

Abstract
Elevated levels of estradiol enhance learning in mammals, including humans, likely a result of hormone-induced heightened plasticity at CA3-CA1 synapses. The increase in long-term potentiation (LTP) magnitude is considered to be a consequence of the estradiol-induced increase in dendritic spine density and NMDA receptor (NMDAR)-mediated transmission; however, direct evidence linking these changes together is lacking. Alternatively, alterations in GABAergic inhibition or presynaptic release probability could contribute. Here, we show in time course studies using hippocampal slices from estradiol-treated ovariectomized rats that the LTP magnitude is increased only when spine density is increased simultaneously with an increase in NMDAR transmission relative to AMPA receptor (AMPAR) transmission, with no role for alterations in GABAergic inhibition or release probability. With time after hormone treatment, AMPAR transmission gradually increases during the maintained increase in spine density and NMDAR transmission. Eventually, the balance between NMDAR and AMPAR transmission is reestablished, and the LTP magnitude is no longer increased. Blocking genomic estrogen receptors prevents the heightened spine density, NMDAR transmission, and LTP magnitude, suggesting a tight mechanistic coupling between these morphological and functional changes. Thus, we propose that the hormone-induced increase in functional synapse density alone is not sufficient to support heightened plasticity. Rather, estradiol increases LTP via enhancing NMDAR transmission, likely through receptor insertion into newly formed or preexisting synapses. Later, when excitability in the circuit is at its highest and spine density remains elevated, the LTP magnitude is no longer increased, probably as a consequence of the delayed increase in AMPAR transmission that resets the balance between NMDAR and AMPAR transmission.
Introduction
Memory processing in mammals, including humans, varies across the estrous cycle, such that elevated levels of estrogen are positively correlated with enhanced short-term memory, and when estrogen levels fall, there is a temporary decline in memory processing (Phillips and Sherwin, 1992a,b). Permanent loss of estrogen, a consequence of normal aging, is associated with a decline in memory acquisition, which in some cases can be rescued with estrogen replacement therapy (Fader et al., 1999; Gibbs, 1999; Frick et al., 2002; Rapp et al., 2003). The precise cellular mechanisms underlying these behavioral effects of estrogen are unknown but likely reside in the profound effects of this hormone on the function of the hippocampus, a brain region required for normal learning and memory.
Elevated levels of 17β-estradiol enhance the magnitude of long-term potentiation (LTP) (Warren et al., 1995; Cordoba Montoya and Carrer, 1997; Good et al., 1999), a cellular correlate of memory (Malenka and Bear, 2004). Elucidating the mechanisms underlying this enhanced plasticity could provide important clues about the process whereby estradiol heightens learning and memory. Estradiol increases apical dendritic spines that are opposed by presynaptic terminals on hippocampal CA1 pyramidal cells via a tamoxifen-sensitive mechanism that requires activation of a classical genomic estrogen receptor, likely to be estrogen receptor α (ERα) (Gould et al., 1990; Woolley et al., 1990, 1996; Woolley and McEwen, 1993; Murphy and Segal, 1996; Woolley, 1998; Hao et al., 2003; MacLusky et al., 2005). Furthermore, estradiol decreases GABAergic inhibition, which is thought to trigger the increase in spine density (Murphy et al., 1998; Rudick and Woolley, 2001) and selectively increases NMDA receptor (NMDAR) expression and transmission (Weiland, 1992; Warren et al., 1995; Gazzaley et al., 1996; Cordoba Montoya and Carrer, 1997; Woolley et al., 1997; Good et al., 1999; Cyr et al., 2000, 2001; Bi et al., 2001; Rudick and Woolley, 2001). It has been assumed that the hormone-induced morphological change and enhanced NMDAR transmission are necessary for the elevation in LTP, but this has not been tested directly.
Alternatively, the genomically mediated increase in spine density may not be required for the enhancement in LTP, because acute application of estradiol to naive hippocampal slices in vitro increases NMDAR and AMPA receptor (AMPAR) transmission and LTP expression within minutes (Wong and Moss, 1992; Foy et al., 1999; Kim et al., 2002; Bi et al., 2003). These effects of estradiol are believed to be mediated by the yet-to-be-identified membrane-associated estrogen receptor, which may couple through G-proteins to increase Src and mitogen-activated protein kinase (MAPK) signaling (Kuroki et al., 2000; Bi et al., 2003; Qiu et al., 2003). The rapid onset of these functional changes implies that they do not require the hormone-induced increase in spine density. Thus, these data raise important questions about whether the increase in spine density contributes to the estradiol-induced heightened LTP magnitude.
In this study, we assessed what mechanisms underlie the observed increase in plasticity after estradiol treatment in vivo. Using Golgi impregnation staining and extracellular dendritic field potential recordings in area CA1 in slices from ovariectomized female rats treated with proestrous levels (80-120 pg/ml) (Woolley and McEwen, 1993) of 17β-estradiol benzoate (or oil vehicle), we find that the magnitude of LTP is increased only when spine density is elevated together with an increase in NMDAR transmission relative to AMPAR transmission. This enhanced LTP magnitude is not dependent on alterations in GABAergic inhibition or in presynaptic glutamate release. Furthermore, these functional synaptic changes require activation of genomic estrogen receptors and NMDARs during estradiol treatment similarly to the increase in spine density, indicating a tight mechanistic coupling between these changes. With time after hormone treatment, although spine density remains elevated, the LTP magnitude is normalized when the enhanced NMDAR transmission is accompanied by a delayed increase in AMPAR transmission that reestablishes the balance between NMDAR and AMPAR transmission. Thus, the increase in spine density may be important for the increase in plasticity observed, but alone this alteration in morphology is not sufficient to increase LTP magnitude. Our data indicate that the selective increase in NMDAR transmission, which occurs on either existing spines or possibly only on the newly developed spines as “silent” (NMDAR-only) synapses, is critical for the enhanced plasticity. Thus, we propose that a shift in the normal balance of glutamate transmission toward increased NMDAR transmission heightens the LTP magnitude. Accordingly, when the relative contribution of NMDAR to AMPAR transmission is reset with time after hormone treatment, the plasticity is restored to control levels despite the heightened excitability in the circuit and an increased density of synapses.
Materials and Methods
Animal treatments. All procedures used in this study were approved by the University of Alabama Institutional Animal Care and Use Committee and follow the guidelines outlined by the National Institutes of Health.
Young adult (7- to 9-week-old) female Sprague Dawley rats were ovariectomized (OVX) under 80 mg/kg ketamine and 5 mg/kg xylazine or 2.5% isoflurane in 100% oxygen, using aseptic conditions. Ten to 12 d after surgery, animals were given two subcutaneous injections (24 h interval) of either 10 μg of 17β-estradiol dissolved in 100 μl of cottonseed oil or oil alone and divided into the following experimental groups: oil vehicle-treated control, estradiol 24 (E24), E48, E72, and E120, where the number corresponds to hours after the second estradiol injection. The postsurgery time frame, although slightly different from that of Woolley et al. (Woolley and McEwen, 1992; Woolley et al., 1997), was chosen to effectively and evenly deplete endogenous circulating estradiol, because animals were at different stages in the estrous cycle when they were ovariectomized. Our model is also similar to that of Zamani et al. (2000), who used a postoperative window of 6-10 d. The slight differences in the postoperative window between research groups are not likely to cause a major problem when comparing our findings to current literature, because the alterations in spine density, NMDAR transmission, and LTP magnitude we report compare very well with those already published when similar time points after estradiol administration are compared. Because we have done a comprehensive time course that has not appeared previously in the literature, much of the data presented here is being reported for the first time.
