

Abstract
Overproduction of the amyloid β (Aβ) peptide is a key factor in the pathogenesis of Alzheimer's disease (AD), but the mechanisms of its pathogenic effects have not been defined. Patients with AD have cerebrovascular alterations attributable to the deleterious effects of Aβ on cerebral blood vessels. We report here that NADPH oxidase, the major source of free radicals in blood vessels, is responsible for the cerebrovascular dysregulation induced by Aβ. Thus, the free-radical production and the associated alterations in vasoregulation induced by Aβ are abrogated by the NADPH oxidase peptide inhibitor gp91ds-tat and are not observed in mice lacking the catalytic subunit of NADPH oxidase (gp91phox). Furthermore, oxidative stress and cerebrovascular dysfunction do not occur in transgenic mice overexpressing the amyloid precursor protein but lacking gp91phox. The mechanisms by which NADPH oxidase-derived radicals mediate the cerebrovascular dysfunction involve reduced bioavailability of nitric oxide. Thus, a gp91phox-containing NADPH oxidase is the critical link between Aβ and cerebrovascular dysfunction, which may underlie the alteration in cerebral blood flow regulation observed in AD patients.
Introduction
There is increasing evidence that Alzheimer's disease (AD) is mediated by overproduction of the amyloid β (Aβ) peptide, a cleavage product of the amyloid precursor protein (APP), but the mechanisms by which Aβ leads to the disease have not been established (Selkoe and Schenk, 2003). Converging lines of evidence suggest that cerebrovascular factors play a key role in the pathogenesis of AD (Iadecola, 2004). Cerebral blood flow (CBF) is reduced in the early stages of AD, and the reactivity of cerebral blood vessels is impaired (Prohovnik et al., 1988; Hock et al., 1997; Mentis et al., 1998; Jagust, 2000; de la Torre, 2004). AD and cerebrovascular diseases share common risk factors (Stewart et al., 1998; Breteler, 2000; Casserly and Topol, 2004), suggesting similarities in their pathogenic mechanisms. Furthermore, AD patients have increased cerebrovascular atherosclerosis and exhibit white matter abnormalities resembling ischemic lesions (Brun and Englund, 1986; Roher et al., 2003, 2004; Van Dijk et al., 2004). These observations have been complemented by studies in mouse models of AD demonstrating that Aβ disrupts the regulation of the cerebral circulation, rendering the brain more susceptible to injury (Zhang et al., 1997). Thus, mice overexpressing APP have a profound attenuation of the increase in CBF produced by neural activity, a critical homeostatic mechanism by which the brain regulates substrate delivery and removes deleterious byproducts of metabolism (Niwa et al., 2000a, 2002b). Furthermore, APP mice have an impairment in cerebrovascular responses mediated by the release of vasoactive factors from brain endothelial cells, such as nitric oxide (NO), which protect the brain by maintaining CBF during hypotension (Huang et al., 1996; Iadecola et al., 1999; Jones et al., 1999; Niwa et al., 2002b). These cerebrovascular alterations are attributable to direct effects of Aβ on cerebral blood vessels and not to neural dysfunction (Niwa et al., 2000b, 2001b).
The mechanisms by which Aβ alters cerebrovascular regulation have not been elucidated. Aβ-induced vascular inflammation, activation of scavenger receptors, and upregulation of endothelin-1 have all been implicated (Crawford et al., 1998; Deane et al., 2003; Paris et al., 2003). Central to these mechanisms is increased production of reactive oxygen species (ROS) leading to vascular oxidative stress (Thomas et al., 1996; Niwa et al., 2000b, 2001b; Paris et al., 2003; Park et al., 2004a). However, the link between Aβ and vascular ROS production has been missing (Stamler, 1996; Iadecola, 2004). A major sources of vascular ROS is NADPH oxidase (Griendling et al., 1994; Mohazzab et al., 1994; Li and Shah, 2004), a multiunit enzyme first described in neutrophils (Babior, 2004; Lambeth, 2004). After activation, the membrane-bound subunits, p22phox and the catalytic subunit gp91phox, assemble with the cytoplasmic subunits (p47phox, p40phox, p67phox, and RAC), leading to transfer of electrons from NADPH to molecular oxygen and production of the ROS superoxide (Lambeth, 2004). Here, we report that NADPH oxidase is responsible for the ROS production mediating the cerebrovascular dysfunction induced by Aβ, an effect attributable in part to a reduction in NO bioavailability.
