

Abstract
We examined the effect of glutamate transporter blockade at the calyx of Held synapse. In immature synapses [defined as postnatal day 8 (P8) to P10 rats], transporter blockade causes tonic activation of NMDA receptors and strong inhibition of the AMPA receptor-mediated EPSC amplitude. EPSC inhibition was blocked with a metabotropic glutamate receptor (mGluR) antagonist [1μm LY341495 (2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid)], suggesting that elevated resting glutamate concentration specifically activates group II and group III mGluRs. Using mGluR subtype-specific agonists and antagonists, we determined that increased glutamate activates presynaptic mGluR2/3 and mGluR8 receptors but not mGluR4, although this receptor is present. Surprisingly, in older animals (P16–P18), transporter blockade had no effect on EPSC amplitude because of a developmental downregulation of group II/III mGluR activation in rats and mice. In contrast to other CNS synapses, we observed no effect of transporter blockade on EPSC decay kinetics, although expression of glutamate transporters was strong in nearby glial processes at both P9 and P17. Finally, using a low-affinity AMPA receptor antagonist (γ-d-glutamylglycine), we show that desensitization occurs at P8–P10 but is absent at P16–P18, even during trains of high-frequency (100–300 Hz) stimulation. We suggest that diffusion and transporter activation are insufficient to clear synaptically released glutamate at immature calyces, resulting in significant desensitization. Thus, mGluRs may be expressed in the immature calyx to help limit glutamate release. In the more mature calyx, there is a far smaller diffusional barrier attributable to the highly fenestrated synaptic terminal morphology, so AMPA receptor desensitization is avoided and mGluR-mediated inhibition is not necessary.
- auditory brainstem
- MNTB
- development
- glutamate transporters
- desensitization
- AMPA and NMDA receptors
- γ-DGG
- diffusion modeling
- group II mGluR
- mGluR8
- TBOA
Introduction
Rapid clearance of glutamate from the synaptic cleft after exocytosis is a prerequisite for fine temporal and spatial discrimination of signaling among neighboring synapses during high-frequency firing. The kinetics of transmitter clearance are controlled by the interplay of passive diffusion, receptor affinity, and active translocation. Diffusion depends on the morphology of the synapse, which may change during development. Transmitter translocation depends on the subtypes and density of glutamate transporters (Diamond, 2005) and their proximity to release sites and, thus, also depends on glial and synaptic geometry.
The mechanisms and speed of glutamate clearance are synapse specific and may depend on stimulation frequency (Rusakov and Kullmann, 1998; Bergles et al., 1999). Bouton-like terminals, such as those found in the hippocampus, allow rapid diffusion of transmitter. Thus, the synaptic glutamate transient is terminated by escape of neurotransmitter out of the synaptic cleft (Hestrin et al., 1990; Isaacson and Nicoll, 1993; Sarantis et al., 1993). At the large climbing fiber–Purkinje cell synapse of the cerebellum, formed by multiple bouton-like terminals, postsynaptic and glial transporters are a major mechanism for glutamate clearance (Huang and Bergles, 2004), and, at parallel fiber synapses, glutamate transporters limit the activation of metabotropic glutamate receptors (mGluRs) (Brasnjo and Otis, 2001). In contrast, at the morphologically tortuous mossy fiber–unipolar brush cell synapse of the cerebellum, diffusional exits are limited, and extracellular glutamate clears very slowly. Here, EPSC termination is also mediated by AMPA receptor (AMPAR) desensitization (Rossi et al., 1995; Kinney et al., 1997), and there is an additional effect of transporters on rapid glutamate clearance (Overstreet et al., 1999). In some calyx-type synapses, in which there are both multivesicular release and limited diffusion out of the synaptic cleft, AMPA receptor desensitization is a major factor shaping fast synaptic transmission (Trussell et al., 1993; Isaacson and Walmsley, 1996; Otis et al., 1996b), with an additional role of transporters in terminating synaptic transmission (Otis et al., 1996a; Turecek and Trussell, 2000).
The calyx of Held is a large glutamatergic nerve terminal that can fire action potentials at high frequencies (up to 1 kHz), which result in precisely timed postsynaptic responses (Guinan and Li, 1990; Wu and Kelly, 1993). Presumably, mechanisms for terminating the glutamate transients must be exquisitely tuned and robust. In immature animals, the presynaptic terminal envelops 40–50% of the postsynaptic soma in a contiguous manner [postnatal day 9 (P9 rats)] (Satzler et al., 2002). However, by postnatal day 14, the terminal becomes highly fenestrated, with many diffusional exits (Kandler and Friauf, 1993; Taschenberger et al., 2002). What is the functional significance of these dramatic morphological changes? Does the rate of glutamate clearance change during development, and does this lead to differing degrees of AMPA receptor desensitization? What role do glutamate transporters play during high-frequency firing? Previous reports showed they are not present in either presynaptic or postsynaptic compartments (Palmer et al., 2003). We study here the mechanisms underlying termination of the synaptic glutamate transient in immature and morphologically mature calyces of Held and discern the subtypes of mGluRs activated by transporter blockade.
Materials and Methods
Slice preparation. Sprague Dawley rat pups (Charles River Laboratories, Wilmington, MA) aged P5–P18 or CD1/CD57 mice aged P6–P18 were used in this study. After rapid decapitation, the brainstem was quickly removed from the skull and immersed in ice-cold (for P5–P14) or warm (for P16–P18) saline containing the following (in mm): 125 NaCl, 2.5 KCl, 3 MgCl2, 0.2 CaCl2, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 0.4 ascorbic acid, 3 myo-inositol, and 2 Na-pyruvate, pH 7.3–7.5 when bubbled with carbogen (95% O2, 5% CO2; osmolality of 310–320 mOsm). Cyanoacrylic glue (World Precision Instruments, Sarasota, FL) was used to glue the brainstem to the stage of a vibratome slicer (VT1000; Leica, Bannockburn, IL), and 180- to 200-μm-thick transverse slices were made of the region containing the medial nucleus of the trapezoidal body (MNTB). Slices were then transferred to an incubation chamber containing normal saline (P5–P14) or a low-sodium, high-magnesium incubation saline (P16–P18) bubbled with carbogen (95% O2,5%CO2), maintained for 30–45 min at 35°C and thereafter at room temperature (RT) (22–25°C). Normal saline was the same as slicing saline but with 1 mm MgCl2 and 2mm CaCl2. Incubation saline for P16–P18 contained 85 mmNa, 7 mm MgCl2, and 0.5 mm CaCl2 to inhibit endogenous synaptic activity; sucrose (75 mm) was added to maintain osmolarity (310–320 mOsm) (Hefft et al., 2002).
Electrophysiology. Whole-cell patch-clamp recordings were performed in normal saline at room temperature (22–24°C) unless otherwise specified. Slices were perfused at 1–3 ml/min and visualized using infrared-differential interference contrast microscopy (Leica) and a 40× water-immersion objective. The pipette internal solution for postsynaptic recordings contained the following (in mm): 130 Cs-gluconate, 10 CsCl, 5 Na2-phosphocreatine, 10 HEPES, 5 EGTA, 10 tetraethylammonium (TEA)-Cl, 4 Mg-ATP, and 0.3 GTP, pH adjusted to 7.3 with CsOH. Presynaptic recordings of capacitance and Ca2+ currents used a pipette solution containing the following (in mm): 130 Cs-gluconate, 15 CsCl, 5 Na2-phosphocreatine, 10 HEPES, 0.2 EGTA, 20 TEA-Cl, 4 Mg-ATP, and 0.3 GTP, pH adjusted to 7.3 with CsOH. d-APV (50 μm) and MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] (5μm) were present in most experiments to isolate AMPA receptor currents, and strychnine (0.5 μm) was added to eliminate inhibitory transmission present especially in older MNTB (Awatramani et al., 2004), unless specified otherwise. For presynaptic recordings, TEA (5 mm) and tetrodotoxin (1 μm) were added to the bath to block voltage-activated potassium and sodium channel currents, respectively.
Pipettes were pulled from borosilicate glass (World Precision Instruments) with a Sutter P-97 electrode puller (Sutter Instruments, Novato, CA) to open tip resistances of 1.6–2.5 MΩ for postsynaptic recordings and 3.5–4.5 MΩ for presynaptic recordings. Access resistance (Rs) was ≤6 MΩ for postsynaptic recordings and ≤20 MΩ for presynaptic recordings. Rs was compensated >85% for postsynaptic recordings and ∼50% for presynaptic recordings. Principal cells were voltage clamped at a holding potential of –70 mV, and presynaptic terminals were held at –80 mV, unless otherwise noted. Occasionally, the EPSC amplitude in older (P16–P18) principal cells exceeded –20 nA, saturating the recording amplifier. In these instances, the holding potential was changed to –30 mV to reduce driving force, or the cells were rejected from analysis.
EPSCs were stimulated with a bipolar platinum/iridium electrode (Frederick Haer Company, Bowdoinham, ME) placed near the midline spanning the afferent fiber tract of the MNTB. An Iso-Flex stimulator driven by a Master 8 pulse generator (A.M.P.I., Jerusalem, Israel) delivered pulses <15 V (direct current at constant voltage) triggered by computer. Data was acquired at 10–25 μs sampling rate, using an EPC-9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany) controlled by Pulse 8.4 software (Instrutech, Port Washington, NY) and filtered on-line at 2.9 kHz. Software was controlled by a Power Macintosh G3 computer (Apple Computers, Cupertino, CA). Data were analyzed off-line and presented using Igor Pro (Wavemetrics, Lake Oswego, OR). All traces for kinetic analysis and display were corrected off-line for series resistance errors (Schneggenburger et al., 1999). Data presented were the average of several (usually three to five) sweeps under the conditions described.
Capacitance recordings were calculated from a 1 kHz, 20 mV amplitude sine wave on the holding potential of –80 mV using the software lock-in capability of the EPC-9 amplifier (sine + DC method, as per Gillis, 1995). The reversal potential was assumed to be 0 mV. Membrane capacitance (Cm) was not measured during step depolarizations (Taschenberger et al., 2002; Sun and Wu, 2001), and 30–60 s was allowed between depolarizations to allow complete recovery of exocytosis. Baseline was measured for 500–1000 ms before step depolarization and corrected for baseline drift in Cm. ΔCm was calculated 500–700 ms after depolarization to avoid depolarization-induced changes in membrane conductance (Yamashita et al., 2005). Presynaptic calcium currents were leak subtracted using the P/n method.
Immunohistochemistry. Slices of MNTB were made as for electrophysiology but fixed 30 min in 4% paraformaldehyde in 0.1 m PBS immediately after cutting, washed repeatedly, and incubated 20 min at RT in antibody blocking solution consisting of 3% goat serum, 0.025% sodium azide, and 0.5% Triton X-100 in 0.1 m PBS. Slices were incubated in blocking solution with primary antibodies raised against Rab3a (mouse anti-Rab3a, 1:1000 dilution; PharMingen-Transduction Labs, San Diego, CA) and mGluR8 (guinea pig anti-mGluR8, 1:1000 dilution; Chemicon, Temecula, CA) for 48 h at 4°C. Primary antibody was detected with appropriate fluorescent Alexa dye-labeled secondary antibodies (Molecular Probes, Eugene, OR), and slices were coverslipped on glass slides under Gel/Mount (Biomeda, Foster City, CA) for microscopy. Confocal fluorescent images were taken on Zeiss (Oberkochen, Germany) LSM510 microscope. Images were analyzed with Zeiss LSM Browser and prepared for presentation with Adobe Photoshop (Adobe Systems, San Jose, CA).