In some experiments (see Fig. 2), animals were treated with only a single hormone or oil injection and were killed at either 24 h after the injection [E24 single (E24S)] or at 48 h [E48 single (E48S)]. The concentration of estradiol used in all experiments yields circulating estradiol concentrations of 80-120 pg/ml in vivo (Woolley and McEwen, 1993). To ensure animals in estradiol-treated groups had received successful hormone injections and to ensure that the ovaries had been completely removed from vehicle-treated control animals, uteri were removed, examined for vascularization and the presence of fluid, patted dry, and weighed (Hall et al., 1992). Uteri from estradiol-treated animals, on average, weighed double that of uteri obtained from oil vehicle-treated animals and were highly vascularized and filled with uterine fluid (uterine weight; vehicle, 0.09 ± 0.01 g; E24, 0.23 ± 0.02 g; E48, 0.19 ± 0.01 g; E72, 0.17 ± 0.01 g; E120, 0.14 ± 0.01 g; E24S, 0.18 ± 0.01 g; E48S, 0.17 ± 0.01 g). An animal was not included in the study if the uterine weight was not consistent with the average uterine weight from the corresponding group. In some experiments, OVX female rats were treated with an intraperitoneal injection of either 2 mg/kg tamoxifen (genomic estrogen receptor antagonist) (see Fig. 6) or 0.2 mg/kg NMDAR antagonist dizocilipine maleate (MK-801) (see Fig. 7) given just before estradiol when specified.
Hippocampal slice preparation. Treated OVX female rats were decapitated after deep halothane anesthesia, and brains were removed and placed in ice-cold artificial CSF (aCSF) containing the following (in mm): 119 NaCl, 26 NaHCO3, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, and 10 glucose, saturated with 95% O2 and 5% CO2, pH 7.4. Coronal slices (400 μm) were cut from dorsal hippocampus in the above solution and were held for 1-4 h in a submersion chamber at room temperature and continuously bubbled with the oxygen/carbon dioxide mixture. Recordings were performed in a submersion chamber perfused continuously with aCSF (26-28°C) at 2-3 ml/min. In some experiments, the noncompetitive GABAA receptor antagonist picrotoxin (100 μm) was added to the aCSF, and the Ca2+/Mg2+ ratio was increased from 2.5:1 to 4:4. In these experiments, it was important to remove CA3 pyramidal cell bodies from the slice to prevent epileptic activity.
Extracellular recordings. To stimulate Schaffer collateral axons, a bipolar stimulating electrode was placed in stratum radiatum within 200-300 μm of a recording electrode. Baseline field EPSPs (fEPSPs) were obtained by delivering 0.1 Hz stimulation at 300-400 μA to elicit baseline fEPSPs of 0.6-0.7 mV in amplitude (∼50% of maximal response) for 20 min with ≤8% variance. Raw fEPSP slopes recorded during baseline transmission were compared between experimental groups before the data were normalized to ensure that any findings observed were not an artifact resulting from differences in starting baseline transmission. Data were collected using an Axopatch 1D amplifier (Molecular Devices, Union City, CA) in current-clamp mode, and signals were magnified 1000×, filtered at 2 kHz, and acquired using software written in Labview (gift from Dr. Richard Mooney, Duke University, Durham, NC). LTP was induced by delivering high-frequency tetani [high-frequency stimulation (hfs); four trains at 100 Hz; 0.5 s duration; 20 s interval] at 1.5 times the baseline stimulation intensity. Experiments were performed for at least 40 min after hfs. The amount of potentiation was calculated at 30 min after tetanus. Significance from baseline was determined using Student's t test (p < 0.05), and values reported are the means ± SEMs. All experiments were included in the averaged data set regardless of the amount of potentiation generated. Only those experiments in which the baseline drifted by >5% or in which there was an increase in fiber volley amplitude after hfs were discarded from analysis. The steady-state dendritic depolarization during the tetanus was analyzed during all four trains by measuring the deviation from baseline at 130 ms (time at which tetani delivered in slices from vehicle-treated control reached a steady state) and statistics are represented as a deviation from baseline during the fourth train.
Stimulus-response curves were generated by incrementally (50 μV) increasing the stimulus intensity from a threshold stimulus until a maximal fEPSP was generated. AMPAR-mediated fEPSPs were isolated by bath-applying 100 μm d,l-APV (NMDAR antagonist) and NMDAR-mediated fEPSPs were isolated by bath-applying 10 μm DNQX (AMPAR antagonist) in nominally Mg2+-free aCSF. Two stimulus-response curves were collected from each slice, and all events (at least eight fEPSPs) at each stimulus intensity were averaged and plotted as a single value. An experiment was considered acceptable only when the variance between the two stimulus-response curves from a single slice was ≤10% at each stimulus intensity, and statistical significance was determined using Student's t test at each stimulus intensity (p < 0.05).
Because of the inherent variability between the stimulating electrodes, it was important for new control data to be collected each time a new stimulating electrode was used. This was particularly important in generating stimulus-response curves, because there is also a great deal of inherent variability between slices. This practice ensured that appropriate and accurate comparisons are made between control and treated experimental groups. With respect to the stimulus-response curves in Figure 5, it is important to note that different control data were obtained at each time point. (Note: the same electrode was used for E72 and E120 so that data could be compared with the same control data.) Therefore, accurate comparisons can be made only between stimulus-response curves generated in slices from vehicle-treated control versus estradiol-treated within each time point and not across time points, because maximal transmission stimulated can be affected by the stimulating electrode used. Thus, maximal transmission cannot be compared across groups, because, in most cases, different stimulating electrodes were used to collect the data.
Histology. Treated animals were perfused with 4% paraformaldehyde (PFA) in 0.1 m PBS with 1.5% (v/v) picric acid and postfixed for 1 week in the PFA-picric acid perfusate. We processed 100 μm sections using a modified version of the rapid Golgi procedure as described previously (Gabbott and Somogyi, 1984). Sections were cleared, mounted on slides, and coverslipped. Animal groups were coded, and spine counts were performed blind by imaging three to five sections, one to three dendritic segments per section, per animal (n = 3-6 animals per group) using confocal microscopy (100×; 1.4 numerical aperture objective). Images were deconvolved, and only obvious spine-like structures were counted per 10 μm segment of dendrite. Statistical significance was determined using Student's t test (p < 0.05). Images shown in Figure 4 are light microscopic images of a single focal plane.