Materials and Methods
Mice
All experiments were performed in male mice ranging in age from 2 to 3 months. Breeder pairs of B6.129-Cybbtm1Din/J mice lacking the gp91phox subunit of NADPH oxidase (gp910/0) (Pollock et al., 1995) were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained by brother (hemizygous for the null allele)-sister (homozygous null) matings (Park et al., 2004b). The Tg2576 transgenic line overexpresses the Swedish mutant (K670N M671I) human APP695 driven by hamster prion protein regulatory sequences (Hsiao et al., 1996). The Tg2576 transgene array was maintained on a mixed B6/SJL background and on the inbred 129S6 background (Niwa et al., 2000a). PCR was used for genotyping the APP transgene array, as described previously (Niwa et al., 2000a). To produce APP transgenic mice (henceforth designated Tg2576), with or without gp91phox, gp910/0 females were crossed with Tg2576 males; because the gene encoding gp91phox (Cybb) is on the X chromosome, male offspring do not express gp91phox. To produce Tg2576-positive or -negative mice expressing gp91phox, C57BL/6J females were crossed with Tg2576 males; only male offspring were used. CBF results did not differ between the offspring of B6/SJL-Tg2576 or 129S6.Tg2576 males. Comparisons were among age-matched animals. gp910/0 mice were back-crossed with C57BL/6J strain six times and genotyped.
General surgical procedures
All experimental procedures were approved by the Institutional Animal Use and Care Committee. As described in detail previously (Iadecola et al., 1999; Niwa et al., 2001a; Park et al., 2004a), mice were anesthetized with isoflurane (induction, 5%; maintenance, 1-2%). One of the femoral arteries was cannulated for recording of arterial pressure and collection of blood samples. Mice were intubated and artificially ventilated with an oxygen-nitrogen mixture adjusted to provide an arterial pO2 of 120-140 mmHg (supplemental Table 1, available at www.jneurosci.org as supplemental material). Rectal temperature was maintained at 37°C using a thermostatically controlled rectal probe connected to a heating pad. After surgery, isoflurane was gradually discontinued, and anesthesia was maintained with urethane (750 mg/kg; i.p.) and α-chloralose (50 mg/kg; i.p.). Throughout the experiment, the level of anesthesia was monitored by testing corneal reflexes and motor responses to tail pinch.
Monitoring of cerebral blood flow
A small craniotomy (2 × 2 mm) was performed to expose the somatosensory cortex, the dura was removed, and the site was superfused with a modified Ringer's solution (37°C; pH 7.3-7.4) (Iadecola et al., 1999). CBF was continuously monitored at the site of superfusion with a laser-Doppler probe (Vasamedic, St. Paul, MN) positioned stereotaxically on the cortical surface and connected to a computerized data acquisition system. CBF values were expressed as percentage increase relative to the resting level. Zero values for CBF were obtained after the heart was stopped by an overdose of isoflurane at the end of the experiment.
Detection of ROS by hydroethidine
Hydroethidine (HE; dihydroethidium; 2 μm; Molecular Probes, Eugene, OR) was dissolved in DMSO and further diluted in Ringer's solution to a final DMSO concentration of <0.5%, which does not affect cerebrovascular responses in this preparation (Niwa et al., 2000a,b). HE was topically superfused on the somatosensory cortex for 60 min (Park et al., 2004b). At the end of the superfusion, the brain was removed and frozen (-30°C). Brain sections (thickness, 20 μm) were cut serially in a cryostat starting from the beginning to the end of the corpus callosum and collected at 100 μm intervals. This area includes the cortex underlying the cranial window. ROS production was assessed using methods from our laboratory that have been published previously (Park et al., 2004a,b). Briefly, brain sections were examined under an Eclipse E800 fluorescence microscope (Nikon, Melville, NY) equipped with an ethidium filter set (number 31014; Chroma Technology, Brattleboro, VT). Images were acquired with a computer-controlled digital monochrome camera (Coolsnap; Roper Scientific, Trenton, NJ). Image acquisition was performed with the same fluorescence settings in all cases. Ethidium fluorescence was assessed in the brain area superfused with HE. The analysis of ROS production in the different conditions studied (see below, Experimental protocol for CBF experiments) was performed in a blinded manner using IPLab software (Scanalytics, Fairfax, VA). After background subtraction of the camera dark current, pixel intensities of ethidium signals were quantified. Fluorescent intensities of all sections (15-20 per animal) were added, divided by the total number of pixels analyzed, and expressed as relative fluorescence units. To study the role of NADPH oxidase in Aβ1-40-induced ROS production, Ringer's solution containing HE alone (vehicle), HE plus gp91ds-tat (1 μm), or HE plus sgp91ds-tat (1 μm) was superfused for 30 min, followed by coapplication of Aβ (5 μm) for 30-40 min. In experiments with Tg2576 with or without gp91phox, Ringer's solution containing HE was superfused for 60 min. At the end of superfusion, brains were removed and processed for ROS assessment as described above.