Immunoblotting, perfusion fixation of Wistar and Sprague Dawley rats, and preembedding light and electron microscopic immunocytochemistry for glutamate transporters [glutamate transporter 1 (GLT1) and glutamate–aspartate transporter (GLAST)] were performed as described previously (Lehre et al., 1995), with the following modifications. The GLAST labeling shown in Figure 11 was done with 1.5% normal goat serum in PBS. For immunoblots, 10–20% gradient gels and a chemiluminescent detection system were used (Supersignal West Pico; Pierce, Rockford, IL).
Drugs and reagents. All salts and serum were purchased from Sigma (St. Louis, MO). All pharmacological reagents were purchased from Tocris Cookson (Ellisville, MO). Tetrodotoxin was purchased from Alomone Labs (Jerusalem, Israel). A lexicon of the mGluRs examined in this study and specific pharmacological agents used to target them has been provided for clarity, as supplemental Table 1 (available at www.jneurosci.org as supplemental material).
G-protein-coupled receptor inhibition during postnatal development
Analysis. Curve fits of normalized EPSC decay kinetics were performed using a modified single- or double-exponential function in Igor (Wavemetrics). The single-exponential function was x(t) = y0 + A1exp(–x/τ1), and the double-exponential function was of the form x(t) = y0 + A1exp(–x/τ1) + A2exp(–x/τ2), where x was the time of the EPSC peak amplitude, τ1 was the fast rate of decay, and τ2 was the slow rate of decay. The fit was constrained such that A < 0. The fit was made from the EPSC peak to baseline when possible. All fits returned χ2 ≤ 0.10, and A1 + A2 ≤ 1.10. Weighted mean time constant (τm) is reported as A1τ1 + A2τ2.
Statistical tests and curve fits were calculated using Prism 4.0 software (GraphPad Software, San Diego, CA). Wilcoxon's nonparametric matched rank test was used for internally matched controls, and one-way ANOVA was used for multiple age comparisons, unless otherwise stated. Significance is indicated as *p < 0.05, **p < 0.01, and ***p < 0.001. Data are shown as mean ± SEM. Curve fit for dose–response curves used the following equation: Y = Ymin + (Ymax–Ymin)/(1 + 10^(log10(EC50)–X)), where X is log10[drug], and Y is percentage inhibition. Curves were constrained as defined in Results.
Modeling. Computer simulations of glutamate diffusion were performed by adjusting a three-dimensional compartmental model of the calyx (Rusakov, 2001) to the structural data obtained from EM reconstructions in 9-d-old rats (Satzler et al., 2002). General parameters of the model are as follows: calyx radius, 10 μm; cleft width, 30 nm; 106 glutamate molecules (∼200 release sites, 5000 molecules each, D = 0.3 μm2/ms) released synchronously over the ∼90% cleft area (Fig. 12, red shadow), with an a-function time course (σ = 39 ms–1). For additional details on modeling and transporter kinetics, see previous work by Rusakov (2001). In agreement with the present findings (see Figs. 10, 11), glutamate transporters were placed in the space compartments representing glial protrusions (an ∼200-nm-wide layer) that surround the calyx and principal cell, with an extracellular transporter concentration of 1mm. The latter corresponds to the total glutamate transporter level in glia of the adult hippocampus (Lehre and Danbolt, 1998).
Results
Transporter blockade inhibits transmission in the immature MNTB
To study the role of glutamate transporters on fast glutamatergic transmission at the calyx of Held synapse, we applied various concentrations of the high-affinity nontransportable glutamate transporter blocker dl-threo-h-benzyloxyaspartate (TBOA) to brainstem slices from P8–P10 or P16–P18 rats and recorded EPSCs in the principal cells of the MNTB. Concentrations of TBOA ≥100 μm inhibited AMPAR-mediated EPSCs significantly after drug application in immature MNTB (P8–P10) (Fig. 1). We also recorded NMDA receptor (NMDAR)-mediated EPSCs because NMDA receptors have a high affinity for glutamate and desensitize very slowly and are thus ideal for sensing glutamate concentration increases. Recording in 0 Mg2+ bath conditions, the EPSC was composed of both AMPAR and NMDAR components (Fig. 1A). When measured in the same cell, AMPAR and NMDAR EPSCs were inhibited by TBOA (300 μm) to a similar extent (75 ± 3% inhibition; n = 29), indicative of a presynaptic locus of inhibition (Fig. 1A,B). Application of TBOA likely increases basal glutamate concentration throughout the slice. If so, the increase in ambient glutamate might result in tonic NMDA receptor activation and could be observed as an increase in holding current (Ihold). When the principal cell was voltage clamped at Vhold of –70 mV, addition of 300 μm TBOA increased Ihold (54 ± 13 pA; n = 9 cells), and this increase could be completely reversed by addition of 50 μm APV (Fig. 1C). Thus, blockade of glutamate transporters by TBOA increases basal glutamate concentration in the MNTB and can be detected by NMDA receptor activation, as an increase in holding current.

Glutamate transporter blockade by TBOA elevates extracellular glutamate concentration, detectable by NMDA receptors in Mg2+-free saline. A, Sample traces showing inhibition of EPSC by application of TBOA in 0 Mg2+ saline. B, Expanded timescale of sweeps in A, illustrating similar level of inhibition of NMDA and AMPA receptor-mediated EPSCs by TBOA. Note that addition of 50 μm APV abolished NMDA receptor current but had no effect on peak AMPA current. C, Bath application of TBOA resulted in a reliable increase in Ihold at low-frequency stimulation (0.05 Hz) and could be reversed by coapplication of 50 μm APV.
In normal saline (1 mm Mg2+, 2 mm Ca2+), 200 μm TBOA strongly inhibited EPSCs in P8–P10 rats (49 ± 6% inhibition; n = 8; p < 0.0001) (Fig. 2A) but had little effect on transmission at P16–P18 (9 ± 7% inhibition; n = 5; p = 0.249) (Fig. 2B). At this concentration of TBOA, previous studies predict that 83–97% of all transporters should be blocked (Shimamoto et al., 1998). The inhibitory effect of TBOA in young animals could be reversed by washout (data not shown). Addition of 1 μm 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495), a selective antagonist for mGluR2/3 and mGluR8, also reversed the inhibitory effect of TBOA (Fig. 2A). This result indicates that inhibition was likely attributable to endogenous elevation of glutamate concentrations in the slice and activation of mGluR receptors at the calyx of Held. Application of LY341495 alone had a small but significant effect on EPSC amplitude in P8–P10 MNTB (107 ± 2%; n = 11; p = 0.011) and no effect by P16–P18 (104 ± 3%; n = 7; p = 0.303), indicating there may be a small endogenous activation of group II/III mGluRs in the young MNTB.
The inhibitory effect of TBOA could be attributable to direct activation of mGluRs because of the relatively high concentrations used. Previous studies showed that 100 μm TBOA did not directly activate mGluR1 or mGluR5 expressed in CHO cells (Shimamoto et al., 2004). Another study excluded 200 μm TBOA from direct mGluR activation in cerebellum Purkinje cells (Takayasu et al., 2004). However, there have been no studies directly assessing the possibility that TBOA can act as a ligand for group II/III mGluRs. We therefore used a structurally different glutamate transporter blocker to study mGluR activation at the calyx of Held synapse. Dihydrokainate (DHK) is a kainate derivative, as opposed to TBOA, which is an aspartate derivative. Application of 800 μm DHK significantly inhibited AMPAR EPSC amplitude in young (P8–P10) MNTB (27.1 ± 6.3% inhibition; n = 4; p = 0.0229) and was reversed by 1 μm LY341495 (10.7 ± 10.8% residual inhibition; n = 3; p = 0.3325). This result indicates that glial transporter blockade by TBOA or DHK likely acts to raise the extracellular glutamate concentration. A similar conclusion was also reached using l-trans-pyrrolidine-2,4-dicarboxylic acid (a transportable glutamate transporter blocker) on P8–P10 rats (Taschenberger and von Gersdorff, 2001). These results suggest that transporter block indirectly activates mGluRs in young (P8–P10) but not older (P16–P18) brainstem by increasing interstitial concentrations of glutamate throughout the slice.

TBOA inhibited EPSC amplitude in immature (P8–P10) but not older (P16–P18) calyx of Held synapses. Ai, TBOA inhibited EPSCs in young (P10) calyx of Held synapses. Cells were stimulated at 0.1 Hz, and 200 μm TBOA was added to the bath solution when indicated. This effect was completely reversible by 1 μm LY341495, an antagonist of group II (mGluR2/3) and mGluR8 receptors. Aii, Sample EPSCs (average of 5 sweeps taken at the points indicated in Ai) show robust inhibition of EPSC peak amplitude. Bi, Bii, An example of a similar experiment as in A, performed at P16, showed no response to TBOA.
Multiple trains of stimulation were delivered at 10 Hz with a 30 s interval between sweeps, to assess the effect of TBOA on repeated synaptic responses. EPSCs normally depress quickly during high-frequency stimulation in the P8–P10 MNTB (von Gersdorff et al., 1997). Stimulation at 10 Hz does not cause desensitization in control conditions of P8–P10 synapses in the MNTB (Wong et al., 2003) but does depress the EPSC by ∼70% before reaching a steady state of 31 ± 3% of the initial EPSC by 1 s (n = 30) (Fig. 3A,E). This depression recovers within 20 s and has a small contribution (∼10%) from presynaptic group III mGluRs (von Gersdorff et al., 1997). Application of 200 μm TBOA depressed the first EPSC and significantly reduced pairedpulse depression by 20% (EPSC2/EPSC1, 51 ± 6% in control; 79 ± 10% in TBOA; n = 8; p = 0.016) (Fig. 3E). This effect on paired-pulse depression is a common indicator of a presynaptic locus of inhibition (Zucker and Regehr, 2002). Furthermore, TBOA significantly relieved steady-state depression (44 ± 5% of initial EPSC amplitude; n = 8; p = 0.039, steady state vs control), as expected if enhanced presynaptic inhibitory mGluR activation in the presence of this drug reduced release probability (Fig. 3E). Presynaptic inhibition by 1 μm (S)-3,4-dicarboxyphenylglycine (DCPG), an agonist for mGluR8, also reduced the initial EPSC by ∼50% and also significantly relieved steady-state depression relative to the first stimulus (DCPG steady state, 50.91 ± 9% of initial EPSC amplitude; p = 0.0312 vs control; n = 6) (Fig. 3E). DCPG reduced the relative degree of synaptic depression from 70 to 50% probably by reducing synaptic vesicle pool depletion and ICa2+ inactivation (Xu and Wu, 2005). Additionally, these effects could be fully reversed to control values by 1 μm LY341495, supporting presynaptic activation of mGluRs by TBOA-induced glutamate accumulation in the brainstem slice. Note that glutamate increases within the synaptic cleft would presumably lead to AMPA receptor desensitization and would not be reversed by LY341495. LY341495 at 1–5 μm alone had no significant effect on the steady-state EPSC at 10 Hz (28 ± 7% of control; n = 5; p = 0.8125 vs control; data not shown). This result may indicate that mGluR4, but not mGluR2/3 and mGluR8, are activated by synaptically released glutamate (see below).