Results
Time-dependent changes in the estradiol-induced increase in LTP magnitude
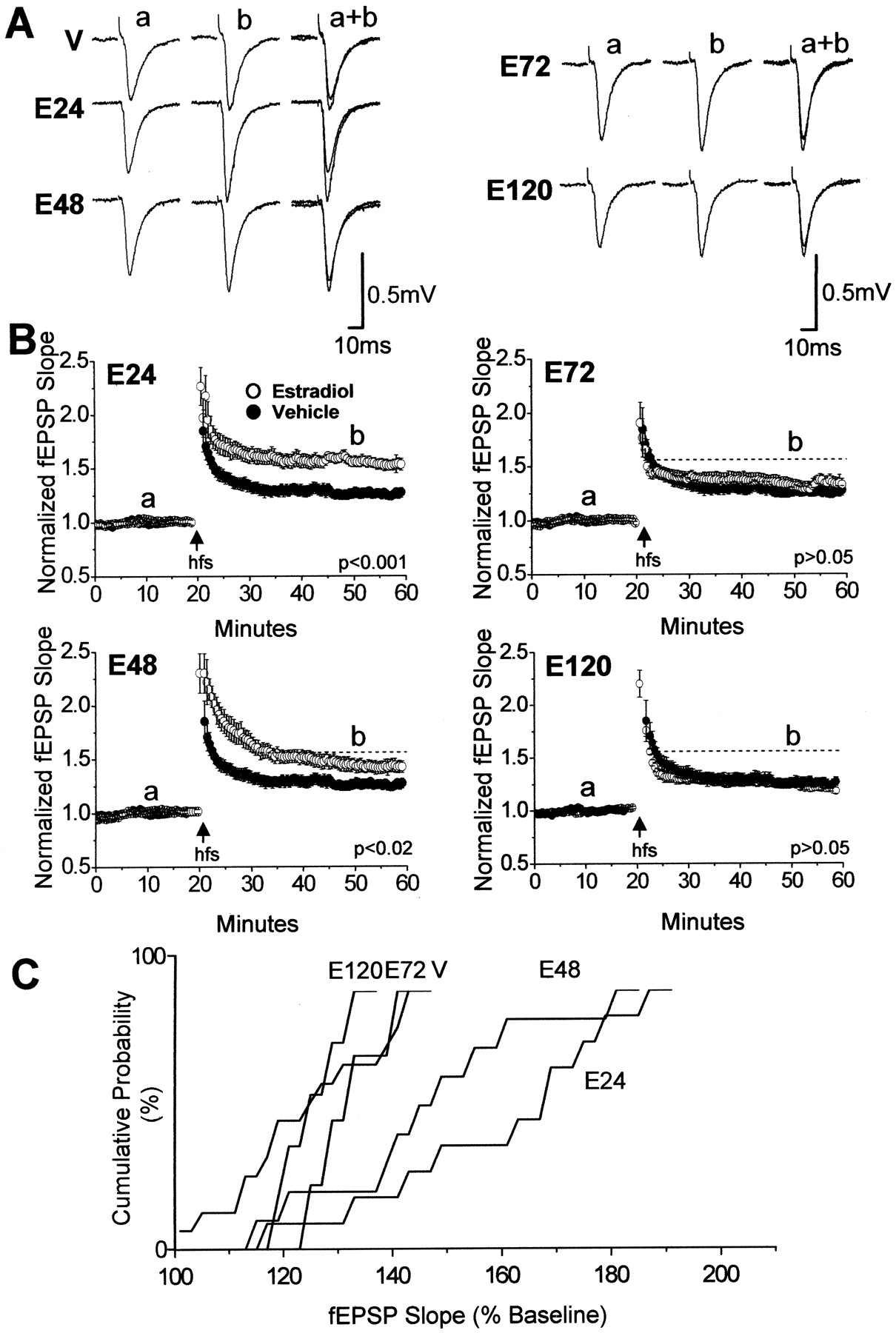
In this study, we used extracellular dendritic field potential recordings (fEPSPs) in area CA1 of acute hippocampal slices prepared from OVX female rats to determine whether hormone-induced alterations in GABAergic inhibition, presynaptic glutamate release, or changes in spine density and glutamate transmission contribute to the enhanced plasticity. We first investigated the time course over which the LTP magnitude is altered after estradiol administration to OVX rats (80-120 pg/ml) (Woolley and McEwen, 1993). In control experiments, we tested whether stress from injections could artificially affect LTP magnitude induced by comparing the LTP magnitude measured in acute slices prepared from oil vehicle-treated OVX animals (24 h after the last injection) to the LTP magnitude measured in slices from uninjected OVX animals. This was important because the effects of hormone on LTP magnitude are measured at varying time points distant from the injection time and are compared with the same control data. We find no significant difference in the LTP magnitude between oil vehicle-injected and uninjected control groups and have therefore pooled the data as a single control (126 ± 6% of baseline fEPSP slope in slices from vehicle-injected control animals, n = 8 slices/7 animals; vs 125 ± 4% of baseline fEPSP slope in slices from uninjected control animals, n = 5 slices/5 animals; p > 0.05).
We next compared the amount of LTP generated in slices from estradiol-treated animals to the pooled control data. At 24 (E24) and 48 (E48) h after the last injection of hormone (two injections separated by 24 h; 10 μg/100 μl), the magnitude of tetanus-induced LTP is significantly increased relative to that measured in slices from vehicle (oil)-treated and uninjected control animals (E24 vs control, p < 0.001; E48 vs control, p < 0.02) (Fig. 1A,B). However, in slices prepared 72 h (E72) after hormone treatment, the LTP magnitude is no longer significantly elevated above control and the same was true at the latest time point measured (E120) (E72 vs control, p > 0.05; E120 vs control, p > 0.05) (Fig. 1B). The temporal change in LTP magnitude after estradiol treatment is summarized in the cumulative probability plot shown in Figure 1C. Estradiol significantly shifts the distribution of LTP magnitude toward enhanced potentiation at E24 and E48 compared with control, E72, and E120 (p < 0.05 using Kolmogorov-Smirnov test). To ensure that these differences in LTP magnitude observed are not an artifact of differences in baseline transmission (stimulus intensity was set to generate baseline fEPSPs of consistent size between experimental groups) (see Materials and Methods), we compared the raw fEPSP slope values between groups before normalizing the data, as shown in Figure 1. We find no statistical difference in baseline transmission between control and E24 (0.18 ± 0.008 mV/ms fEPSP slope in slices from E24 vs 0.16 ± 0.008 mV/ms fEPSP slope in slices from control animals; p > 0.05), indicating that increased LTP at E24 cannot be explained by a difference in magnitude of baseline transmission. Furthermore, the time-dependent decrease in LTP magnitude induced at E24, E48, E72, and E120 is also not a result of differences in baseline transmission recorded from slices in each group, because baseline fEPSP slope values are not different between any of the hormone-treated groups (0.18 ± 0.008 mV/ms fEPSP slope in slices from E24; 0.18 ± 0.01 mV/ms fEPSP slope in slices from E48; 0.20 ± 0.008 mV/ms fEPSP slope in slices from E72; 0.18 ± 0.01 mV/ms fEPSP slope in slices from E120; p > 0.05 between all groups). Therefore, differences in LTP magnitude measured at time intervals distant to the time of hormone injection cannot be explained by differences in baseline transmission.
Alterations in GABAergic inhibition are not a major contributor to the heightened LTP magnitude
Disinhibition of pyramidal cells facilitates activation of NMDARs, which could facilitate induction of NMDAR-dependent LTP. Proestrous levels of estradiol depress GABAergic inhibition and disinhibit pyramidal cells as early as 24 h after a single injection of hormone (E24 single) (at the same concentration used in our two-injection model) (Rudick and Woolley, 2001). Therefore, if the hormone-induced increase in LTP magnitude is a direct consequence of decreased inhibitory drive onto pyramidal cells, then the LTP magnitude should be heightened by 24 h after a single injection of estradiol. Thus, we tested this idea and found no differences in LTP magnitude at this time compared with control slices (E24 single vs control; p > 0.05) (Fig. 2A). We considered the possibility that the mechanisms required for the increase in LTP magnitude could take longer to develop than the decrease in inhibition, so we waited an additional 24 h [48 h after the single hormone injection (E48S)] before killing the animals and testing for an increase in LTP magnitude. Still, the LTP magnitude was not different between groups (E48S vs control; p > 0.05) (Fig. 2B). These findings indicate that the estradiol-induced decrease in GABAergic inhibition is not likely responsible for the increase in plasticity and, furthermore, that estradiol must be elevated for a longer period of time (as in our standard two-injection protocol) for the heightened LTP expression to occur.

Time course of the estradiol-induced increase in LTP magnitude. A, Representative fEPSP waveforms at baseline (a) and 30 min after tetanus (b) recorded in slices from oil vehicle-treated control animals [vehicle (V)], and 24 h (E24), 48 h (E48), 72 h (E72), and 120 h (E120) after estradiol treatment are shown. B, Summary plots of the magnitude of LTP induced with a high-frequency tetanus (hfs; 100 Hz; 0.5 s duration; repeated 4 times with a 20 s interval between each tetanus; stimulus intensity during tetanus, 1.5× baseline) in slices from E24, E48, E72, E120, and vehicle-treated/uninjected control animals [○, estradiol; •, control (vehicle)]. Control experiments (125 ± 4%) were performed in n = 13 slices/12 animals, E24 (158 ± 7%) in n = 10 slices/9 animals, E48 (145 ± 6%) in n = 10 slices/9 animals, E72 (131 ± 6%) in n = 6 slices/4 animals, and E120 (125 ± 3%) in n = 5 slices/5 animals. LTP magnitude is increased compared with control (V) at E24 (p < 0.001) and E48 (p < 0.02) but not at E72 (p > 0.05) or E120 (p > 0.05). The dotted line represents the magnitude of LTP measured at E24. Control LTP is replotted on each graph for comparison. C, Cumulative probability plot shows potentiation in all animal groups. The plot shows enhanced potentiation at E24 and E48 compared with E72, E120, and vehicle. Error bars represent SEM.