Measurement of Aβ
Aβ was measured using an ELISA-based assay, as described previously (Scheuner et al., 1996). Briefly, the left hemispheres from the mice used for CBF studies were sonicated in 1% SDS with protease inhibitors and centrifuged. The supernatant contained SDS-soluble Aβ peptides. The pellet was sonicated in 70% formic acid and centrifuged as described above. The formic acid extract was neutralized by a 1:20 dilution into 1 m Tris phosphate buffer, pH 8.0. Aβ1-40 and Aβ1-42 concentrations were determined in supernatant (SDS soluble) and in the formic acid extract of the pellet (SDS insoluble) using the BAN-50/BA-27 and BAN-50/BC-05 sandwich ELISA as described previously (Scheuner et al., 1996). Concentrations in picomoles per gram of brain tissue were calculated by comparing the sample absorbance with the absorbance of known concentrations of synthetic Aβ1-40 and Aβ1-42.
Experimental protocol for CBF experiments
CBF recordings were started after arterial pressure and blood gases were in a steady state (supplemental Table 1, available at www.jneurosci.org as supplemental material). All pharmacological agents studied were dissolved in Ringer's solution unless otherwise indicated.
Effect of topical application of Aβ1-40 on functional hyperemia and endothelium-dependent and -independent responses. The cranial window was first superfused with Ringer's solution, and CBF responses to whisker stimulation were recorded. The whiskers were gently stroked for 60 s with a cotton-tipped applicator. In some mice, the endothelium-dependent vasodilators acetylcholine (ACh; 10 μm; Sigma), bradykinin (BK; 50 μm; Sigma), or the calcium ionophore A23187 (3 μm; Sigma) were superfused topically for 3-5 min. CBF responses to the NO donor S-nitroso-d-penicillamine (SNAP; 50 μm; Sigma) or to the NO-independent vasodilator adenosine (400 μm; Sigma) were also tested (Yang et al., 2003). After testing CBF responses during Ringer's solution superfusion, the superfusion solution was changed to Ringer's solution containing Aβ1-40 (5 μm; rPeptide, Athens, GA). This concentration of Aβ1-40 was previously found to produce maximal cerebrovascular effects (Niwa et al., 2000a,b, 2001b). As in previous studies (Niwa et al., 2000a,b, 2001b; Park et al., 2004a), to minimize aggregation of the peptide during the experiment, Aβ1-40 was freshly solubilized in DMSO and then diluted in normal Ringer's solution. The final DMSO concentration was <0.5%. The CBF response to whisker stimulation, ACh, BK, A23187, SNAP, or adenosine was tested 30-40 min after Aβ1-40 superfusion. This time interval was selected on the basis of previous studies in which the time course of the cerebrovascular effects of Aβ1-40 was investigated (Niwa et al., 2000a,b).
Effect of gp91ds-tat. In some experiments, the effects of the NADPH oxidase peptide inhibitor gp91ds-tat (YGRKKRRQRRRCSTRIRRQL-NH2) (Rey et al., 2001) on the cerebrovascular alterations induced by Aβ were studied. The scrambled sequence (sgp91ds-tat) (YGRKKRRQRRRCLRITRQSR-NH2) served as control. To facilitate entry into cells, the gp91phox sequence is linked to the human immunodeficiency virus-tat peptide, an amino acid sequence that is internalized by all cells. The peptides were synthesized (Bio-Synthesis, Lewisville, TX) with or without a fluorescent tag (carboxytetramethylrhodamine). Cerebrovascular effects were assessed 40 min after gp91ds-tat (1 μm) or sgp91ds-tat (1 μm) superfusion. After the experiment, the brain was removed and frozen, and the area under the window was sectioned in a cryostat (thickness, 20 μm). The penetration of the peptide in the cortex underlying the cranial window was verified in all cases by using a fluorescence microscope. In experiments using HE, peptides without a fluorescent tag were used.
Cerebrovascular reactivity in Tg2576, gp910/0, and crosses between Tg2576 and gp910/0. In these studies, CBF responses to whisker stimulation, ACh, BK, A23187, SNAP, and adenosine were tested in Tg2576. Transgene negative littermates served as controls. CBF responses were also examined in crosses between Tg2576 and gp910/0. Four groups of mice were studied: transgene-negative mice with wild-type gp91phox (gp91wt/wt), Tg2576 mice with wild-type gp91phox (Tg2576/gp91wt/wt), Tg2576 mice lacking gp91phox (Tg2576/gp910/0), and transgene-negative mice lacking gp91phox (gp910/0).