Blockade of glutamate uptake relieved depression in P8–P10 synapses. A, Application of TBOA considerably reduced the peak EPSC amplitude throughout a 10 Hz train in the MNTB of a P10 rat. Samples shown for each treatment are averages of five sweeps taken 30 s apart. Inset shows first and last EPSC. Calibration:2 nA, 1ms. Calibration is the same for all insets in A and B. LY, LY341495. B, At P16, there was no effect of TBOA on EPSC size or kinetics of transmission at 10Hz. The sample shown is an average of five sweeps for each treatment. Ci, The first EPSC in P8–P10 MNTB showed negligible slowing of EPSC decay in the presence of 200μmTBOA. Decay in young rats was well fit by a double exponential, corresponding to fast and slow components of decay. Addition of 200 μm TBOA did not affect kinetics of fast or slow phases of AMPAR decay versus control. Cii, The 20th pulse in a 10 Hz train was also unaffected by 200μmTBOA. Di, Dii, EPSC decay in older MNTB was well fit by a single exponential, comparable with τfast at P8–P10. Neither the first nor the 20th EPSC in P16–P18 (from B) showed any effect of TBOA on kinetics of fast transmission. Arrows point to noncalyceal inputs that were often present in older animals. E, Summary of cells at P8–P10, normalized to peak amplitude of the first EPSC in a train. Control synapses reached a steady state of ∼30% by 1 s (10 stimuli). Application of 200 μm TBOA significantly reduced depression of the second pulse and resulted in a striking relief from steady-state depression. DCPG, an mGluR8 agonist, similarly reduced depression and increased the steady state. The inhibitory effect of TBOA was fully reversible to control values by 1 μm LY341495, an antagonist for group II mGluRs and mGluR8. F, At P16–P18, there was no effect of TBOA or DCPG on synaptic depression, although the synapse showed less depression overall compared with younger synapses.
By P16, there is little or no effect of the transporter blocker (TBOA) on EPSCs during a 10 Hz train on either initial or steady-state amplitude (Fig. 3B,F). This lack of effect suggests that the increase in extracellular glutamate concentration does not activate additional AMPA receptors in the synaptic cleft and is also insufficient to desensitize AMPA receptors in the MNTB. Thus, transporters do not appear to play a prominent role in glutamate clearance during 10 Hz trains. Passive diffusion may therefore suffice to terminate the synaptic glutamatergic transient at the functionally mature calyx of Held synapse.
Dissecting the identity of mGluRs activated by extrasynaptic glutamate
All three classes of mGluRs are present at the MNTB, and all have been shown to inhibit EPSCs (Barnes-Davies and Forsythe, 1995; Takahashi et al., 1996; von Gersdorff et al., 1997; Elezgarai et al., 1999, 2001; Kushmerick et al., 2004). Which mGluR subtype is activated after transporter block by TBOA?
It is unlikely that group I mGluRs are involved in the observed TBOA effect, because 1 μm LY341495 should not significantly block their activation. In non-neuronal cells expressing mGluRs, LY341495 has been shown effective at blocking group I mGluR activation by quisqualate, with IC50 of 7–8 μm (Kingston et al., 1998). Other studies have used 20–100 μm LY341495 as a broad-spectrum mGluR antagonist (Watabe et al., 2002; Howson and Jane, 2003; Palhalmi et al., 2004). However, in the MNTB, only 200 μm LY341495 was capable of blocking activation by 100 μm DHPG, a group I mGluR agonist (Kushmerick et al., 2004).
Group III mGluRs (mGluR4, mGluR7, and mGluR8) are present presynaptically at the young calyx of Held (Barnes-Davies and Forsythe, 1995; Takahashi et al., 1996; Leão and von Gersdorff, 2002). mGluR4 and mGluR8 are activated by 50 μm l-2-amino-4-phosphonobutyrate (L-AP-4), resulting in inhibition of presynaptic Ca2+ current (Takahashi et al., 1996) and EPSC (Barnes-Davies and Forsythe, 1995). Antibody staining for mGluR4a revealed that it is developmentally regulated at the rat MNTB, with levels peaking at 9 d after birth and then falling off dramatically by P14 (Elezgarai et al., 1999). No mGluR7 protein was detected in the MNTB during this time (Elezgarai et al., 1999), and it is doubtful whether the protein is expressed in the adult MNTB (Kinoshita et al., 1998). Expression of mGluR8 mRNA or protein has not been reported in the MNTB, but this nucleus has not been looked at specifically in previous studies (Duvoisin et al., 1995; Kinoshita et al., 1996; Saugstad et al., 1997; Wada et al., 1998).
Until recently, there were no pharmacological tools capable of distinguishing between group III mGluR subtypes, specifically mGluR4 and mGluR8 (Schoepp et al., 1999). The development of DCPG, an agonist specific for mGluR8 over mGluR4 or mGluR7 (Thomas et al., 2001), allows a pharmacological discrimination of these receptors. In addition, the potent group II antagonist LY341495 has unique properties that result in selective potency for mGluR8 over mGluR4 or mGluR7 and thus can aid in further elucidating the physiological role of these group III mGluR receptors (Kingston et al., 1998). We applied these drugs to slices containing MNTB to dissect the presence and contribution of group III mGluR subtypes to EPSC inhibition (Fig. 4).
Application of DCPG resulted in up to 80% inhibition of AMPAR EPSCs in P8–P10 MNTB (Fig. 4A). We began to see significant inhibition at 100 nm DCPG (Fig. 4A–C), suggesting mGluR8 activation. The dose–response curve described by DCPG inhibition was fitted well by a sigmoid curve (R2 = 0.939) (Fig. 4C, black line); however, the fit returned an EC50 of 393 nm, a much higher value than that reported in transfected cell lines expressing only mGluR8 or from spinal cord (EC50 of 31 nm) (Thomas et al., 2001). This may be attributable to mGluR4 expression in MNTB at this age, supported by immunohistochemistry (Elezgarai et al., 1999), which would be activated by higher concentrations of DCPG (EC50 of 8.8μm for mGluR4) (Thomas et al., 2001). Thus, the affinity data from transfected cell culture experiments together with the data from the present work suggest independent contributions from both mGluR4 and mGluR8 at this synapse.
Using the maximal inhibition attributable to mGluR4 alone (60.6% in 50 μm l-AP-4 and 1 μm LY341495) (Fig. 5) and the EC50 of mGluR4 for DCPG reported by Thomas et al. (2001), we calculated a hypothetical dose–response curve to DCPG in the MNTB by mGluR4 alone (Fig. 4C, gray trace) (see Materials and Methods). This curve shows a negligible contribution to EPSC inhibition by mGluR4 at concentrations ≤1 μm. We then calculated a similar hypothetical curve for mGluR8, constraining the equation with the reported EC50 from transfected cells, and used the difference in DCPG inhibition at 1 μm and the hypothetical mGluR4 curve to determine maximum inhibition from mGluR8 alone (Fig. 4C, hatched trace). At high concentrations of DCPG, the effects of these two receptors do not linearly sum. This is likely attributable to saturation of the presynaptic inhibitory machinery (Leão and von Gersdorff, 2002). However, this result suggests that DCPG can effectively be used at 1 μm to discriminate between mGluR4 and mGluR8 at a mixed receptor synapse.
Similarly, we investigated the ability of low doses of LY341495 to inhibit mGluR8 activation by DCPG in the MNTB (Fig. 4D,E). Nanomolar concentrations of LY341495 strongly antagonized 1 μm DCPG-mediated inhibition, and LY341495 fully blocked inhibition at concentrations ≥1 μm (Fig. 4D). Figure 4E shows a dose–response curve for LY341495 antagonism of 1 μm DCPG. Curve fit returned an IC50 of 16.1 nm (R2 = 0.990; see Materials and Methods), consistent with the reported affinity of LY341495 for mGluR8 in spinal cord (14–57 nm) (Howson and Jane, 2003) and much less than that reported for mGluR4 (22 μm) (Kingston et al., 1998). Thus, low micromolar concentrations of LY341495 can be used to completely and specifically block mGluR8-mediated inhibition of transmission in the MNTB.
In addition, we used antibodies specific for mGluR8 to localize the receptor at the calyx of Held in P8–P10 rats (Fig. 4F). Signal for mGluR8 was restricted to the calyx terminal and was not present in the axon at this age. By comparison, the synaptic vesicle-associated protein Rab3a signal is strong in the terminal and is also present in the axon for some distance (∼20 μm). Minimal cross-reactivity or nonspecific staining of secondary antibodies was observed at the MNTB in control experiments (data not shown).
Group II mGluRs (mGluR2 and mGluR3) have also been localized in the MNTB via immunohistochemistry and are reported to be located in the presynaptic calyx membrane, apposed to glial cells and also occasionally facing the postsynaptic principal cell (Elezgarai et al., 2001). Previous reports on the physiology of these receptors have used 50 μm (1S,3S)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD) or 5 μm (2′S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV) as agonists for group II mGluRs at the MNTB and reported EPSC inhibition by 40–50% (von Gersdorff et al., 1997). However, neither of these agonists is clearly specific for group II mGluRs; ACPD at 50 μm can also activate group I and III mGluRs (Cartmell and Schoepp, 2000), and DCG-IV could also activate NMDA receptors (Breakwell et al., 1997). We used instead the agonist (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate (APDC), which shows specificity for group II mGluRs over all other mGluRs by almost two orders of magnitude (Cartmell and Schoepp, 2000). APDC (50 μm) reversibly inhibited the AMPAR EPSC peak amplitude by 55% (Figs. 5, 6, Table 1). The level of inhibition by group II mGluRs is similar to that seen by 200 μm TBOA. Activation of the high-affinity group II mGluRs may thus account for the majority of the effect on EPSCs of transporter blockade by TBOA.
The effect of group II activation by APDC (50 μm) did not occlude the effect of group III mGluRs, because DCPG (10 μm) further inhibited the EPSC amplitude (Fig. 5A). Likewise, DCPG (1 μm) did not occlude the effect of transporter block by TBOA (200 μm) (Fig. 5B). However, APDC (50 μm) did occlude a majority of the inhibitory effect of TBOA, indicating that transporter block may preferentially activate group II mGluRs (n = 4 cells) (Fig. 5C). There was some additional inhibition of the EPSC by TBOA, likely attributable to mGluR8 activation. In summary, these results suggest that increased glutamate concentrations attributable to transporter blockade predominately activate group II (subtype 2/3) mGluRs and also may activate mGluR8 at the calyx of Held but has no detectable effect on group I mGluR-mediated modulation.