Using a second strategy to test the role of altered inhibition, we reasoned that if disinhibition is responsible for the difference in LTP magnitude between E24 (in our two-injection model) and vehicle-treated control, then pharmacologically blocking GABAA receptors (GABAARs) with picrotoxin (100 μm) should prevent this difference. Interestingly, we noted that slices from estradiol-treated animals had a much greater tendency to become epileptic, and this was even more pronounced in the presence of picrotoxin, clearly demonstrating the increased excitability induced by hormone treatment. Therefore, in these experiments, CA3 cell bodies were removed from the slice and the Ca2+/Mg2+ ratio was changed from 2.5:1.3 to 4:4 mm to decrease epileptic activity in the presence of picrotoxin. We found that the LTP magnitude generated in slices from vehicle-treated animals in the presence of picrotoxin was still significantly different from that generated in E24 slices (E24 plus picrotoxin vs vehicle plus picrotoxin; p < 0.05) (Fig. 2C), indicating that disinhibition is likely not responsible for the enhanced LTP magnitude.
Finally, we wondered whether the reestablishment of the control level of GABAergic inhibition (which occurs after elevated levels of estradiol) (Rudick and Woolley, 2001) is the reason that the LTP magnitude induced at E72 is no longer significantly elevated above control. If inhibition is more potent at E72 than at E24, then, in theory, the same tetanus delivered would not activate NMDARs as effectively at E72 as at E24; thus, a smaller amount of LTP would be induced. If this is true, then pharmacologically blocking inhibition at E72 should increase the LTP magnitude toward that observed at E24. However, in the presence of picrotoxin, the LTP magnitude at E72 remained at control levels (E72 plus picrotoxin vs vehicle plus picrotoxin; p > 0.05) (Fig. 2D). Together, these findings show that alterations in GABAergic inhibition do not underlie the increase in LTP at E24 or the return of LTP at E72.
Estradiol does not alter the paired-pulse facilitation ratio
We next considered that the hormone-induced increase in plasticity could result from an increase in release of glutamate. To investigate a potential increase in release probability, we measured the paired-pulse facilitation (PPF) ratio, an indirect measure of presynaptic release probability (Dobrunz and Stevens, 1997), at several interstimulus intervals (10-50 ms) in E24 slices (because LTP magnitude is maximal at this time point) and compared them to those measured in vehicle-treated control slices. No differences were found at any of the tested intervals, including the 10 ms interval, which is the stimulus interval during the 100 Hz tetanus (p > 0.05) (Fig. 3A). These findings suggest that estradiol does not cause a dramatic increase in release probability. However, a lack of change in the PPF ratio does not rule out all potential presynaptic effects of estradiol. Interestingly, during the high-frequency tetanus (hfs) delivered to slices from animals treated with estradiol, we observed an increase in the steady-state dendritic depolarization at E24, E48, and E72, but not at E120, compared with vehicle-treated control slices (E24, E48, E72 vs vehicle, all at p < 0.05; E120 vs vehicle, p > 0.05; p values represent comparisons between the fourth tetanus; measurements were made when the depolarization reached a steady state in control slices at 130 ms) (Fig. 3B). The increase in depolarization was observed during all four tetani used to induce LTP (fourth tetani are shown), and no differences in the magnitude of the depolarization were found when E24 was compared with E48 and E72 (E24 vs E48, p > 0.05; E24 vs E72, p > 0.05).

Alterations in GABAergic inhibition are not responsible for the estradiol-induced increase in LTP magnitude. A, B, Summary plots show that a single estradiol injection is not sufficient to increase the magnitude of LTP measured at either 24 or 48 h after the estradiol injection [○, E24 single, 126 ± 7%, n = 3 slices/3 animals; ○, E48 single, 128 ± 3%, n = 9 slices/6 animals; •, control (vehicle), 125 ± 4%, n = 13 slices/12 animals; E24 single vs control (vehicle), p > 0.05; E48 single vs control, p > 0.05]. The dotted line indicates the magnitude of LTP measured 24 h after two estradiol injections. Control LTP in A and B is replotted from Fig. 1 for comparison. C, Summary plot shows that the magnitude of LTP remains greater in slices 24 h after two injections of estradiol (E24) (○; 146 ± 4%; n = 12 slices/10 animals) compared with oil vehicle-treated control (•; 129 ± 5%; n= 6 slices/4 animals; p < 0.05) during blockade of GABAA receptors with 100 μm picrotoxin (picro). D, Summary plot shows that blockade of GABAA receptors with picrotoxin does not increase the LTP magnitude recorded in slices 72 h after two injections of estradiol (E72; ○, 131 ± 4%, n = 8 slices/8 animals; vehicle-treated control; •; p > 0.05) with respect to vehicle-treated control slices. LTP from vehicle plus picrotoxin in C is replotted in D for comparison. Error bars represent SEM. E, Estradiol; V, vehicle; picro, picrotoxin; hfs, high-frequency tetanus.
Currently, we do not know what mechanism mediates the increase in depolarization during hfs. On close inspection of the traces (Fig. 3B), there appears to be less tetanus-induced depletion of neurotransmitter in slices from hormone-treated animals, but this remains to be investigated further. Importantly, our data indicate that, regardless of the mechanism, the increase in dendritic depolarization achieved during hfs is not the major mechanism driving the heightened LTP magnitude, because at E72 the steady-state depolarization is clearly apparent, precisely when the magnitude of LTP has returned to control levels.
A temporal relationship exists between the hormone-induced increase in LTP magnitude, spine density, and glutamate transmission
The above data suggest that neither a decrease in GABAergic inhibition nor an increase in presynaptic release is responsible for the hormone-induced heightened LTP magnitude. Previous reports have suggested that an important consequence of the estradiol-induced increase in spine density and NMDAR transmission is an increase in plasticity (Woolley, 1998), although this has not been investigated thoroughly. If the increase in spine density and NMDAR transmission are critical contributors to the heightened plasticity, then the time course of these changes should directly overlap in time with the changes in LTP magnitude.
We examined the time course of changes in spine density on CA1 pyramidal cells, using Golgi impregnation staining of hippocampal slices from perfusion-fixed animals (estradiol-treated or vehicle as above). We find that spine density is elevated at E24 with a maximal increase at E48 (E24 vs vehicle, p < 0.001; E48 vs vehicle, p < 0.001) (Fig. 4). The spine density remains elevated at E72, near that measured at E24 (E72 vs vehicle; p < 0.001) (Fig. 4). By E120, the spine density decreased to a level below that measured in vehicle-treated control slices (E120 vs vehicle; p < 0.02) (Fig. 4). Thus, the temporal relationship between the changes in spine density (Fig. 4) and LTP magnitude (Fig. 1) do not exactly correlate.
To evaluate the time course of the changes in glutamate transmission, we performed stimulus-response curves in separate slices, examining pharmacologically isolated NMDAR- and AMPAR-mediated transmission. Experiments were performed at E24, when LTP is at its highest magnitude and spines are elevated; at E48, when LTP is still significantly elevated and spine density is at its maximum; at E72, when LTP has returned to control levels but spines remain elevated; and at E120, when both LTP magnitude and spine density have returned to control levels. To relate changes in NMDAR transmission to changes in AMPAR transmission across the time periods studied, we calculated a ratio by dividing the averaged maximum NMDAR response by the averaged maximum AMPAR response at E24, E48, E72, and E120, and we refer to this as an NMDAR/AMPAR ratio. These ratios were calculated from stimulus-response curves obtained from at least five slices from five animals in each experimental group. At E24, NMDAR transmission is selectively enhanced, with no significant effect on AMPAR transmission (Woolley et al., 1997; Rudick and Woolley, 2001) (Fig. 5A-C, first column) (glutamate transmission at E24 vs vehicle, p > 0.05; isolated AMPAR transmission at E24 vs vehicle, p > 0.05; isolated NMDAR transmission at E24 vs vehicle, p < 0.01). Because NMDAR-mediated transmission is selectively increased, the NMDAR/AMPAR ratio is significantly increased compared with vehicle (Fig. 5D, first column) (p < 0.03). Importantly, the selective increase in NMDAR transmission (Fig. 5C, first column) occurs when spine density (Fig. 4) is increased and when the LTP magnitude is at its highest (Fig. 1).