Effects of neuronal NO synthase inhibitors on the cerebrovascular effects of Aβ1-40. CBF responses to whisker stimulation, ACh, or adenosine were first obtained during Ringer's solution superfusion. Next, the relatively selective neuronal NO synthase (NOS) inhibitor 7-nitroindazole (7-NI; 50 mg/kg; Sigma) (Yang et al., 1999) was administered intraperitoneally, and cerebrovascular responses were tested again 30-40 min later. After 7-NI, Aβ1-40 (5 μm) was superfused on the neocortex, and responses were tested again 30-40 min later. In experiments in which the CBF response to whisker stimulation was tested, the Na+ channel inhibitor tetrodotoxin (TTX; 3 μm; Sigma) was superfused after pretreatment of 7-NI or the nonselective NOS inhibitor Nω-nitro-l-arginine (l-NNA; 1 mm; Sigma).
Effects of NOS inhibitors on cerebrovascular responses in Tg2576. CBF responses to whisker stimulation, ACh, or adenosine were first obtained during Ringer's solution superfusion. Next, the relatively selective neuronal NOS inhibitor 7-NI (50 mg/kg) (Yang et al., 1999) was administered intraperitoneally, and cerebrovascular responses were tested again 30-40 min later. In some experiments, the nonselective NOS inhibitor l-NNA (1 mm; topical superfusion) (Iadecola, 1992) was used instead of 7-NI. In experiments in which the CBF response to whisker stimulation was tested, the Na+ channel inhibitor TTX (3 μm) was superfused after pretreatment of l-NNA or 7-NI.
Data analysis
Data are expressed as means ± SEM. Two-group comparisons were analyzed by the two-tailed t test for dependent or independent samples, as appropriate. Multiple comparisons were evaluated by ANOVA and Tukey's tests. Statistical significance was considered for p < 0.05.
Results
The NADPH oxidase assembly inhibitor gp91ds-tat abrogates the cerebrovascular effects of Aβ
We used gp91ds-tat, a peptide that prevents the binding of p47phox to gp91phox and blocks enzyme assembly and activation (Rey et al., 2001), to investigate whether NADPH oxidase is responsible for the alteration in cerebrovascular regulation produced by Aβ. Direct application of Aβ1-40 onto the mouse somatosensory cortex reduced baseline CBF and attenuated the increase in CBF evoked by whisker stimulation (Fig. 1a,b). In addition, Aβ1-40 attenuated the increase in flow produced by ACh, BK, and the calcium ionophore A23187 (Fig. 1c-e), agents that act by releasing NO and other vasoactive factors from cerebral endothelial cells (Sobey and Faraci, 1997; Niwa et al., 2001a). The response to the NO donor SNAP was also attenuated (Fig. 1f), whereas the increase in CBF produced by adenosine, a response not mediated by NO (Yang et al., 2003), was not affected (Fig. 1g). Superfusion with gp91ds-tat, but not its scrambled version, completely abolished the cerebrovascular effects of Aβ1-40 (Fig. 1a-f). These data implicate NADPH oxidase in the cerebrovascular dysfunction induced by Aβ1-40.

Effect of suffusion of Aβ1-40 (5 μm) on the mouse somatosensory cortex with or without the NADPH oxidase peptide assembly inhibitor gp91ds-tat (gp91-ds) or its scrambled version (sgp91-ds). Aβ attenuates resting CBF (a) and reduces CBF responses to whisker stimulation (b), acetylcholine (c), the calcium ionophore A23187 (d), bradykinin (e), and the NO donor SNAP (f). Aβ did not alter the vasodilation produced by the NO-independent vasodilator adenosine (g). The cerebrovascular effects of Aβ were abrogated by pretreatment with gp91ds-tat but not sgp91-ds. LDU, Laser-Doppler perfusion units.*p < 0.05; ANOVA and Tukey's test; n = 6 per group. Error bars represent SEM.
Aβ does not impair cerebrovascular regulation in mice lacking the NADPH oxidase subunit gp91phox
To provide additional evidence implicating NADPH oxidase in the cerebrovascular dysfunction induced by Aβ1-40, we investigated the effect of Aβ1-40 in gp910/0 (Pollock et al., 1995). CBF responses to functional activation, ACh, A23187, and SNAP were normal in gp910/0 (Fig. 2a-e). However, in gp910/0, Aβ1-40 did not alter resting CBF and the increase in CBF produced by functional hyperemia or by endothelium-dependent (ACh, A23187) and -independent (SNAP, adenosine) vasodilators (Fig. 2a-e). These findings provide additional support to the hypothesis that a gp91phox-containing NADPH oxidase is responsible for the cerebrovascular effects of Aβ1-40.