DCPG-mediated inhibition is attributable to both mGluR8 and mGluR4 at the rat MNTB in vitro. Peak EPSC amplitude attributable to a nondepressing (0.1 Hz) stimulation protocol was challenged by various concentrations of DCPG. A, Example of data collected showing incremental and reversible inhibition of EPSC by DCPG. Drug was applied as indicated by bars, and EPSC amplitude was monitored at 0.1 Hz. Submicromolar concentrations of DCPG resulted in inhibition and was maximal by 1 μm. B, Sample EPSCs taken at the times (A–E) shown in A. Increasing DCPG concentration resulted in greater inhibition of EPSC amplitude, without affecting kinetics. C, The dose–response curve described by DCPG was well fit by a sigmoidal curve. n = 3–8 at each concentration. Using the EC50 values reported from expression systems for mGluR4 or mGluR8, we overlaid the predicted EPSC inhibition attributable to DCPG activation of mGluR4 (gray trace) and mGluR8 (hatched trace) (see Results). At concentrations <1 μm, there is contribution to DCPG-mediated inhibition exclusively by mGluR8. D, Example trace showing concentration dependence of LY341495 (LY) relief of DCPG inhibition. EPSCs were stimulated at 0.1 Hz, and peak amplitude was measured. Addition of 1 μm DCPG inhibited EPSCs by ∼50% in P9 rat MNTB slice. This effect was fully reversible by 100 nm or 1 μm LY341495. LY341495 blocked mGluR8 activation at low micromolar concentration. E, Summary data were well fit by a sigmoid curve. n = 3–4 at each concentration. The open symbol indicates inhibition by 1 μm DCPG alone. F, Immunoreactivity for mGluR8 was located presynaptically at the developing calyx of Held. The calyx from a P10 rat was labeled with antibodies against Rab3a (left panel, green), a synaptic vesicle associated protein. Arrows show calyceal structures in the MNTB. Note that Rab3a is also present in the axon. Immunoreactivity to mGluR8 is shown in the middle panel (red) and colocalized entirely with Rab3a immunoreactivity in the calyx (Merge, especially arrows).

Occlusion experiments between mGluRs and TBOA (glutamate transporter antagonist): inhibitory mechanisms in the MNTB of P8–P10 rats are not saturated by APDC (group II mGluR agonist) or DCPG (mGluR8 agonist). A, The group II mGluR agonist APDC (50μm) inhibits EPSC peak amplitude but does not occlude activation of mGluR8 by DCPG (10 μm) and is fully blocked by 200 nm LY341495 (LY). B, Application of 1 μm DCPG, an mGluR8 agonist, does not occlude inhibition attributable to transporter blockade by 200 μm TBOA. The effect of both DCPG and TBOA can be fully reversed by 1μm LY341495. C, Group II activation via 50 μm APDC occludes most of the inhibitory effect of 200 μm TBOA.
Developmental loss of mGluRs at the calyx of Held
We determined the presence of group II and group III mGluRs in the MNTB during postnatal maturation of the calyx of Held synapse between P5 and P18 to see whether the loss of inhibition attributable to transporter block by TBOA could be correlated with a loss of mGluR activity (Fig. 6). There are multiple inhibitory G-protein-coupled receptors (GPCRs) present presynaptically at the calyx of Held, linked to Gi/o activation (Barnes-Davies and Forsythe, 1995; Takahashi et al., 1996, 1998; Isaacson, 1998; Leão and von Gersdorff, 2002; Kimura et al., 2003). Previous studies have shown strong inhibition of the EPSC by group II and group III mGluRs at the MNTB, up to age P14 (Barnes-Davies and Forsythe, 1995; von Gersdorff et al., 1997; Leão and von Gersdorff, 2002). Activation of group II mGluRs with 50 μm APDC resulted in >50% inhibition but disappeared by P14 (Fig. 6, Table 1). Activation of either mGluR4 or mGluR8 (50 μml-AP-4 plus 1 μm LY341495, or 1 μm DCPG, respectively) also resulted in >50% inhibition in P5–P7 and P8–P10. Simultaneous activation of both receptors by 50 μm l-AP-4 inhibited transmission by >80%, less than that expected by additive inhibition. Because mGluR4 and mGluR8 likely share the same second-messenger pathway (for review, see Cartmell and Schoepp, 2000), it is possible that we may be saturating the presynaptic inhibitory mechanisms present at the calyx with 50 μml-AP-4. After the onset of hearing (P12–P14) (Blatchley et al., 1987), the effect of mGluR4 or mGluR8 activation was greatly reduced to ∼25% for both receptors (Fig. 6C). At this age, activation of both mGluRs (by 50 μm l-AP-4) results in ∼50% inhibition, equivalent to summation of an independent effect of the two receptors. Application of 1 mm l-AP-4, which activates mGluR7 in addition to mGluR4 and mGluR8, did not have any additional inhibitory effect on transmission, indicating that mGluR7 is unlikely to be expressed in the MNTB at this age. By P16–P18, only activation of mGluR4 and mGluR8 in combination (50 μm l-AP-4) resulted in significant inhibition (Fig. 6B), and this was considerably less than at earlier ages (Fig. 6C, Table 1). However, similar to the result observed in younger animals, mGluR7 did not contribute significantly to synaptic inhibition at P16–P18. The developmental loss of functional group III mGluRs was also seen in the mouse MNTB, with a slightly accelerated time course (supplemental data, available at www.jneurosci.org as supplemental material). Even a large dose, l-AP-4 (100 μm) had no inhibitory effect at a P18 mouse calyx of Held EPSC, suggesting a complete absence of group III mGluRs at this age.
GABAB receptors, also Go-coupled metabotropic receptors, are present in the mouse and rat MNTB throughout the first 2 weeks of development (Takahashi et al., 1998). In contrast to mGluRs, activation of these receptors with baclofen (20 μm) resulted in strong inhibition of excitatory transmission in rats and mice throughout the developmental time period studied here (Fig. 6, Table 1) (supplemental data, available at www.jneurosci.org as supplemental material). However, note that the effect of baclofen was reduced by one-half at P18 compared with P9–P10 mouse slices (supplemental data, available at www.jneurosci.org as supplemental material). This indicates that the transduction cascade, which is shared between these two classes of metabotropic receptors, has not been disrupted during the course of postnatal development.
mGluR8 directly inhibits Ca2+ influx and exocytosis
The occlusion experiments in Figure 5 indicated that mGluR8 was only mildly activated by TBOA. We used voltage-clamp recordings from the calyx of Held to determine the magnitude and precise mechanism of inhibition by mGluR8. Recordings were made in P8–P10 calyces, in which DCPG exerts a very strong effect, and presynaptic recordings are more feasible (Borst and Sakmann, 1996). To independently confirm that mGluR8 activation inhibits synaptic transmission, we recorded directly from the presynaptic terminal to examine the effect of 1 μm DCPG on synaptic vesicle exocytosis (Fig. 7A). Brief depolarizations (to 0 mV, 2 ms) resulted in fast, reliable increases (jumps) in Cm relative to baseline, representing the addition of synaptic vesicle membrane to the terminal via exocytosis (see Materials and Methods). Application of 1 μm DCPG caused a significant 38 ± 0.1% decrease in capacitance jump (control, 88 ± 16 fF; DCPG, 57 ± 16 fF; p = 0.0312 vs control; n = 6) and was reversed by 1μm LY341495 (DCPG plus LY341495, 126 ± 33 fF). Capacitance jumps were measured 500–700 ms after the end of the depolarization (Fig. 7A, gray bar) to avoid contamination by Ca2+-dependent conductances (Yamashita et al., 2005). Summarized results from serial application of DCPG and LY341495 are shown in Figure 7B and show that capacitance is significantly decreased by activation of mGluR8. The capacitance jump amplitude after a 2 ms depolarization to 0 mV is consistent with previous experiments (Sun and Wu, 2001; Taschenberger et al., 2002). Inhibition of capacitance by DCPG was similar at 2 or 5 ms depolarization to 0 mV (data not shown). This result shows that presynaptic activation of mGluR8 acts to decrease exocytosis and glutamate release and explains the effect of DCPG on EPSCs.

Group II and group III mGluR-mediated inhibition was strikingly reduced in morphologically mature MNTB synapses (P16–P18). Addition of group II agonists and combinations of group III mGluR agonists and subtype-specific antagonists showed robust and reversible inhibitory effects on peak EPSC amplitude in young (P8–P10) but not older (P16–P18) MNTB synapses. APDC, APDC at 50 μm, specific for group II mGluRs. l-AP-4,l-AP-4 at 50 μm, specific form GluR4 and mGluR8. LY, LY341495 at 5 μm, a group II- and mGluR8-specific antagonist at this concentration; thus, l-AP-4 plus LY341495 is specific for mGluR4 activation. DCPG, DCPG at 10μm, an mGluR8-specific agonist. Baclofen, Baclofen at 20μm, an agonist for GABAB, is shown here to compare differentially coupled Gi/o-dependent inhibitory mechanisms and illustrate that the GPCR mechanism has not rundown over the course of the recording. A, An example recording from the MNTB of a P7 rat. Many Gi/o-coupled GPCRs are present: group II, mGluR4, mGluR8, and GABAB all cause dramatic inhibition. This inhibition was, in all cases, reversible. There was no apparent endogenous activation of mGluR8 because 5μm LY341495 did not significantly increase the EPSC over that of control amplitude; however, some run-up of the size of the response was present on occasion. Timescale is shared between A and B. B, By P16, the MNTB was essentially resistant to group II and group III mGluR agonists. Inhibition attributable to GABAB activation was still very strong and reversible, indicating that the GPCRGi/opath way is still in tact at this age. C, Inhibition of synaptic transmission by group II and group III mGluRs is developmentally regulated. Combinations of specific agonists and antagonists were used to pharmacologically dissect the presence and contribution of group III mGluRs to synaptic inhibition at the rat MNTB throughout postnatal development (see also Table 1). Amplitudes of EPSCs were recorded at 0.1 Hz and are shown relative to control amplitude.
Brief depolarizations of the presynaptic terminal (2 or 5 ms from –80 to 0 mV, 30 s apart) result in influx of Ca2+; we used the integral of the current to measure charge transfer across the membrane (2 ms; control, 1.46 ± 0.11 pC; n = 8) (Fig. 7C,D). Ca2+ current responses were very stable between sweeps, indicating that the interstimulus interval was long enough to buffer intraterminal [Ca2+]i to baseline levels (Borst and Sakmann, 1998). Takahashi et al. (1996) showed that activation of group III mGluRs (by 50 μm l-AP-4) in the MNTB inhibits presynaptic Ca2+ currents. As shown in Figure 7, C and D, the Ca2+ current was also significantly inhibited by 1 μm DCPG (1.07 ± 0.08 pC; p < 0.01 vs control), indicating that mGluR8 is acting presynaptically to inhibit Ca2+ influx. Similar to capacitance results, this inhibition could be fully recovered by the addition of 1 μm LY341495 (1.34 ± 0.1 pC). The Ca2+ flux attributable to 2 ms depolarization was inhibited by 26 ± 5% (Fig. 7D), similar to that seen for application of 50 μm l-AP-4 (25 ± 4%) (Takahashi et al., 1996). Consistent with reports using l-AP-4, the inhibitory effect of DCPG was similar for 2 or 5 ms depolarization and did not alter the Ca2+ channel current–voltage relationship (data not shown).