At E48, when LTP magnitude is still significantly elevated above control, and spine density is maximally increased, the heightened NMDAR-mediated transmission is then accompanied by an increase in AMPAR-mediated transmission (Fig. 5A-C, second column) (glutamate transmission at E48 vs vehicle, p < 0.01; isolated AMPAR transmission at E48 vs vehicle, p < 0.02; isolated NMDAR transmission vs vehicle, p < 0.001). Importantly, however, the NMDAR/AMPAR ratio, as at E24, is increased compared with vehicle (Fig. 5D, second column) (p < 0.01), and, at both time points, the magnitude of LTP is significantly increased.

Estradiol does not alter the PPF ratio. A, Overlaid averaged pairs of fEPSP waveforms (8-10 events) elicited at 10, 20, 30, 40, and 50 ms interstimulus intervals obtained from vehicle-treated control and E24 slices show no difference in paired-pulse facilitation (top). Bar chart shows averaged paired-pulse facilitation ratios recorded at the various interstimulus intervals in slices from vehicle-treated control and E24 animals (bottom). At least 8-10 trials at each interval were averaged per slice and at least five slices were recorded from at least five animals from each group. Error bars represent SEM. No significant differences were found at any interval (p>0.05). B, Representative fEPSP waveforms during high-frequency tetanus (hfs) from E24, E48, E72, and E120 compared with control. E, Estradiol; V, vehicle-treated control. The steady-state dendritic depolarization during hfs is elevated at E24, E48, and E72 (p < 0.05) but returns to vehicle-treated control by E120 (p > 0.05). Measurements for statistical analysis were made once steady state was reached in slices from vehicle-treated control animals (i.e., 130 ms from stimulus artifact).
At E72, both NMDAR- and AMPAR-mediated transmission remain elevated above control (Fig. 5A-C, third column). However, in contrast to that at E48, the NMDAR/AMPAR ratio at E72 is not different from that measured in vehicle-treated control slices (Fig. 5D, third column), and, likewise, the LTP magnitude is also not different from control. Thus, at this time, although the maximum amount of transmission mediated by each receptor subtype is significantly increased, the relative contribution of each receptor is normalized, which is correlated with the return of the LTP magnitude to control (Fig. 5A-D, third column) (glutamate transmission at E72 vs vehicle, p < 0.01; isolated AMPAR transmission at E72 vs vehicle, p < 0.04; isolated NMDAR transmission vs vehicle, p < 0.05; NMDAR/AMPAR ratio at E72 vs vehicle, p > 0.05).
At E120, the system has completely reset. The NMDAR/AMPAR ratio at this time point, as at E72, is not different from the control ratio; however, unlike that at E72, the maximum transmission mediated by both NMDARs and AMPARs has now returned to the level measured from vehicle-treated control slices (Fig. 5A-D, fourth column) (glutamate transmission at E120 vs vehicle, p > 0.05; isolated AMPAR transmission at E120 vs vehicle, p > 0.05; isolated NMDAR transmission at E120 vs vehicle, p > 0.05; NMDAR/AMPAR ratio at E120, p > 0.05). The reestablishment of the NMDAR- and AMPAR-mediated transmission to control levels occurs precisely when spine density has also returned to the density measured in vehicle-treated control slices (Fig. 4), indicating that the density of functional synapses at E120 and in vehicle treated-control slices is similar.
The above data show that the elevation in LTP magnitude at E24 is temporally correlated with an enhancement in NMDAR transmission and an increase in the NMDAR/AMPAR ratio, which occurs when spine density is also elevated, suggesting estradiol may upregulate NMDAR-only-containing synapses. Interestingly, at E48, when LTP is still elevated and spine density is at its maximum, an increase in AMPAR transmission accompanies the increase in NMDAR transmission, but the NMDAR/AMPAR ratio, as at E24, is still significantly elevated above control. Lastly, at E72, the tetanus-induced LTP and the NMDAR/AMPAR ratio have been normalized to that measured in slices from vehicle-treated control. Whether the alterations in glutamate transmission are a result of receptor insertion into new or existing spines is not clear. However, the simultaneous return of spine density and evoked glutamate transmission to control levels at E120 argues that the enhanced transmission is caused by receptor insertion into new synapses.
Tamoxifen prevents the effects of estradiol on spine density, NMDAR transmission, and LTP magnitude
The estradiol-induced increase in spine density requires activation of a tamoxifen-sensitive genomic estrogen receptor, likely ERα (Murphy and Segal, 1996). If the changes in morphology and function are mechanistically coupled, then it is predictable that the hormone-induced increase in NMDAR transmission and LTP magnitude would also be sensitive to blockade by tamoxifen. Ovariectomized animals were concurrently treated with estradiol and tamoxifen (2 mg/kg), and as predicted, the estradiol-induced increase in spine density was completely prevented, as was the increase in NMDAR transmission, and LTP magnitude (LTP, E24 vs vehicle, p < 0.001; E24 vs vehicle plus tamoxifen, p < 0.001; E24 plus tamoxifen vs E24, p < 0.01; E24 plus tamoxifen vs vehicle plus tamoxifen, p > 0.05) (spine density, E24 vs vehicle plus tamoxifen, p < 0.04; E24 vs E24 plus tamoxifen, p < 0.001; E24 plus tamoxifen vs vehicle plus tamoxifen, p > 0.05) (isolated NMDAR transmission, E24 vs vehicle, p < 0.001; E24 vs vehicle plus tamoxifen, p < 0.003; E24 plus tamoxifen vs E24, p < 0.02; E24 plus tamoxifen vs vehicle plus tamoxifen, p > 0.05) (Fig. 6A-C). Importantly, in control experiments, tamoxifen alone had no effect of its own on the magnitude of LTP, spine density, or NMDAR transmission (LTP, vehicle vs vehicle plus tamoxifen, p > 0.05; spine density, vehicle vs vehicle plus tamoxifen, p > 0.05; NMDAR-mediated transmission, vehicle vs vehicle plus tamoxifen, p > 0.05) (Fig. 6 A-C). Together, these data suggest that the increase in NMDAR transmission and LTP magnitude require activation of genomic estrogen receptors, similarly to the increase in spine density (Murphy and Segal, 1996), rather than to activation of the unidentified membrane-associated estrogen receptor (Foy et al., 1999; Fugger et al., 2001). In support of this idea, we tested whether the increase in NMDAR transmission induced by acute application of estradiol (1 nm) to naive slices could persist in the presence of tamoxifen (100 nm) and found that transmission was still significantly enhanced (our unpublished observation). This supports our interpretation of the involvement of genomic receptors in mediating the increase in NMDAR transmission we observe. Thus, the increase in spine density, NMDAR transmission, and LTP magnitude are similarly dependent on activation of genomic estrogen receptors, although it is not clear whether these changes are mediated by a single cellular pathway that induces sequential changes in spine density and synaptic function or via independent but parallel pathways that all require activation of ERα for their initiation.

Time course of estradiol-induced changes in spine density. A, Deconvolved confocal images of Golgi-stained apical dendrites from vehicle-treated control, E24, E48, E72, and E120. Scale bar, 10 μm. B, Bar chart displays the number of dendritic spines/10 μm of apical dendrite from each experimental group (vehicle-treated control slices, 6.9 ± 0.4 spines/10 μm, 29 sections of dendrite from 3 animals; E24, 10.6 ± 0.3 spines/10 μm, 42 sections from 6 animals; E48, 14.2 ± 0.7 spines/10 μm, 25 sections from 4 animals; E72, 12.0 ± 0.6 spines/10 μm, 28 sections from 4 animals; E120, 5.4 ± 0.3 spines/10 μm, 23 sections from 7 animals were analyzed). E24, E48, and E72 versus vehicle, p < 0.001; E120 versus vehicle, p < 0.02; E24 versus E48, p < 0.001; E24 versus E72, p < 0.04. Asterisks denote significance as indicated. Error bars represent SEM.