Effect of suffusion of Aβ1-40 (5 μm) on the somatosensory cortex of wild-type mice and mice lacking gp91phox, the catalytic subunit of NADPH oxidase. In gp91wt/wt, Aβ1-40 reduced resting CBF (a) and attenuated the increase in CBF elicited by whisker stimulation (b), acetylcholine (c), A23187 (d), and SNAP (e). Aβ1-40 was devoid of cerebrovascular effects in gp910/0. LDU, Laser-Doppler perfusion units. *p < 0.05; ANOVA and Tukey's test; n = 6 per group. Error bars represent SEM.
Tg2576 mice lacking gp91phox have normal cerebrovascular reactivity
In the experiments described above, relatively high concentrations of exogenous Aβ1-40 were applied acutely to the neocortex. We next examined whether NADPH oxidase also plays a role in the cerebrovascular dysfunction induced by chronic elevations in endogenous Aβ comparable with those observed in AD. In these studies, we used Tg2576 in the presence or absence of gp91phox. Tg2576 were used at an age at which Aβ concentrations are beginning to increase as a result of aggregate formation, but before Aβ deposition in amyloid plaques is observed (Hsiao et al., 1996; Kawarabayashi et al., 2001; Park et al., 2004a). CBF responses to whisker stimulation, ACh, A23187, BK, and SNAP were attenuated in Tg2576 expressing gp91phox (Fig. 3a-e). However, cerebrovascular responses were normal in Tg2576 lacking gp91phox (Fig. 3a-e), implicating NADPH oxidase in the mechanism of the vascular dysfunction induced by Aβ. To rule out that the lack of gp91phox affected APP processing and Aβ production, Aβ levels were measured in Tg2576 carrying wild-type gp91phox or lacking gp91phox. As illustrated in Figure 4, levels of both SDS-soluble and SDS-insoluble forms of Aβ1-40 and Aβ1-42 did not differ between Tg2576 carrying wild-type gp91phox or lacking gp91phox. Therefore, the absence of cerebrovascular alterations in Tg2576 lacking gp91phox cannot be attributed to reduced levels of Aβ.

Cerebrovascular function in crosses between Tg2576 and gp910/0. Four groups of mice were studied: mice without the transgene and carrying wild-type gp91phox, mice carrying Tg2576 and wild-type for gp91phox, mice carrying the transgene and lacking gp91phox, and mice without the transgene and lacking gp91phox. Tg2576/gp91wt/wt mice exhibited attenuation of the increase in CBF produced by whisker stimulation (a), acetylcholine (b), A23187 (c), bradykinin (d), and the NO donor SNAP (e). The CBF response to adenosine was not altered (f). These cerebrovascular abnormalities were not observed in Tg2576/gp910/0 (a-e). gp910/0 mice had normal cerebrovascular reactivity (a-f). *p < 0.05; ANOVA and Tukey's test; n = 6 per group. Error bars represent SEM.
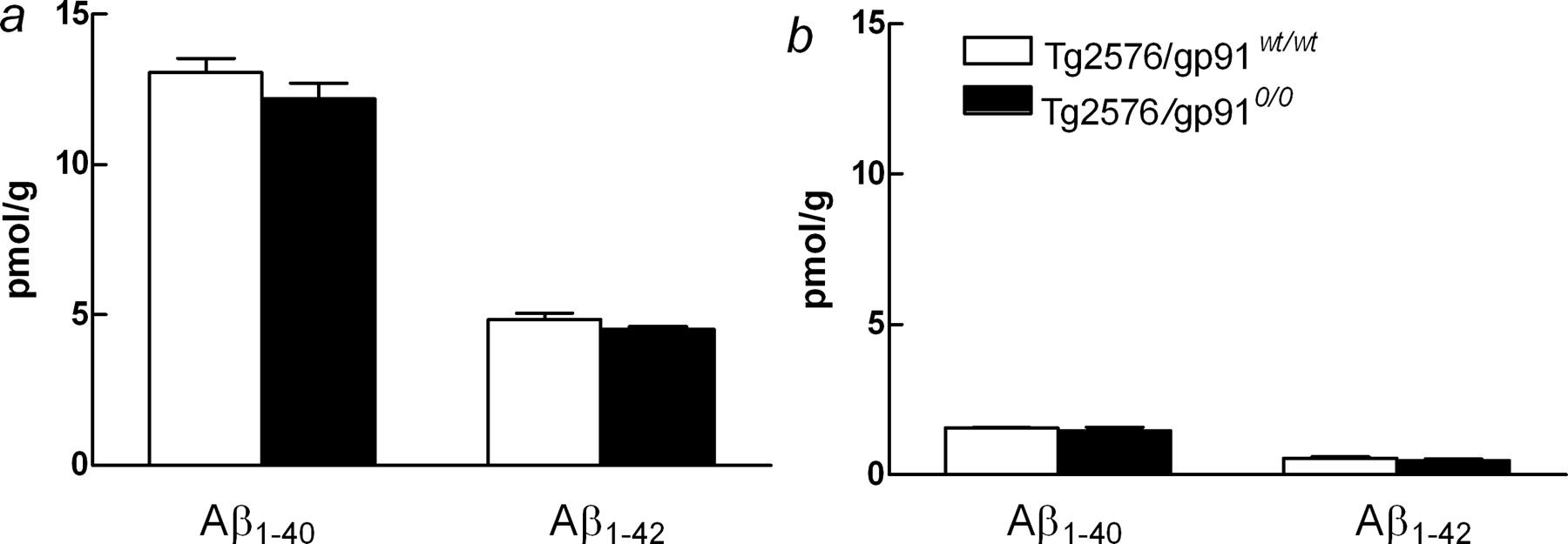
Brain levels of Aβ1-40 and Aβ1-42 in Tg2576 with (Tg2576/gp91wt/wt) and without (Tg2576/gp910/0) gp91phox. Both SDS-soluble (a) and SDS-insoluble (b) Aβ were examined. n = 6 per group. Error bars represent SEM.