A double-pulse depolarization protocol showed that the mechanism of inhibition by DCPG is attributable to direct interaction of G-protein βγ subunits with Ca2+ channels (Fig. 7C). Prepulse depolarization to +100 mV has been shown to alleviate inhibition of Ca2+ channels by βγ subunits (Bean, 1989; Kajikawa et al., 2001; Leão and von Gersdorff, 2002). A test depolarization (0 mV, 2 ms; first pulse) was followed 50 ms later by “prepulse” depolarization (+100 mV, 10 ms) and then a second test depolarization (0 mV, 2 ms; second pulse) 10 ms later. Consistent with a βγ-dependent mechanism of action, strong depolarization relieved the inhibition of ICa2+ by DCPG (Fig. 7C,D). This prepulse depolarization did not change the relative current in control cells or after addition of 1 μm LY341495 (p = 0.65 vs control first pulse), indicating that mGluR8 is likely not endogenously activated. These results suggest that mGluR8 is an inhibitory mGluR receptor present in the calyx of Held, and mGluR8 activation acts directly through βγ subunits to inhibit Ca2+ influx.
TBOA indirectly inhibits presynaptic Ca2+ current
We have shown that the transporter blocker TBOA (200 μm) inhibits excitatory transmission in the immature MNTB, which can be reversed by an mGluR antagonist (Figs. 1, 2, 3). The reversal by 1 μm LY341495 is indicative of presynaptic group II and mGluR8 receptors, which disappear with a similar time course as the loss of TBOA effect (Figs. 4, 5, 6), and mGluR activation can fully explain the inhibitory effect of TBOA on transmission. To test this hypothesis further, we recorded presynaptic Ca2+ currents directly from the presynaptic terminal in the presence of 200 μm TBOA (Fig. 7E). Blockade of transporters resulted in a small but significant decrease in Ca2+ current, inhibiting Ca2+ influx by 12% (control, 844 ± 55 fC; TBOA, 746 ± 60 fC; p = 0.039; n = 8) (Fig. 7F), which could then be reversed by 1 μm LY341495 to control amplitude (851 ± 13 fC). The effect of TBOA on Ca2+ current is relatively small. However, because of the power relationship of 3–4 on exocytosis, this inhibition is sufficient to explain EPSC inhibition (Schneggenburger et al., 1999). A 12% decrease in Ca2+ influx thus results in 32–40% inhibition of exocytosis, roughly similar to the ∼50% inhibition of EPSC by TBOA. Therefore, this result can account for the inhibitory effect of TBOA on EPSCs, via activation of presynaptic mGluRs.
TBOA does not affect the EPSC kinetics
The rate of glutamate clearance from CNS synapses is often a critical determinant of the EPSC. In addition, glutamate transporters help limit the amount of mGluR activation and provide a buffer between adjacent neurons to allow independent processing by nearby synapses (for review, see Bergles et al., 1999; Huang and Bergles, 2004). We thus wanted to know whether transporters aid in the termination of fast EPSCs at the calyx of Held, making it well suited for high-frequency transmission. We first examined the effect of transporter blockade on the decay kinetics of EPSCs when mGluRs were blocked by 1 μm LY341495 and also when AMPA receptor desensitization was additionally blocked by a low-affinity antagonist.
Previous reports showed that the decay kinetics of fast AMPAR EPSCs at RT in the MNTB are well fit by a double exponential in young animals [τfast ≤ 1 ms, τslow ≈ 2–4 ms (Barnes-Davies and Forsythe, 1995; Chuhma and Ohmori, 1998; Taschenberger and von Gersdorff, 2000)] but better fit by a single fast exponential in older animals (P12–P14) (Taschenberger and von Gersdorff, 2000; Iwasaki and Takahashi, 2001). In our hands, EPSCs from P8–P10 animals showed similar fast and slow components (τfast = 0.64 ± 0.09 ms, carried 77 ± 4%% of decay; τslow = 3.07 ± 0.46; n = 11). By P16–P18, >98% of a double-exponential fit in control was attributable to the fast decay constant, so only single-exponential values are reported (τ = 0.29 ± 0.02 ms; n = 12). There was no effect of 1 μm LY341495 on EPSC kinetics at either age (n = 3). Surprisingly, we saw no effect of nearly complete transporter blockade (200 μm TBOA) on the decay kinetics of fast glutamatergic EPSCs, in young or older synapses (Fig. 3C,D). Neither fast nor slow components of EPSC decay were affected (n = 10 cells at P8–P10; n = 5 at P16–P18).
At physiological temperature (35–36°C), we still did not see any effect of the transporter blocker (TBOA) on decay kinetics or EPSC amplitude in the presence of 1 μm LY341495 (data not shown; n = 3 at P8–P10; n = 4 at P16–P18). These results indicate that glutamate transporters have little or no role in shaping the synaptic waveform and that AMPA receptor kinetics plus diffusion and/or receptor desensitization likely sculpt the EPSC waveform, in both young and older calyceal synapses of the MNTB.
AMPA receptor desensitization in the MNTB
At some auditory synapses, postsynaptic receptor desensitization aids in determining the shape and amplitude of EPSCs (Trussell et al., 1993; Otis et al., 1996b). Several groups have used the low-affinity competitive AMPA antagonist γ-d-glutamylglycine (γ-DGG) to effectively block receptor desensitization and saturation (Wadiche and Jahr, 2001; Wong et al., 2003; Takayasu et al., 2004). In particular, modeling studies at the calyx of Held have confirmed that 4 mm γ-DGG is effective at blocking AMPA receptor desensitization (Wong et al., 2003). The EPSC rise time is slowed because of displacement of γ-DGG by synaptically released glutamate (Wadiche and Jahr, 2001), but it is unclear how the antagonist affects decay rates. We used 4 mm γ-DGG to block AMPA receptor desensitization and examined the effect of transporter blockade on the resulting EPSCs in young and older animals.
Addition of γ-DGG reversibly reduced the EPSC amplitude to ∼10% of control, in both young and old animals (P8–P10, 87 ± 3% inhibition, n = 11; P16–P18, 90 ± 2% inhibition, n = 9). Scaling the EPSCs to peak amplitude reveals a small but significant effect of γ-DGG on the fast component of EPSC decay kinetics relative to control in both immature (τfast = 0.96 ± 0.17 ms; n = 11; p = 0.0137) and older (τfast = 0.39 ± 0.04 ms; n = 10; p = 0.0391) synapses (Fig. 8Aiii,Biii). This effect could be attributable to endogenous desensitization of AMPA receptors, especially in younger animals (Joshi et al., 2004). More likely, however, the effect on decay kinetics could be an artifact of the drug used. Competitive binding of AMPA receptors by γ-DGG and glutamate results in delayed rise time (Wadiche and Jahr, 2001), which would then amplify any inherent asynchronicity of synaptic vesicle release in an EPSC, resulting in prolonged decay. Decreasing the EPSC amplitude with γ-DGG did not occlude the inhibitory effect of transporter block (by 200 μm TBOA) in P8–P10 animals (Fig. 8A). Addition of TBOA still significantly reduced the EPSC amplitude in P8–P10 (51 ± 6% relative to γ-DGG; n = 10; p < 0.0001). The inhibitory effect of TBOA could be reversed by blocking mGluR activation (1 μm LY341495; 106 ± 52% of γ-DGG peak amplitude; n = 9), reinforcing the hypothesis that activation of the mGluRs mGluR2/3/8 are attributable to tonic increases in glutamate concentration. In older animals, transporter block had no effect on EPSC peak amplitude (89 ± 9% relative to γ-DGG; n = 6; p = 0.2840) (Fig. 8Bi,Bii), consistent with a loss of mGluR expression.

Presynaptic recordings show that the inhibitory effect of DCPG and TBOA is attributable to reduced presynaptic Ca2+ currents.A,Activation of presynaptic mGluR8 inhibited exocytosis at the calyx of Held. Brief depolarization decreased synaptic exocytosis in the presence of DCPG. Capacitance of the calyceal terminal was monitored using the sine + DC method (see Materials and Methods), and the terminal was depolarized for 2 ms to 0 mV. The resulting capacitance change was measured 500–700 ms after depolarization (gray bar). The depolarizing pulse produced a capacitance jump (black circles). Application of 1 μm DCPG (gray circles) decreased the capacitance jump,and the control response could be fully recovered by the addition of 1 μmLY341495 (open circles). All traces were taken serially from the same cell over the course of 20 min. B, DCPG significantly decreased exocytosis at the calyx of Held. Pooled data from multiple experiments show a significant decrease in exocytosis by 1μm DCPG (*p<0.05 vs control). C, Ca2+ currents were inhibited by mGluR8 via βγ subunits. DCPG at 1 μm (gray trace) reduced Ca2+ influx in the calyx terminal by ∼25%. This effect was reversible by 1 μm LY341495. Inhibition of Ca2+ channels by mGluR8 was attributable to βγ subunit interaction with voltage-dependent Ca2+ channels,as revealed by double-pulse protocol. Double-pulse protocol and sample trace are shown from P9 rat and are averages of three to five sweeps 20–30 s apart for each treatment. Depolarization prepulse (+100 mV, 10 ms) removed βγ-mediated inhibition and reversed DCPG-induced inhibition completely. DCPG-induced inhibition was recovered to control values by adding 1μm LY341495. D, Summary of double-pulse protocol results in P8–P10MNTB. DCPG(1μm)very significantly inhibited Ca2+ charge transfer (current area during depolarization) during 2 ms depolarization (**p < 0.01). Adding 1μm LY341495 completely reversed inhibition (DCPG + LY). Second pulse resulted in Ca2+ charge transfer that was similar to control in all cases. Transporter blockade inhibited presynaptic Ca2+ current. Ca2+ currents attributable to 2 ms depolarization were recorded from the calyx terminal, as in A. Perfusion of 200 μm TBOA inhibited Ca2+ current and could be relieved by serial addition of 1 μm LY341495, a group II and mGluR8 antagonist. E, Example of experimental data showing small but reliable inhibition of Ca2+ current by TBOA (gray trace) attributable to 2 ms depolarization to 0 mV relative to control (black trace) and reversal to control amplitude by addition of 1 μm LY341495 (hatched trace). F, Summary data from eight cells show significant inhibition of Ca2+ current by 200μm TBOA (*p < 0.05 vs control).