The NMDAR antagonist MK-801 prevents the hormone-induced changes in spine density, NMDAR transmission, and LTP magnitude
In an attempt to separate the functional synaptic changes from the morphological changes, we treated animals concurrently with estradiol and MK-801 (0.2 mg/kg), an NMDAR antagonist known to prevent the estradiol-induced increase in spine density (Woolley and McEwen, 1994; Murphy and Segal, 1996) and asked whether the elevation in LTP magnitude and NMDAR transmission could persist in the absence of an increase in spine density. First, to ensure MK-801 is successfully cleared from the animal and does not artificially influence the experimental out-come, animals were treated with MK-801, and LTP experiments were performed at 6 or 12 h after MK-801 treatment. By 12 h after the injection, we found the amount of LTP induced after a single MK-801 treatment is not different from that observed in control animals (125 ± 4% of baseline fEPSP slope in control, n = 13 slices/12 animals, vs 133 ± 5% of baseline fEPSP in MK-801-treated, n = 3 slices/3 animals; p > 0.05) (data not shown), indicating that the antagonist has been successfully cleared from the animal and that this treatment regime will not interfere with our results. In other control experiments, we found that the 2 d treatment of animals with MK-801 also had no significant effect of its own on LTP magnitude, spine density, or NMDAR transmission compared with vehicle-treated controls (LTP, vehicle vs vehicle plus MK-801, p > 0.05; spine density, vehicle vs vehicle plus MK-801, p > 0.05; NMDAR-mediated transmission, vehicle vs vehicle plus MK-801, p > 0.05) (Fig. 7A-C).
Similar to tamoxifen, MK-801 completely prevented the increase in spine density, NMDAR transmission, and LTP magnitude (LTP, E24 vs vehicle, p < 0.001; E24 plus MK-801 vs E24, p < 0.01; E24 plus MK-801 vs vehicle, p > 0.05) (spine density, E24 vs vehicle plus MK-801, p < 0.001; E24 plus MK-801 vs E24, p < 0.001; E24 plus MK-801 vs vehicle, p > 0.05) (isolated NMDA transmission, E24 vs vehicle, p < 0.01; E24 plus MK-801 vs E24, p < 0.01; E24 plus MK-801 vs vehicle, p > 0.05) (Fig. 7A-C). Furthermore, we find a single dose of MK-801 given on either day 1 or day 2 of estradiol treatment is sufficient to prevent the functional synaptic changes, indicating that NMDAR activation is needed on both days of hormone treatment to induce these functional effects [NMDAR transmission, E24 plus MK-801 on day 1 (0.05 ± 0.007 mV/ms) or day 2 (0.05 ± 0.007 mV/ms) vs vehicle (0.05 ± 0.006 mV/ms), both with p > 0.05; LTP, E24 plus MK-801 on day 1 (128 ± 4%) or day 2 (124 ± 4%) vs vehicle (125 ± 4%), both with p > 0.05]. Thus, our findings indicate that both the functional and morphological changes require activation of NMDARs during hormone treatment, which provides additional evidence that these changes are tightly coupled to one another mechanistically.
Discussion
Estradiol increases the magnitude of LTP at CA3-CA1 synapses, which may be critical to the memory-enhancing effects of this hormone (Phillips and Sherwin, 1992a,b). Many studies, both in vitro and in vivo, have demonstrated that LTP is heightened after an increase in circulating hormone (Warren et al., 1995; Cordoba Montoya and Carrer, 1997; Good et al., 1999; Bi et al., 2001). Others have shown that synaptic efficacy also can be increased by a rapid nongenomic mechanism coupled to extracellular signal-regulated kinase (ERK)/MAPK signaling that is stimulated during acute estradiol application to naive slices in vitro (Foy et al., 1999; Fugger et al., 2001). Given these reports, several mechanisms could contribute to the increase in LTP magnitude.

Time-dependent modulation of glutamate transmission by estradiol. Summary plots show stimulus-response curves of glutamate transmission (A, glutamate), pharmacologically isolated AMPAR transmission (B, AMPAR only), or NMDAR transmission (C, NMDAR only). AMPAR transmission was isolated by blocking NMDARs with 100 μm d,l-APV, and NMDAR transmission was isolated by blocking AMPARs with 10 μm DNQX in nominally Mg2+-free aCSF. Open circles (○) represent data obtained in slices from estradiol (E)-treated animals, and filled circles (•) represent data from vehicle (V)-treated control animals. A, Glutamate transmission is unchanged at E24 (first column, E24, 0.35 ± 0.04 mV/ms; V, 0.28 ± 0.04 mV/ms, p > 0.05), is increased at E48 (second column, E48, 0.45 ± 0.04 mV/ms; V, 0.29 ± 0.02 mV/ms; p < 0.01) and E72 (third column, E72, 0.55 ± 0.05 mV/ms; V, 0.32 ± 0.05 mV/ms, p < 0.01), and is normalized by E120 (fourth column, E120, 0.30 ± 0.04 mV/ms; V, 0.32 ± 0.05 mV/ms, p > 0.05). B, Pharmacologically isolated AMPAR-mediated transmission is unchanged at E24 (first column, E24, 0.33 ± 0.05 mV/ms; V, 0.32 ± 0.04 mV/ms, p > 0.05), is increased at E48 (second column, E48, 0.43 ± 0.04 mV/ms; V, 0.30 ± 0.02 mV/ms, p < 0.02) and E72 (third column, E72, 0.47 ± 0.03 mV/ms; V, 0.37 ± 0.03 mV/ms, p < 0.05), and is normalized by E120 (fourth column, E120, 0.33 ± 0.04 mV/ms; V, 0.37 ± 0.03 mV/ms, p > 0.05). C, NMDAR-mediated transmission is selectively increased at E24 (first column, E24, 0.1 ± 0.01 mV/ms; V, 0.05 ± 0.006 mV/ms, p < 0.03) and remains increased at E48 (second column, E48, 0.07 ± 0.004 mV/ms; V, 0.03 ± 0.004 mV/ms, p < 0.001) and at E72 (third column, E72, 0.12 ± 0.01 mV/ms; V, 0.08 ± 0.01 mV/ms, p < 0.04) and is normalized by E120 (fourth column, E120, 0.07 ± 0.01 mV/ms; V, 0.08 ± 0.01 mV/ms, p > 0.05). Asterisks denote significance from vehicle-treated control (p < 0.05). D, Bar graphs show the maximum amplitude of evoked AMPAR and NMDAR transmission (measured at 800 μA) and the NMDAR:AMPAR ratio calculated by dividing the maximal NMDAR response by the maximal AMPAR response measured in slices from E24 (first column, E24, 0.51± 0.09; V, 0.16 ± 0.02, p < 0.03), E48 (second column, E48, 0.17 ± 0.01; V, 0.11 ± 0.01, p < 0.01), and E72 (third column, E72, 0.28 ± 0.05; V, 0.21 ± 0.06, p > 0.05), and E120 (fourth column, E120, 0.21 ± 0.05; V, 0.21 ± 0.06. p > 0.05) compared with vehicle-treated control. Each point represents the averaged responses obtained from at least five slices from five animals in each group. Calibration: 0.5 mV, 5 ms (for glutamate and AMPAR transmission); 0.4 mV, 10 ms (for NMDAR transmission). Error bars represent SEM.