gp91phox is required for Aβ-induced free radical production
The studies presented above suggest that a gp91phox-containing NADPH oxidase is involved in the cerebrovascular alterations induced by Aβ1-40. To determine whether NADPH oxidase is the source of the ROS underlying the cerebrovascular dysfunction, we investigated ROS production using hydroethidine fluoromicrography (Park et al., 2004a,b). In wild-type mice, neocortical application of Aβ1-40 increased ROS production (Fig. 5a,b,h). Similarly, ROS production was elevated in the neocortex of Tg2576 (Fig. 5g,h). Aβ1-40-induced ROS-production was blocked by the peptide inhibitor gp91ds-tat (Fig. 5d,h) but not by its scrambled version (Fig. 5e,h) and was not elevated in Tg2576 lacking gp91phox (Fig. 5f,h). These findings demonstrate that a gp91phox-containing NADPH oxidase is the source of the ROS induced by Aβ1-40.

ROS production in somatosensory cortex assessed by hydroethidine microfluorography. Aβ1-40 neocortical superfusion (5 μm) increases ROS production in wild-type mice (a, b) but not in mice lacking gp91phox (a, c). The increase in ROS production produced by Aβ is blocked by the NADPH oxidase assembly inhibitor gp91ds-tat (gp91ds) (d) but not by its scrambled version (sgp91-ds) (e). ROS overproduction is present in Tg2576 with wild-type gp91phox (g) but not in Tg2576 lacking gp91phox (f). The bar graphs in h represent group data corresponding to a-g. Scale bar, 50 μm. RLU, Relative fluorescent light units. *p < 0.05; ANOVA and Tukey's test; n = 6 per group. Error bars represent SEM.
Aβ does not disrupt functional hyperemia if NO synthesis is inhibited
We then sought to examine the mechanisms by which Aβ-derived ROS attenuate cerebrovascular responses. One possibility is that ROS inactivate NO and reduce its bioavailability (Beckman et al., 1990; Droge, 2002). This possibility is suggested by our observation that responses to the NO donor SNAP are attenuated by exogenous Aβ1-40 (Figs. 1f, 2e) and in Tg2576 (Fig. 3e). We reasoned that if the vascular dysfunction is related to scavenging of NO by free radicals, inhibition of NO synthesis would prevent the effect of Aβ1-40. The neuronal NOS inhibitor 7-NI attenuated resting CBF and the CBF increase produced by whisker stimulation, but not ACh, a response mediated by endothelial NOS (Fig. 6a-c). After 7-NI, Aβ1-40 superfusion failed to attenuate resting CBF and the response to whisker stimulation (Fig. 6a,b). Furthermore, in Tg2576, 7-NI did not alter resting CBF or the increase in CBF produced by whisker stimulation (Fig. 7a,b). Similar results were obtained with the nonselective NOS inhibitor l-NNA. To exclude that the lack of attenuation in Tg2576 was attributable to the fact that the response was maximally reduced and could not be attenuated further, we used TTX, a Na+ channel blocker that attenuates functional hyperemia by inhibiting neural activity (Yang et al., 1999). As illustrated in Figures 6b and 7b, application of TTX after 7-NI attenuated the response to whisker stimulation further, attesting to the fact that the CBF response was not maximally reduced. These findings indicate that NO scavenging by ROS contributes to the attenuation in functional hyperemia and ACh responses induced by Aβ1-40.

