb expression induces impaired neuronal differentiation and marked reduction in the expression of TrkA and p75NTR in developing DRGs in vivo (Lee et al., 1994). In contrast, the present study has shown that the expression patterns and levels of TrkA and p75NTR are normal in necdin-null DRGs (Fig. 1C-E). Thus, it seems likely that necdin is not required for the differentiation of NGF-responsive nascent neurons but for their NGF-dependent survival at later stages.
The present study has also shown that necdin interacts with a kinase-active form of TrkA. The association between necdin and TrkA may not be mediated via the specific phosphotyrosine residues that are recognized by known TrkA-interacting signaling proteins (Fig. 3D). However, we cannot rule out the possibility that necdin binds indirectly to TrkA via an as-yet-unidentified molecule that links necdin and activated TrkA. Intriguingly, necdin binds to the structurally unrelated receptors TrkA and p75NTR to promote their association (Fig. 3C). Many previous studies have shown that TrkA and p75NTR receptors cooperate to increase NGF responsiveness (Roux and Barker, 2002; Chao, 2003). Thus, it is possible that necdin is an intrinsic regulator of the association between TrkA and p75NTR to enhance NGF-mediated activation of TrkA and its downstream signal transduction. This may account for the present findings that ectopic expression of necdin in NGF-stimulated PC12 cells induces sustained phosphorylation of TrkA and MAPK (Fig. 4C,D). Furthermore, NGF-induced phosphorylation of TrkA and MAPK was significantly reduced in necdin-deficient DRG explants (Fig. 5C,D). These findings support the idea that necdin facilitates the conversion of short-lived TrkA signaling events into long-lasting ones to sustain the NGF signal transduction. It is noteworthy that NRAGE (MAGE-D1; Dlxin-1), another p75NTR-interacting MAGE family protein with proapoptotic activity, disrupts the association between TrkA and p75NTR (Salehi et al., 2000). Thus, the MAGE family proteins necdin and NRAGE can modulate NGF signaling for neuronal survival by regulating the association between these receptors.
In this study, we mainly investigated the roles of necdin in the association between TrkA and p75NTR in NGF-dependent neurons. However, necdin may also modulate the proapoptotic function of p75NTR in certain settings. Necdin interacts directly with the intracellular domain of p75NTR (Tcherpakov et al., 2002; Kuwako et al., 2004) and suppresses the apoptotic activities of the transcription factors E2F1 and p53 (Taniura et al., 1998, 1999; Kobayashi et al., 2002), both of which are thought to function as intrinsic proapoptotic nuclear factors in neurons (Yoshikawa, 2000). In addition, overexpression of p75NTR antagonizes the inhibitory effect of necdin on E2F1-induced apoptosis (Kuwako et al., 2004). Necdin is translocated to the cytoplasm near the plasma membrane and the nucleus when coexpressed with p75NTR and E2F1, respectively (Kuwako et al., 2004). These findings suggest that necdin serves as a signaling protein for neuronal death/survival events through its interactions with p75NTR and nuclear proapoptotic proteins. The proform of NGF (proNGF) is a high-affinity ligand for p75NTR and induces p75NTR-dependent neuronal apoptosis, which requires sortilin as a coreceptor (Lee et al., 2001; Nykjaer et al., 2004). Furthermore, the intracellular domain of p75NTR is translocated into the nucleus after the ligand-induced cleavage (Frade, 2005). Therefore, we speculate that necdin is also involved in the proNGF/p75NTR signaling pathway to modulate neuronal apoptosis.
Necdin is abundantly expressed in central and peripheral neurons at early stages of development (Uetsuki et al., 1996). The absence of necdin expression is likely to augment apoptosis in neurotrophin-responsive neurons during the critical period for life-and-death decisions. Necdin may modulate the survival signals of neurotrophins such as NGF, brain-derived neurotrophic factor (BDNF), and NT-3. We found that necdin is expressed in large neurons as well as TrkA-positive small neurons in DRGs (Fig. 2A). These large neurons are thought to be a subpopulation of NT-3-dependent neurons that express TrkC (Mu et al., 1993). Because the intracellular domains of Trk family receptors (TrkA, TrkB, and TrkC) are highly conserved, it is possible that necdin also modulates the differentiation and survival of these large DRG neurons (proprioceptive cells) through its interaction with TrkC. In fact, we found that necdin binds to the intracellular domain of TrkB tyrosine kinase (K. Kuwako and K. Yoshikawa, unpublished observations). Thus, necdin deficiency may generally impair neurotrophin/Trk family signal transduction systems. The BDNF/TrkB signal transduction plays important roles in the development and function of central neurons (Bibel and Barde, 2000). Intriguingly, BDNF and TrkB are coexpressed in specific hypothalamic nuclei associated with satiety and locomotor activity, and heterozygous BDNF+/- mice develop behavioral abnormalities and hyperphagia accompanied by significant weight gain in early adulthood (Lyons et al., 1999; Kernie et al., 2000). These phenotypes resemble the hypothalamic defects seen in PWS. We speculate that necdin potentiates the BDNF/TrkB signaling in developing central neurons that are most affected by PWS. Additional studies on the modulatory roles of necdin in neurotrophin signaling systems will lead to a better understanding of the mechanisms underlying neuronal abnormalities in PWS and genomic imprinting-associated regulation of neuronal development.
Footnotes
This work was supported by a grant-in-aid (Scientific Research B2, No. 16300118) from the Japan Society for the Promotion of Science to K.Y. We thank Drs. E. M. Shooter, L. F. Reichardt, and T. Yamashita for research materials and Drs. T. Yagi, C.-H. Kuo, H. Taniura, and M. Niinobe for helpful discussions.
Correspondence should be addressed to Kazuaki Yoshikawa, Institute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: yoshikaw{at}protein.osaka-u.ac.jp.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257090-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}