The goal of this study was to elucidate the mechanisms underlying the heightened LTP magnitude after elevated levels of estradiol in vivo. Our studies show that a decrease in GABAergic inhibition mediated by GABAARs (Rudick and Woolley, 2001; Rudick et al., 2003) or an increase in presynaptic release probability are not major contributors. However, at this time, we cannot rule out a potential contribution of decreased inhibition mediated by postsynaptic GABAB receptors to the enhanced plasticity. Our time course studies strongly suggest a critical role for specific and timely alterations in glutamate transmission. We find that the LTP magnitude is increased only when NMDAR transmission is increased relative to AMPAR transmission, which occurs during a time when spine density is also elevated. Later, although spine density remains high, the magnitude of tetanus-induced LTP and the ratio of NMDAR to AMPAR transmission have been normalized. Thus, reestablishing the relative contribution of NMDAR transmission to AMPAR transmission in the circuit returns a homeostatic balance in excitatory drive while synapse density remains elevated.

Tamoxifen blocks the estradiol-induced increase in spine density, the selective increase in NMDAR transmission, and LTP magnitude. A, Summary plot of LTP experiments obtained in slices from vehicle-treated control (V) (•; 125 ± 4%; n = 13 slices/12 animals replotted from Fig. 1), V plus tamoxifen (T), a selective estrogen receptor modulator (2 mg/kg) [V + T (□); 123 ± 4%; n = 7 slices/7 animals], E24 (○; 158 ± 7%; n = 10 slices/9 animals; replotted from Fig. 1), and E24 + T (▵; 122 ± 5%; n = 7 slices/7 animals). Tamoxifen blocks the estradiol-induced increase in LTP magnitude (V + Tvs E24 + T, p > 0.05; V + Tvs V, p > 0.05). B, Bar chart displays the number of dendritic spines/10 μm of apical dendrite from each experimental group [V (6.9 ± 0.4 spines/10 μm; n = 29 sections of dendrite from 3 animals), V+T (5.9 ± 0.4 spines/10 μm; n = 11 sections of dendrites from 4 animals), E24 (10.6 ± 0.3 spines/10 μm; n = 42 sections from 6 animals), and E24+T animals (5.4 ± 0.2 spines/10 μm; n = 30 sections of dendrites from 5 animals)]. Both V and E24 are replotted from Figure 4 for comparison. C, Summary plot showing stimulus-response curves of pharmacologically isolated NMDAR transmission (10 μm DNQX; nominally Mg2+ free) obtained in slices from V (•; 0.06 ± 0.002 mV/ms), V + T (□; 0.06 ± 0.003 mV/ms), E24 (○; 0.08 ± 0.002 mV/ms), and E24 + T (▵; 0.06 ± 0.005 mV/ms) (E24 vs E24 + T, p < 0.02; E24 + Tvs V + T, p > 0.05; V + Tvs V, p > 0.05). Asterisk denotes significance as indicated. Each point within this plot represents averaged responses obtained from at least five slices from five animals in each group. *p < 0.04, **p < 0.001. Error bars represent SEM.
Clearly, changes in glutamate transmission are correlated with the alterations in LTP magnitude, but what is the role of the increase in dendritic spine density? Our findings do not show definitively that the increase in synapse density contributes to the heightened plasticity. A contribution of the increase in spine density is an attractive mechanism, however, because changes in the density of excitatory glutamate synapses alter memory acquisition (Leuner et al., 2003) and learning, and memory is enhanced during the time frame when estradiol induces an increase in synapse density (Leuner et al., 2003). If the increase in spine density is required, then there must be an accompanying increase in glutamate transmission, because these new spines must form functional synapses to be involved in heightened plasticity. Accordingly, the increase in NMDAR transmission we observe at E24 could result from insertion of newly expressed NMDARs into the newly developed synapses, an idea supported by binding studies that show increased NMDAR expression (Weiland, 1992; Daniel and Dohanich, 2001) and electrophysiological recordings (performed during the time frame that spines are elevated), which indicate that at least a portion of these receptors are synaptically located (Woolley et al., 1997; this study). However, no study to date (including this study) has determined directly at the level of single synapses whether the new NMDARs are inserted into preexisting synapses, are exclusively inserted into the newly developed spines, or some combination of the two. Selective insertion into new spines would increase the density of highly plastic silent (NMDAR-only-containing) synapses whose conversion to active (AMPAR- and NMDAR-containing) synapses after an LTP-inducing stimulus could be responsible for the enhanced plasticity (Isaac et al., 1995; Liao et al., 1995). Thus, based on the temporal relationship we observe between the estradiol-induced changes in spine density, glutamate transmission, and LTP magnitude, a plausible scenario compatible with our results is that estradiol creates new synapses that are initially silent (contain only NMDARs) at E24, thereby increasing the magnitude of LTP that can be induced. Later, at E72, the newly developed silent synapses have been converted to active synapses through the insertion of AMPARs. This delayed increase in AMPAR transmission reestablishes the NMDAR/AMPAR ratio and the ratio of silent to active synapses, thereby resetting the magnitude of LTP that can be induced to control levels. This idea is consistent with the observation that, at E72, the maximal amount of AMPAR-mediated transmission is now increased along with a maintained increase in NMDAR transmission (Fig. 5A-C, right) and spine density (Fig. 4). Furthermore, at E120, the simultaneous return of spine density and maximal evoked glutamate transmission to basal levels suggests that the enhanced transmission is the result of receptor insertion into the new synapses.

MK-801 blocks the estradiol-induced increase in spine density, the selective increase in NMDAR transmission, and LTP magnitude. A, Summary plot of LTP experiments obtained in slices from vehicle-treated control (V) (•; 125 ± 4%; n = 13 slices/12 animals replotted from Fig. 1), V + MK-801 (M), an NMDAR antagonist (0.2 mg/kg) (□; 130 ± 7%; n = 5 slices/5 animals), E24 (○; 158 ± 7%; n = 10 slices/9 animals replotted from Fig. 1), and in E24 + M (▵; 128 ± 4%; n = 8 slices/5 animals). MK-801 blocks the estradiol-induced increase in LTP magnitude (V + Mvs E24 + M, p > 0.05; V + Mvs V, p > 0.05). B, Bar chart displays the number of dendritic spines/10 μm of apical dendrite from each experimental group [V (6.9 ± 0.4 spines/10 μm; n = 29 sections of dendrite from 3 animals), V + M (6.1 ± 0.5 spines/10 μm; 17 sections of dendrites from 5 animals), E24 (10.6 ± 0.3 spines/10 μm; n = 42 sections from 6 animals), and E24 + M (6.0 ± 0.4 spines/10 μm; 21 sections from 4 animals)]. Both V and E24 are replotted from Figure 4 for comparison. C, Summary plot showing stimulus-response curves of pharmacologically isolated NMDAR transmission (10 μm DNQX; nominally Mg2+ free) obtained in slices from V (•; 0.05 ± 0.006 mV/ms; replotted from Fig. 5), V + M (□; 0.06 ± 0.009 mV/ms), E24 animals (○; 0.1 ± 0.01 mV/ms; replotted from Fig. 5), and in E24 + M (▵; 0.05 ± 0.001 mV/ms) (E24 vs E24 + M, p < 0.01; E24 + Mvs V + M, p > 0.05; V + Mvs V, p > 0.05). Each point within this plot represents the averaged responses obtained from five slices from five animals in each group. *p < 0.001. Error bars represent SEM.