Effect of NOS inhibition on the cerebrovascular actions of Aβ peptides. The neuronal NOS inhibitor 7-NI attenuated resting CBF (a) and the CBF response to whisker stimulation (b) but not acetylcholine or adenosine (c, d). Application of Aβ1-40 after 7-NI did not further reduce resting CBF (a) or the responses to whisker stimulation (b). The absence of Aβ1-40 effects could not beat tributed to the fact that the response was already maximally attenuated, because the Na+ channel blocker TTX further attenuated the CBF response to whisker stimulation (b). LDU, Laser-Doppler perfusion units. *p < 0.05 from Ringer's solution; #p < 0.05 from 7-NI; ANOVA and Tukey's test; n = 6 per group. Error bars represent SEM.

Effect of NOS inhibition on the cerebrovascular actions of APP overexpression. In wild-type mice, the nonselective NOS inhibitor l-NNA attenuated resting CBF (a) and the CBF response to whisker stimulation (b) or acetylcholine (c). The CBF increase produced by adenosine was not affected (d). The neuronal NOS inhibitor 7-NI attenuated resting CBF and the CBF response to whisker stimulation but not acetylcholine or adenosine (a-d). In Tg2576, 7-NI or l-NNA did not attenuate resting CBF (a) and the increase in CBF produced by whisker stimulation (b) or acetylcholine (c). Responses to adenosine were not affected (d). The absence of effects on functional hyperemia could not be attributed to the fact that the response was already maximally reduced, because TTX further attenuated the CBF response to whisker stimulation (b). 7-NI enhanced the response to ACh in Tg2576 (c), but the significance of this finding is unclear. LDU, Laser-Doppler perfusion units. *p < 0.05 from Ringer's solution in wild-type mice; #p < 0.05 from 7-NI in wild-type and Tg2576 mice; &p < 0.05 from Ringer's solution in Tg2576 mice; ANOVA and Tukey's test; n = 6 per group. Error bars represent SEM.
Discussion
We found that NADPH oxidase is a key factor in the cerebrovascular dysfunction induced by Aβ1-40. First, the NADPH oxidase assembly inhibitor gp91ds-tat prevented the ROS production induced by exogenous Aβ1-40 and abrogated the associated alteration in functional hyperemia and endothelium-dependent relaxation. Second, exogenous Aβ1-40 did not elicit ROS production and vascular dysregulation in mice lacking the essential subunit of NADPH oxidase gp91phox. Third, the increase in ROS production and the related vascular dysregulation observed in Tg2576 was not observed in Tg2576 lacking gp91phox. The genetic background of the mice was carefully matched to eliminate confounding effects related to background genes. These findings provide the first demonstration that a gp91phox-containing NADPH oxidase is the link between Aβ1-40 and the ROS mediating the vascular dysregulation.
We then examined the potential mechanisms by which ROS alter the regulation of the cerebral circulation. In particular, we investigated whether Aβ1-40-induced ROS inactivate the potent vasodilator NO. NO has emerged as a major factor in the mechanisms regulating the cerebral circulation (Iadecola and Niwa, 2002). NO contributes to the increase in CBF produced by neural activity, a critical homeostatic mechanism that matches energy consumption to delivery of substrates through blood flow (Iadecola, 2004). Furthermore, NO released from endothelial cells plays a critical role in the compensatory response of cerebral blood vessels to hypotension (Jones et al., 1999; Busse and Fleming, 2003) and to vasoactive agents such as ACh (Ayata et al., 1996; Sobey and Faraci, 1997). We found that Aβ1-40 attenuates the increase in CBF produced by the NO donor SNAP, an effect abolished by blocking NADPH oxidase-dependent ROS production. This observation supports the hypothesis that NO scavenging by ROS is involved in the attenuation in NO-dependent responses induced by Aβ1-40. To provide additional evidence for this hypothesis, we assessed the effects of Aβ1-40 after inhibition of NO synthesis. We found that Aβ1-40 failed to attenuate the responses after administration of NOS inhibitors. These findings implicate NO as a target for the effects of NADPH oxidase-derived radicals. The vasodilatatory action of NO is potently antagonized by ROS, a process that is thought to be mediated by multiple mechanisms (Soriano et al., 2001; Droge, 2002; Adachi et al., 2004; Iadecola, 2004). NO reacts with the ROS superoxide to form peroxynitrite, a reaction that reduces the amount of NO available for vasodilation (Beckman et al., 1990; Adachi et al., 2004). During functional hyperemia, NO produced by neuronal NOS diffuses to local arterioles to mediate vasodilation (Ayata et al., 1996; Yang and Iadecola, 1997). The present findings, in concert with previous data demonstrating the peroxynitrite