When mGluR2/3/8 and AMPAR desensitization were blocked with LY341495 and γ-DGG, respectively, TBOA could be used to evaluate the role of transporters in glutamate clearance. In this environment, there was a surprising lack of effect of transporter blockade on the EPSC time course in young or older calyceal synapses (Fig. 8Aiii,Biii). These results indicate that, even in the absence of AMPA receptor desensitization, the EPSC generated by the calyx of Held is not significantly shaped by glutamate transporter activity during postnatal synaptic maturation, and that glutamate clearance at this synapse is probably dependent on passive diffusion from the synaptic cleft.
Previous reports have shown that there is significant desensitization in young MNTB, especially during high-frequency stimulation (Scheuss et al., 2002); however, it is controversial whether this desensitization is developmentally regulated or whether it persists in the functionally mature MNTB (Joshi and Wang, 2002; Taschenberger et al., 2002; Wong et al., 2003). We stimulated the MNTB of P8–P10 and P16–P18 rats at 100 and 300 Hz in the absence of desensitization (blocked by 4 mm γ-DGG) and mGluR activation (blocked by 1 μm LY341495) and examined the effect of transporter blockade on synaptic transmission and EPSC kinetics. At 100 Hz, significant desensitization occurred early in the train and reduced EPSC amplitudes in younger animals; however, there was apparently no desensitization in the MNTB of P16–P18 rats (Fig. 9). Blocking desensitization very significantly increased the pairedpulse ratio (PPR = EPSC2/EPSC1) at 100 Hz in P8–P10 MNTB (Figs. 8Aiii, 9A), consistent with previous findings (Wong et al., 2003). PPR was 0.37 ± 0.09 in control and 0.88 ± 0.18 in 4 mm γ-DGG (p = 0.001; n = 11). In fact, depression was significantly relieved by γ-DGG until the fifth pulse at 100 Hz, consistent with covariance analysis (Scheuss et al., 2002). However, at MNTB synapses of P16–P18 rats, there was far less depression, and we saw no effect of blocking AMPA receptor desensitization on EPSC amplitude (Figs. 8Biii, 9B).
PPR in older animals was 0.89 ± 0.08 in control (n = 11) and 0.86 ± 0.05 by 4 mm γ-DGG (n = 10). When transporters were also blocked (with 200 μm TBOA) in addition to blocking desensitization (by 4 mm γ-DGG) in P8–P10 MNTB, the initial EPSC is further inhibited and results in facilitation (PPR of 1.24 ± 0.22; p = 0.002 v control; n = 10) (Fig. 8Aiii). This effect was fully reversed by the mGluR antagonist LY341495 (1 μm), and the response was identical to controls in which γ-DGG blocked desensitization (PPR of 0.94 ± 0.20; n = 9). In the older MNTB, addition of 200 μm TBOA no longer inhibited the EPSC, nor did it affect PPR (PPR of 0.96 ± 0.11; n = 9). This result supports the idea that the major effect of transporter blockade in young MNTB is to activate presynaptic mGluRs. Furthermore, as shown in the absence of receptor desensitization, glutamate transporters do not seem to affect the rapid clearance of residual glutamate from the synaptic cleft, even during high-frequency activity.

Blocking desensitization supports absence of transporter role in synaptic inhibition. In the presence of 4 mm γ-DGG, used to block AMPA receptor desensitization, there was no additional effect of glutamate transporter blockade (200 μm TBOA) on AMPA receptormediated EPSCs in P16–P18 rat MNTB synapses. Ai, Example of EPSC results to drug application in immature (P8) MNTB. γ-DGG competitively inhibited AMPAR EPSCs but did not occlude the inhibitory effect of TBOA, which could be recovered fully by 1 μm LY341495 (LY). Shown are EPSC peak amplitudes at 0.1 Hz stimulation. Aii, Example traces of P10 MNTB response to stimulation at 100 Hz.γ-DGG (4 mm) inhibited the initial EPSC (green trace), and addition of 200μm TBOA (red trace) further suppressed EPSC amplitude. In control conditions, EPSCs dramatically and quickly depressed to < 10% of initial amplitude after five stimuliin all drug conditions.Aiii, AMPA receptors quickly desensitized during high-frequency transmission in young MNTB. The first three stimulations are shown for the sweeps in Aii and scaled to the peak amplitude of the first EPSC for control and inγ-DGG (desensitization blocked). There was a dramatic decrease in paired-pulse depression of the second EPSC when AMPAR desensitization was blocked. Application of 4 mm γ-DGG (green trace) did significantly slow EPSC decay, but addition of TBOA did not lengthen the decay phase of the EPSC any further when mGluR activation was blocked by LY341495 (blue trace), indicating that there is no apparent prolongation of glutamate in the synaptic cleft attributable to blockade of glutamate uptake transporters. However, TBOA further inhibited the first EPSC and eliminated paired-pulse depression. Bi, P18 MNTB was impervious to transporter blockade by TBOA. Addition of 4mm γ-DGG (green trace) inhibited the peak EPSC amplitude similar to the effect in P8–P10 animals (see A). In contrast, 200 μm TBOA (red trace) had no additional effect on EPSC peak amplitude. The effect of γ-DGG was completely reversible with washout of drug(black dotted trace). Stimulation protocol and calibration is the same as in A. Bii, By P18, the synapse is much more reliable at 100 Hz, but γ-DGG still inhibited EPSC amplitude. Biii, First three EPSCs in the train shown in Bii, scaled to peak of the first EPSC inγ-DGG. There was no apparent kinetic difference attributable toγ-DGG versus control conditions (green and black traces, respectively). Further addition of TBOA also had no effect on EPSC profile at this age.
Steady-state transmission at high frequencies
Prolonged stimulation depresses EPSCs in a train, and eventually they reach a plateau, or steady-state level (for review, see von Gersdorff and Borst, 2002). We investigated the potential role of transporters and desensitization on the steady state after 20–50 stimuli at high frequency in P8–P10 animals. At 100 Hz, the EPSC amplitude reaches a steady state of 4.8 ± 1.4% of the initial EPSC after 10 stimuli in young MNTB synapses (n = 11) (Fig. 9A). This steady-state level, at 20–50 stimuli, appears unaffected by blocking AMPA receptor desensitization (4 mm γ-DGG) or blocking glutamate transporters in the absence of mGluR activation (200 μm TBOA plus 1 μm LY341495). However, see the analysis below (for review, see Schneggenburger et al., 2002).
In P16–P18 synapses, prolonged stimulation at 100 Hz also resulted in depression but to a proportionally smaller degree than in young animals; steady state was 25 ± 3% of the initial EPSC amplitude (n = 8) (Fig. 9B). Additionally, there was no effect of either γ-DGG or TBOA on the degree of synaptic depression. At an even higher frequency (300 Hz), there was no effect of desensitization (revealed by γ-DGG) or glutamate transporters (blocked by TBOA) on the synaptic response; however, at this frequency, there was facilitation of the second stimulus in all conditions, likely attributable to AMPAR summation (Fig. 9C). Note that, in older animals, depression did not reach a steady state until after 30 stimuli. This result shows that transmission can be sustained in older calyces for relatively long periods even at high-frequency stimulation. A similar lack of effect was seen in experiments in which TBOA was applied to P16–P18 MNTB in the absence of γ-DGG (data not shown; n = 3). These results support a developmental shift away from AMPA receptor desensitization, as reported previously (Brenowitz and Trussell, 2001; Joshi and Wang, 2002; Taschenberger et al., 2002). Furthermore, these results show there is no apparent role of glutamate transporters in clearance of synaptically released glutamate, even under conditions that maximally drive synaptic vesicle exocytosis. After normalizing the EPSCs in γ-DGG to the control response for each stimulus in the train (i.e., EPSCDGG/EPSCCO-ntrol) (Fig. 9D), it is clear that there is significant desensitization after the first stimulus in P8–P10 at 100 Hz but not at 10 Hz. No evidence for desensitization was found at 10, 100, or 300 Hz in P16–P18 rats (Fig. 9D). Averaging the contribution of desensitization during the train for each cell results in very significant desensitization in P8–P10 at 100 Hz versus all other stimulation conditions and ages shown (p < 0.01 vs P8–P10 at 10 Hz, P16–P18 at 10 Hz, and P16–P18 at 100 Hz; p < 0.05 vs P16–P18 at 300 Hz). This result unambiguously shows that desensitization is present in the MNTB of immature but not morphologically mature synapses.

Transporter blockade only had an effect on high-frequency transmission in immature synapses. A, Blocking transporters in P8–P10 MNTB changed depression at 100 Hz to facilitation in the absence of desensitization. Control sweeps depressed to a steady state of ∼5% initial amplitude by the 10th stimulus. γ-DGG blocked desensitization and significantly relieved depression for the first two stimuli (#p < 0.05 and ##p < 0.01,γ-DGG vs control). Addition of TBOA resulted in significantly more relief from depression in the third and fourth stimuli and completely eliminated depression until the fifth stimulus (*p < 0.05 vs γ-DGG). TBOA-induced relief from depression was completely reversed by 1μm LY341495 (LY). n = 5–8 for each condition. Late in the train, the depressed steady state was unaffected by any treatment relative to control. B, By P16, synaptic depression attributable to 100 Hz stimulation was reduced in both size and onset. Control depressed to a steady state only after 100 ms at 100 Hz (25% of initial amplitude). Furthermore, 4 mm γ-DGG had no effect on depression, indicating that there is no AMPA receptor desensitization in P16–P18 at 100 Hz, and depression is wholly attributable to vesicle depletion. n = 5–6 for each condition. C, At 300 Hz, depression in P16–P18 was biphasic and did not reach a steady state until after 25 stimuli. However, fast synaptic transmission was still independent of receptor desensitization, because γ-DGG had no effect. Furthermore, depression at this frequency was unaffected by transporter blockade, indicating that diffusion is the major mechanism for synaptic glutamate clearance at this age. n = 4–6 for each condition. D, Desensitization of AMPA receptors was only present in young MNTB at high frequency. EPSCs from various frequency trains were divided by control responses to illustrate the frequency dependence of desensitization in young versus older MNTB, in the absence of presynaptic vesicle depletion. Net desensitization (EPSCγ-DGG/EPSCcontrol) was calculated for each EPSC during the train and plotted versus stimulus number. There is apparently no desensitization in P8–P10 at 10 Hz or P16–P18 at 10, 100, or 300 Hz. At 100 Hz in P8–P10, there is a very significant contribution of receptor desensitization throughout the stimulus train.