Of course, mechanisms alternative to the one described above must be considered. Recently published findings challenge the need for an increase in NMDAR expression and spine density in the enhancing effects of estradiol on LTP magnitude. Within minutes of bath application of estradiol to either slices from male rats or untreated OVX female rats, NMDA and AMPA transmission are potentiated, as well as the magnitude of LTP (Wong and Moss, 1992; Foy et al., 1999; Kim et al., 2002; Bi et al., 2003). The rapid time course of these effects suggests that estradiol can alter synaptic function in the absence of an increase in spine density, which requires a genomic mechanism (Murphy and Segal, 1996; this study). Thus, estradiol most likely increases synaptic function via the unidentified membrane-associated receptor. This would potentially lead to stimulation of Src and ERK/MAPK signaling, resulting in an increase in NMDAR phosphorylation and function (Bi et al., 2001). However, alterations in membrane excitability could contribute to the enhanced NMDAR transmission observed by us and others (Woolley et al., 1997), because estradiol reduces the amplitude of the afterhyperpolarization via altering intracellular Ca2+ levels and the function of KCa2+ channels (Kumar and Foster, 2002; Carrer et al., 2003). Increasing NMDAR activation would result in an increase in LTP induction. This potential mechanism would bypass the need for increased synapse density and NMDAR expression. Zamani et al. (2004) have shown recently that pharmacologically enhancing NMDAR transmission, by recording in 0 mm Mg2+ and 10 μm glycine, eliminates the selective increase in NMDAR transmission (with respect to AMPA transmission) after estradiol replacement to OVX animals. These authors suggest that the increase in monosynaptic NMDAR potentials originally reported by Woolley et al. (1997) (and confirmed in this study) result not from increased receptor expression as interpreted (but see Weiland, 1992; Daniel and Dohanich, 2001) but rather are a result of a mechanism involving estradiol-induced changes in membrane depolarization.
However, neither of these potential mechanisms can explain our results. First, all of the morphological and functional effects we observe require 24-48 h to occur after in vivo hormone administration and are prevented by the genomic estrogen receptor antagonist, tamoxifen, indicating that a genomic mechanism rather than the rapid membrane-associated mechanism is required. Furthermore, the combined sensitivity of the morphological and functional changes to blockade by tamoxifen and the NMDAR antagonist MK-801 indicates that these estradiol-induced changes require the same mechanistic pathway, tightly coupling them together. This strengthens the idea that the increase in spine density underlies the changes in synaptic function and plasticity. Unfortunately, we cannot eliminate the possibility that separate NMDAR-dependent and tamoxifen-sensitive mechanisms individually mediate the increase in spines, NMDAR transmission, and LTP magnitude. However, we favor the possibility that these events are sequentially related, because we have been unable to separate them and because of the prolonged period of time over which these changes occur. Second, although an increase in excitability could, in theory, account for the increase in NMDAR transmission we and others observe (Woolley et al., 1997), it is difficult to explain how this mechanism could contribute to the delayed increase in AMPAR transmission we observe first at E48 (which is maintained at E72), because AMPARs are not voltage dependent. An obvious mechanism is an increase in expression of AMPARs, an interpretation that is supported by others (Palomero-Gallagher et al., 2003). Whether NMDAR transmission is elevated by increased excitability or through insertion of NMDARs into newly formed synapses as our data suggest requires additional investigation at the level of single synapses. But regardless of the mechanism, our data clearly indicate that increased NMDAR transmission relative to AMPA transmission is required and that a simple increase in excitability may not be all that is needed. This idea is strongly supported by our finding that tetanus-induced dendritic depolarization remains significantly increased above control at E72 and is similar in magnitude to that at E24, where LTP is at its highest. However, at E72, the LTP magnitude induced is no longer significantly different from control level, indicating that enhanced excitability is not the major contributor to the heightened plasticity.
Estradiol-induced changes are reminiscent of early synaptic development
During early postnatal development, the density of NMDAR-only-containing postsynaptically silent synapses is higher than in a mature hippocampus. This is believed to increase the capacity of immature circuits for plastic changes in synaptic strength (Nusser et al., 1998; Petralia et al., 1999). Synapses at this time predominantly express NMDAR 2B (NR2B)-containing NMDARs (Portera-Cailliau et al., 1996). Estradiol has been shown to selectively increase expression of NR2B subunits (Cyr et al., 2001), suggesting the possibility that this hormone increases the density of synapses that have immature characteristics and are highly capable of plastic changes. Thus, we speculate that estradiol takes advantage of mechanisms available during development to transiently increase the density of silent synapses. Thus, estradiol may temporarily heighten plasticity and learning by modifying a mature synaptic circuit through the addition of highly plastic immature synapses reminiscent of those during early development.
Other mediators of the effects of estradiol on hippocampal function
The estradiol-induced disinhibition of CA1 pyramidal cells is believed to trigger the increase in spine growth (Murphy et al., 1998; Rudick and Woolley, 2001). However, other mechanisms likely also participate in the morphological and functional effects of this hormone. Estradiol increases mRNA levels of choline acetyltransferase, the synthetic enzyme for acetylcholine (ACh), and increases release of ACh in the hippocampus (Gibbs, 1996; Gibbs et al., 1997). ACh depresses release of GABA (Pitler and Alger, 1992), which could contribute to the disinhibition of pyramidal cells and facilitate spine growth and thus LTP. Furthermore, the estradiol-induced increase in NMDAR binding requires activation of muscarinic M2 receptors (Daniel and Dohanich, 2001), and the hormone-induced decrease in GABAergic transmission and increase in spine density are partially prevented by lesion of the medial septum (Leranth, 2000; Rudick et al., 2003). Moreover, enhanced memory acquisition mediated by elevated levels of estradiol requires cholinergic modulation of hippocampus (Gibbs, 1998). Cholinergic modulation of hippocampal circuits is clearly important for synaptic plasticity (Ovsepian et al., 2004) and for normal learning and memory (Hasselmo, 1999); thus, the ability of estradiol to modulate cholinergic transmission could underlie the hormone-induced enhancement in LTP magnitude as well as memory processing.
In addition, it has been shown recently that BDNF immunoreactivity increases in the hippocampus during the estrous cycle, which is correlated with increases in circulating levels of estradiol (Scharfman et al., 2003). The increase in BDNF is also correlated with an increase in neuronal excitability. Because of the well documented ability of BDNF to increase spine growth (Tyler and Pozzo-Miller, 2003; Alonso et al., 2004), the possibility exists that the increase in BDNF after elevations in circulating estradiol could be causal to the hormone-induced increase in spine density. Therefore, each one of these molecules, GABA, acetylcholine, and BDNF has been shown to be important for alterations in hippocampal synaptic plasticity (Murphy et al., 1998; Tyler and Pozzo-Miller, 2001; Alonso et al., 2004; Casasola et al., 2004; Kramar et al., 2004; Ovsepian et al., 2004). Thus, it is probable that estradiol may increase the density of silent synapses and plasticity through one or all of these molecules.
It is clear that spine density, synaptic efficacy, and transmission play vital roles in learning and memory, and it is quite intriguing that estradiol modulates each one of these processes (Phillips and Sherwin, 1992a,b; Warren et al., 1995; Cordoba Montoya and Carrer, 1997; Woolley et al., 1997; Fader et al., 1999; Gibbs, 1999; Good et al., 1999; Rudick and Woolley, 2001; Frick et al., 2002; Rapp et al., 2003). We show that the estradiol-induced alterations in synaptic efficacy require changes in synapse density, which may underlie the estradiol-induced improvements in memory. It is critical to know how estradiol mediates its effects, because this hormone has been shown to have protective effects against debilitating diseases such as Alzheimer's disease (Garcia-Segura et al., 2001). Thus, determining the mechanisms by which estradiol modulates hippocampal morphology and synaptic function could provide insight into potential therapeutic targets against memory deficits that occur during menopause and with disease.
Footnotes
This work was supported by an award from the Evelyn F. McKnight Foundation to L.L.M. and Ruth L. Kirschstein Predoctoral National Research Service Award MH71085 to C.C.S. We thank Drs. Lynn Dobrunz and Suzanne Bausch for helpful comments on this manuscript, Justin Daigre and John Sahawneh for technical assistance with ovariectomy surgeries, Jacqueline Falduto for helpful advice on Golgi impregnation, and Albert Tousson, Shawn Williams, and the University of Alabama at Birmingham High-Resolution Imaging Facility for their help with spine image acquisition and processing.
Correspondence should be addressed to Dr. Lori L. McMahon, University of Alabama at Birmingham, 1918 University Boulevard, McCallum 964, Birmingham, AL 35294-0005. E-mail: mcmahon{at}physiology.uab.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257780-12$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}