Glutamate transporter expression is limited to glial cells in the MNTB
We have shown previously immunoblots from adult rat brainstem (Lehre et al., 1995) and developing rat forebrain (Ullensvang et al., 1997) with our GLAST and GLT glutamate transporter antibodies. However, to determine which glutamate transporter subtype is the prevalent in the developing MNTB and to verify antibody specificity, we microdissected part of the trapezoidal body containing the MNTB from P9 and P17 rats. Immunoblots were labeled with antibodies against the four glutamate transporter subtypes: GLAST, GLT (Lehre et al., 1995), excitatory amino acid carrier (EAAC) (Haugeto et al., 1996), and excitatory amino acid transporter 4 (EAAT4) (Dehnes et al., 1998). Comparison of labeling intensities for MNTB with that for samples from hippocampus and cerebellum, in which we have previously quantified the amounts of GLAST and GLT (Lehre and Danbolt, 1998), EAAC (S. Holmseth, Y. Dehnes, N. C. Danbolt, and K. P. Lehre, unpublished observations), and EAAT4 (Dehnes et al., 1998), indicated that GLAST is the dominating transporter subtype in the MNTB at both 9 and 17 d, with GLAST concentration on the same order of magnitude as that in the adult hippocampus. We also detected GLT and EAAC, but at lower concentrations. In agreement with immunoblots, staining of tissue sections with GLAST antibodies produced strong labeling adjacent to the calyx of Held and principal cell but was not present in neurons of the MNTB at P9 and P17 (Fig. 10). At higher magnification, electron microscopy showed that GLAST was localized in glial cell processes ensheathing the calyceal terminal (Fig. 11). Thus, GLAST is present in glial processes next to the region of the principal cell not contacted by the calyx and in glial processes next to the calyx. This localization of transporters in the MNTB is consistent with previous physiology in the MNTB (Palmer et al., 2003) and similar to other regions of the developing rat CNS (Ullensvang et al., 1997; Kugler and Schleyer, 2004).
Modeling of the glutamate transient
Computational models and experimental data support the escape of significant concentrations of glutamate out of the synaptic cleft, in which it can activate extrasynaptic receptors (Bergles et al., 1999; Scanziani, 2002; Huang and Bergles, 2004) or adjacent synapses (Otis et al., 1996a; Silver et al., 1996; DiGregorio et al., 2002). Extrasynaptic activation of mGluRs can thus act to further modulate synaptic transmission (Rusakov and Lehre, 2002). Using a geometric model of the immature calyx of Held synapse, we used a three-dimensional compartmental model to depict glutamate diffusion in the presence and absence of transporter activity (Fig. 12). Transporter location and concentrations were determined from ultrastructural localization shown in Figure 11 and previous work (see Materials and Methods). Glutamate concentrations were monitored at multiple locations within the synaptic cleft and in the adjacent extrasynaptic space. Consistent with our experimental results, simulations predicted no effect of transporters on the time course of glutamate within or adjacent to the synaptic cleft. However, transporter activity strikingly reduced the concentration of extrasynaptic glutamate fourfold within 20 ms. Thus, glial glutamate transporters suppress basal glutamate levels surrounding the calyx of Held but are likely to have no effect on the clearance of glutamate from the synaptic cleft. In addition, if mGluRs are located outside the synaptic cleft, transporter block may facilitate their activation. These results are wholly consistent with the findings in Figure 5, in which transporter block predominantly activates group II mGluRs. From the model, when transporters are blocked, the peak glutamate concentration at the edge of the calyx reaches ∼6 μm (Fig. 12 B). This concentration would be sufficient to activate most group II mGluRs, which have a high affinity for glutamate (EC50 of 0.04–20 μm) (Cartmell and Schoepp, 2000). This concentration of glutamate would also partially activate mGluR8 receptors, which have an EC50 for glutamate in this range (3–11 μm) (Cartmell and Schoepp, 2000).

Immunocytochemical localization shows GLAST in glia of the MNTB at 17 d. The images show four unlabeled principal cells surrounded by labeled glial processes. Note that the labeling follows the outside of the 1- to 2-μm-thick structures (i.e., large nerve terminals; arrowheads) on the principal cell surface. Also, note the glial-like profiles within nerve fiber bundles (arrows). Scale bar, 15 μm.

Electron micrographs show localization of GLAST in glia of the MNTB in 9-d-old rats. A, The staining forms a broken dark circular profile (arrowheads) surrounding the large principal cell. The inset at higher magnification, taken from the boxed area in the main image, shows that the labeling is present in thin glial processes next to the principal cell (PC). B, C, Two different calyces (Cx) contacting principal cell bodies. Note that the cell membrane of calyces appears unlabeled (arrows), whereas glial processes next to the calyces are heavily stained. Scale bars: A, 500 nm; B, C, 100 nm.
Discussion
The synapse formed by the calyx of Held terminal is a specialization within a sound localization circuit (Oertel, 1999) that can retain precise timing information (<225 μs differences in latency at 10 Hz stimulation in P16–P18 rats) (Leão et al., 2005). We investigated here the contribution of glutamate transporters and AMPA receptor desensitization to evoked synaptic currents. Our results suggest that passive diffusion is the major mechanism used for rapid glutamate extrusion from the cleft in both P8–P10 and P16–P18 calyxes. The EPSC waveform was unaffected by blockers of glutamate transporters in either age group. However, blocking transporters in the immature synapse reduced EPSC amplitude via presynaptic mGluR activation. We suggest that, in young calyces, the glutamate transient in the synaptic cleft is substantially prolonged, because we observed a significant reduction in the peak EPSC attributable to AMPA receptor desensitization. In contrast, diffusion of synaptically released glutamate is likely very rapid in P16–1-P8 calyxes because, even during stimulation at 300 Hz, AMPA receptor desensitization was not observed in the EPSC steady-state amplitude. We thus suggest that transporters play a role in glutamate clearance only after glutamate has left the synaptic cleft. The striking developmental changes in the shape of the calyx terminal during the first 3 postnatal weeks provide a structural underpinning that explains these results: the calyx changes from a contiguous sheath that hinders glutamate diffusion at P8–P10 to a highly fenestrated structure with numerous diffusional exits at P16–P18 (Kandler and Friauf, 1993; Taschenberger et al., 2002).
Glutamate transporters in the MNTB: housekeeping and neuroprotective roles
We detected strong expression of glutamate transporters in glial cell processes contacting the calyx and principal cell throughout the period of structural and functional remodeling of the calyx of Held studied here (Figs. 11, 12). This finding is consistent with a previous report that found an absence of electrogenic transporters at the presynaptic or postsynaptic compartments of this synapse (Palmer et al., 2003). Importantly, glial transporters are excluded from the synaptic cleft and may thus have a primary role in limiting glutamate diffusion between adjacent terminals. Modeling of the effect of transporters on the glutamate transient shows that they only suppress glutamate that has already “spilled out” of the terminal. At high concentrations of TBOA (200 μm), we saw robust inhibition (50%) of the EPSC amplitude in young animals, and the degree of short-term depression at 10 Hz was also significantly attenuated, presumably because global increases in extracellular glutamate activated presynaptic mGluRs. We further show that this inhibition is specific for mGluR2/3 and mGluR8 activation but likely not mGluR4, because it was fully antagonized by 1 μm LY341495. In more mature animals, TBOA had no effect on EPSC kinetics or amplitude, even during high-frequency stimulation. Thus, under certain pathological conditions when extracellular glutamate concentrations rise, synaptic transmission in the mature MNTB may remain essentially unperturbed in vivo. In contrast, excitotoxicity may be attenuated in the immature calyx by inhibition of synaptic vesicle fusion via mGluR activation. Immature brainstem synapses may thus be less prone to ischemic damage than mature synapses.
Autoregulation of glutamate exocytosis: task-specific mGluRs
Activation of specific compartmentalized mGluRs, particularly within groups II and III, is supported by location and affinity data. mGluR2/3 have a high affinity for glutamate and produce similar inhibition as TBOA (50%). mGluR2/3 has been localized to the presynaptic terminal but was not specifically detected at the active zone, in contrast to the synaptic localization of mGluR4 at the MNTB (Elezgarai et al., 1999, 2001). Ultrastructural immunoreactivity at the parallel fiber to Purkinje cell synapses of the cerebellum also supports the presence of presynaptic mGluR4 in the synaptic cleft, in close proximity to docked synaptic vesicles (Mateos et al., 1999). The exact subcellular location of mGluR8 is currently unknown; one report showed reactive product for mGluR8 at the ultrastructural level near presynaptic specializations in piriform cortex (Kinoshita et al., 1996). Our immunohistochemistry results suggest that it is present throughout the calyx terminal.

A, Diagram (not to scale) showing the two-dimensional central section of a three-dimensional compartmental model of the calyx in 9-d-old rats. For parameter details, see Materials and Methods. Arrows depict sampling positions for concentration measurements: 1 and 2, 3 and 6 μm from the central axis; 3, 100 nm inside the cleft; 4, 200 nm outside the cleft (region of glia); 5, 1μm outside the cleft (beyond the glial sheath). B, Simulated glutamate concentration time course at different sites inside (red lines) and outside (blue lines) the synaptic cleft, without glutamate transporters. C, The same as B, but with transporters included. Curve indices 1–5 correspond to monitor positions indicated in B. Glutamate concentrations represent average values over the 100 nm space compartments. Although glutamate concentration could briefly (for <1 ms) peak at 2–5 mm immediately under release sites (data not shown), this would still have a volume-average effect on the extrasynaptic transporters occurring >300 nm away from the nearest release site.
Previous reports from different CNS synapses have shown that synaptic activity elevates glutamate in the synaptic cleft, leading to presynaptic inhibition attributable to group II and III mGluR activation (Scanziani et al., 1997; von Gersdorff et al., 1997; Oliet et al., 2001; Chen et al., 2002; Selkirk et al., 2003). We did not see a significant effect of LY341495 alone on synaptic depression at the calyx of Held, in contrast to the effect reported in a previous study (von Gersdorff et al., 1997) that reported a 6% relief from depression at 10 Hz with (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG), an antagonist for all group II and group III mGluRs at 300 μm. Additionally, recent work also using CPPG showed that group III mGluRs are responsible for speeding recovery from high-frequency activity (Billups et al., 2005). We used LY341495 at a concentration that only antagonizes group II mGluRs, and mGluR8, but not mGluR4 (Losonczy et al., 2003). Our results are consistent with mGluR2/3 and mGluR8 localization on the extrasynaptic face of the calyx and mGluR4 in the synaptic lumen. Thus, mGluR4 may be the first mGluR to sense synaptically released glutamate and may be responsible for the effects seen by von Gersdorff et al. (1997) and Iwasaki and Takahashi (2001), whereas extrasynaptic rises in glutamate may be detected first by group II mGluRs (mGluR2/3). This hypothesis is supported by the independent modeling of glutamate at the edge of the calyx, which reaches peak concentrations sufficient to fully activate group II mGluRs and partially activate mGluR8 (Fig. 12).
We saw no evidence for activation of group I mGluRs when ambient glutamate was increased by transporter blockade. A recent report showed that this receptor is present presynaptically and postsynaptically at this synapse and is located in the synaptic lumen, albeit relatively distant from docked vesicles (Kushmerick et al., 2004). However, these receptors have a much lower affinity for glutamate (∼200 μm) (Cartmell and Schoepp, 2000) and thus may not be activated except under extreme conditions.
Developmental pruning of mGluRs
The developmental downregulation we report for group II and III mGluRs is similar to that reported for other Gi/o-coupled modulators in the MNTB, such as adenosine and norepinephrine (Leão and von Gersdorff, 2002; Kimura et al., 2003). It is less clear whether group I mGluRs are also developmentally restricted (Kushmerick et al., 2004). In contrast, GABAB receptors are present and effective late in postnatal development (Takahashi et al., 1998), although we have observed a significant decline in P18 mice (supplemental data, available at www.jneurosci.org as supplemental material). The downregulation of inhibitory mGluRs in older animals may be an adaptation to further sustain high temporal fidelity during high-frequency activity. Moreover, because the calyx in older animals is highly fenestrated, any putative extrasynaptic receptors might sample higher effective glutamate concentrations and act to attenuate transmission, thus disrupting the efficacy of the synapse.
AMPA EPSCs: termination via diffusion and desensitization
In other calyx-type synapses, receptor desensitization is a major contributor to EPSC termination (Trussell et al., 1993; Isaacson and Walmsley, 1996; Otis et al., 1996b). Kinetic analysis of excised patches from the mouse MNTB principal cell showed that AMPA receptors are normally partially desensitized in immature synapses (Joshi et al., 2004). In the calyx of Held, a typical synaptic event is composed of glutamate liberated from several hundred individual active zones, which may result in transmitter pooling. Additionally, many sites undergo multivesicular release at immature ages (Taschenberger et al., 2002). Older animals have reduced multivesicular release, perhaps because presynaptic action potential waveforms are briefer and active zones have fewer docked vesicles. Here we observed significant postsynaptic AMPA receptor desensitization at P8–P10, consistent with previous reports (Joshi and Wang, 2002; Scheuss et al., 2002). However, in contrast to the report by Wong et al. (2003), which used rats up to P13, we saw very little evidence for desensitization in older animals (P16–P18). Using γ-DGG, an AMPA receptor antagonist, we have shown that no desensitization occurs during 100 Hz stimulus trains at P16–P18 calyx of Held synapses. Even at 300 Hz stimulation frequencies, we saw no evidence for significant desensitization during steady-state EPSC trains. Reduced desensitization in older animals was not attributable to changes in AMPA receptor properties, because AMPA receptors retain their ability to quickly desensitize in older animals (Koike-Tani et al., 2005). More likely this is attributable to less residual glutamate in the synaptic cleft after release at morphologically mature synapses. We also note that hearing onset occurs at approximately P12 in rats, but auditory nerve responses to high-frequency tones (e.g., 40 kHz) are observed consistently only at P16 and become adult-like by P20–P36 (Blatchley et al., 1987). It is therefore important to conduct developmental studies to at least P16–P18 in rodents.
In conclusion, at the immature MNTB, diffusion and AMPA receptor desensitization end synaptic events, and transporter block inhibits transmission via mGluR activation. In the older MNTB, the presence of multiple diffusional exits is sufficient to terminate individual excitatory events, whereas mGluR functional activation is greatly reduced, and this may allow for prolonged high-frequency firing.
Footnotes
The Zeiss LSM510 confocal microscope was partially supported by National Institutes of Health Grant RR16858. R.R. was supported by National Institute on Deafness and Other Communication Disorders Postdoctoral Fellowship DCD-06768. N.P. was supported by a European Union Marie Curie Fellowship (Contract HPMT-CT-2001-00406) and Ministerio de Ciencia y Tecnología Grant BFI2002-01474. D.A.R. was supported by the Wellcome Trust. R.D. was supported by National Eye Institute Grant EY09534. L.-Y.W. was supported by the Burroughs Wellcome Fund, EJLB Foundation, an Individual Operating grant, and “The Synapse” Group grant from the Canadian Institutes of Health Research. K.P.L. was supported by The Research Council of Norway and European Union BIOMED Contract QLG3-CT-2001-02004. H.v.G. was supported by Human Frontier Science Program and National Institute on Deafness and Other Communication Disorders grants. We thank Catherine Morgans (Neurological Sciences Institute/Oregon Health and Science University) for the generous gift of mGluR8 antibody and Craig Jahr, Laurence Trussell, Christopher Kushmerick, and Ko Matsui for discussions.
Correspondence should be addressed to Dr. Henrique von Gersdorff, The Vollum Institute, L-474, Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: vongersd{at}ohsu.edu.
DOI:10.1523/JNEUROSCI.1848-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258482-16$15.00/0





{kind=link}
{kind=link}
{kind=link}
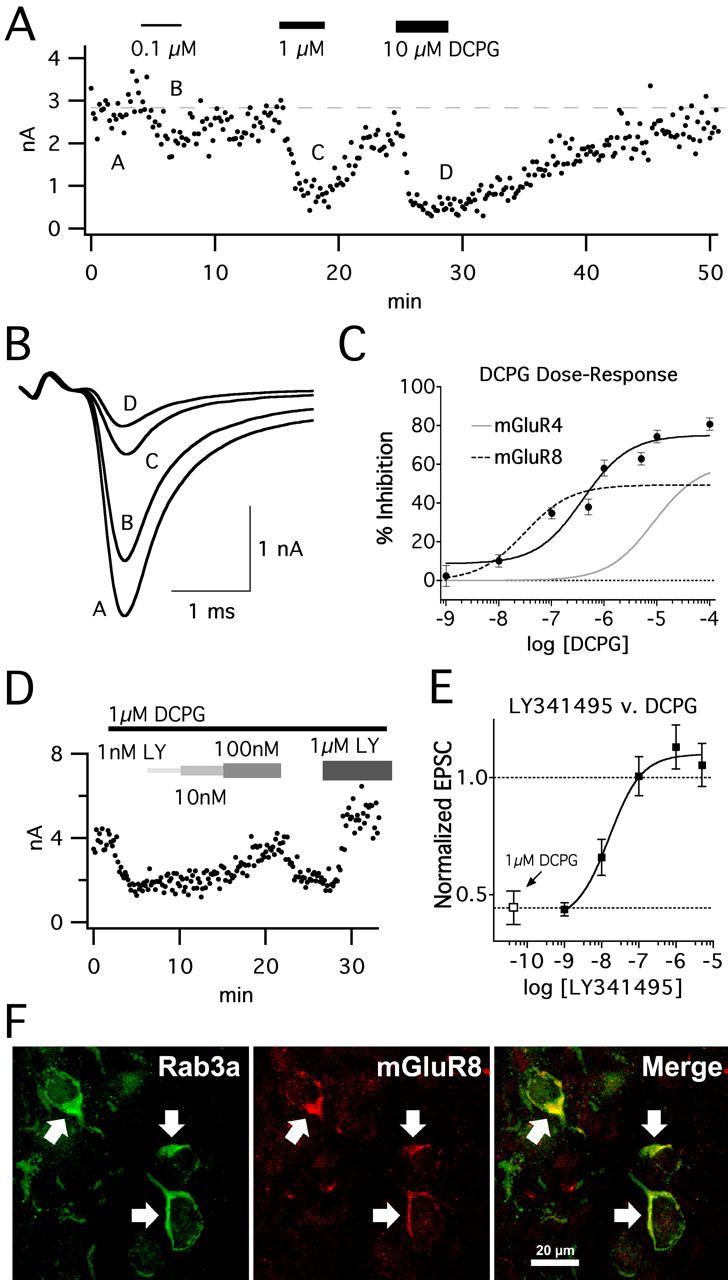{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
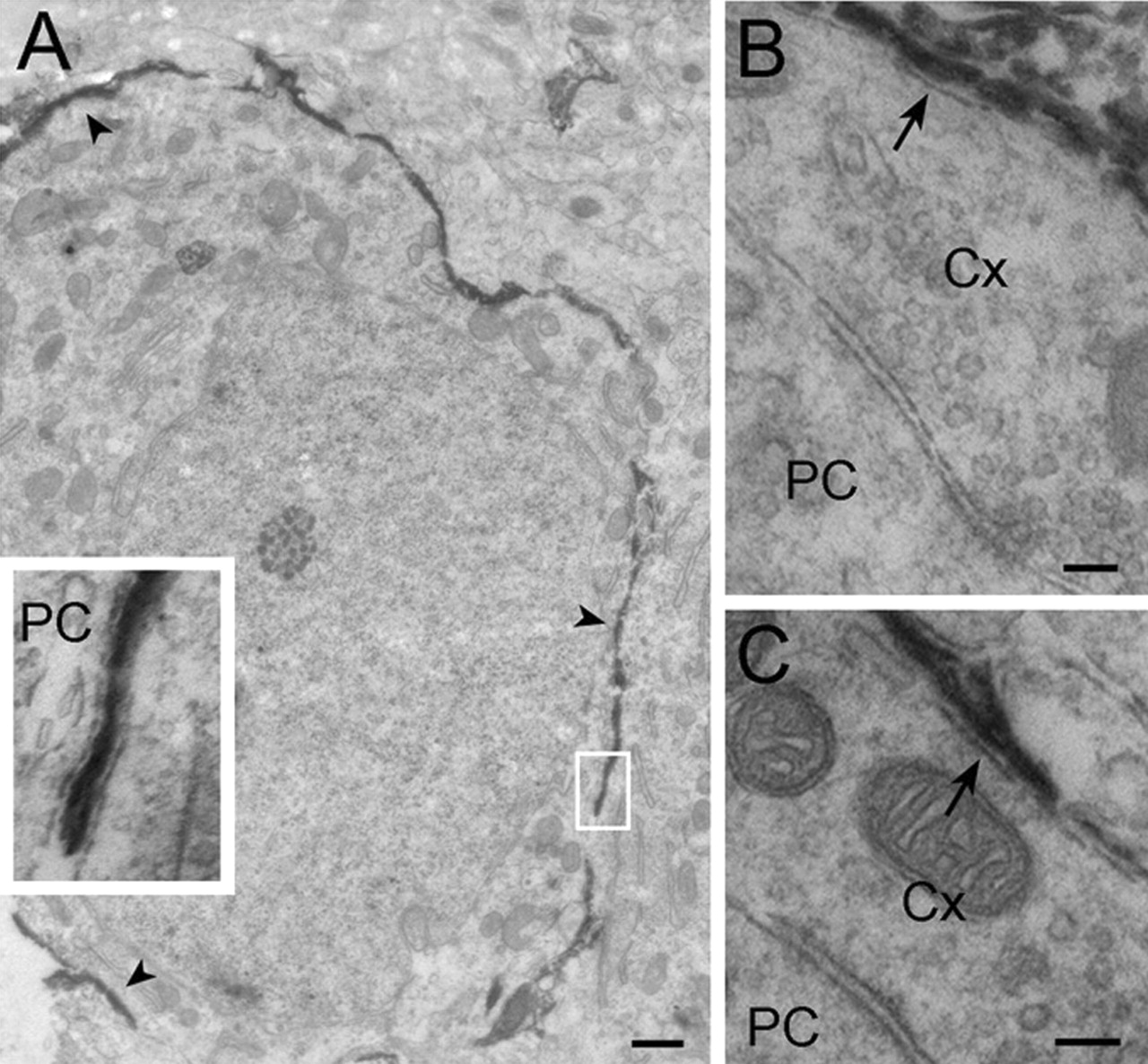{kind=link}
{kind=link}