marker 3-nitrotyrosine in cerebral arterioles of Tg2576 (Park et al., 2004a), suggest that once NO reaches the blood vessels, it reacts with superoxide to form peroxynitrite. In the case of the response to ACh, NO produced in endothelial cells reacts with locally generated superoxide to form peroxynitrite, resulting in impairment of vasodilation. Peroxynitrite also leads to the nitration of other protein involved in vascular regulation. These include the enzyme prostacyclin synthase, which produces the vasodilator prostacyclin, and the free-radical scavenging enzyme superoxide dismutase (Zou et al., 1999; Guo et al., 2003; Ischiropoulos and Beckman, 2003). Furthermore, ROS inactivate the essential NOS cofactor tetrahydrobiopterin and attenuate NO production (Katusic, 2001). Therefore, the disturbance in the vasodilation may results both from the loss of NO and from oxidative/nitrosative stress of vascular proteins. These alterations, collectively, produce vascular dysregulation by increasing oxidative stress and producing vasoconstriction of cerebral vessels. However, the CBF responses evoked by BK and A23187 are not dependent on NO (Rosenblum, 1987; Rosenblum et al., 1989; Niwa et al., 2001a). This finding suggests that the mechanisms of the cerebrovascular dysfunction exerted by Aβ1-40 involve factors other than NO scavenging and that other effects of ROS on proteins involved in vascular function are also likely to play a role (Droge, 2002).
Although there are several enzymatic systems that can produce ROS (Droge, 2002), the enzyme NADPH oxidase has emerged as a major source of ROS in several cell types (Babior, 2004; Lambeth, 2004). In blood vessels, NADPH oxidase is responsible for the ROS production and vascular dysfunction in a wide variety of vascular pathologies, including angiotensin-induced hypertension, hyperlipidemia, atherosclerosis, and hyperhomocysteinemia (Faraci, 2003; Li and Shah, 2004). Although the enzyme is also present in neurons and glia both in the normal brain and in AD (Bianca et al., 1999; Shimohama et al., 2000; Abramov et al., 2004), it is likely that the effects of Aβ1-40 are mediated by vascular NADPH oxidase. ROS are highly reactive chemical species that do not travel long distances (Stamler, 1996). Therefore, the ROS responsible for the vascular effects of Aβ1-40 are most likely generated at the vascular level. Furthermore, oxidative stress is observed in cerebral vessels of Tg2576 at a time when there is no evidence of oxidative stress in other brain cells or presence of amyloid plaques (Park et al., 2004a). This observation suggests that vascular oxidative stress is an early event in the brain dysfunction associated with Aβ overproduction in mouse. Vascular dysfunction is proportional to the amount of Aβ1-40 and occurs even in mice whose Aβ level does not change with age and never develop plaques (Iadecola et al., 1999; Niwa et al., 2000a). Therefore, it is likely that soluble Aβ monomers or small oligomers are the active entity. Future studies in which the cellular source of the ROS is established would help define the vascular and nonvascular components of the oxidative stress.
The mechanisms by which Aβ1-40 activates NADPH oxidase remain to be defined. One possibility is that Aβ activates NADPH oxidase through the scavenger receptors RAGE (receptor for advanced glycation end product) or CD36 (El Khoury et al., 1996, 2003; Yan et al., 1996; Coraci et al., 2002). Activation of these receptors leads to increased ROS production, which is mediated by NADPH oxidase (Wautier et al., 2001; Bamberger et al., 2003; Abramov et al., 2004; Fuhrman et al., 2004). Indeed, evidence of activation of NADPH oxidase by Aβ has been provided in microglial and astrocytic cultures and in AD brains (Bianca et al., 1999; Shimohama et al., 2000; Parvathenani et al., 2003). Another possibility is that the NADPH activation occurs through endothelin receptors. Aβ upregulates endothelin-1 in cerebral blood vessels (Deane et al., 2003), and activation of endothelin receptors leads to production of ROS through NADPH oxidase (Duerrschmidt et al., 2000; Zheng et al., 2003). It is, therefore, conceivable that Aβ leads to activation of NADPH oxidase through endothelin receptors. Regardless of the mechanisms of activation, the findings provide evidence that NADPH oxidase is the missing link between Aβ and the oxidative stress leading to cerebrovascular dysfunction.
What are the implications of Aβ-induced vascular dysfunction for AD? Reduction in resting CBF of a magnitude similar to that reported in Tg2576 (Niwa et al., 2002a) and in AD (Jagust, 2000) are not sufficient to produce ischemic cell dysfunction or death (Hossmann, 1994). However, such reductions in CBF can attenuate protein synthesis (Mies et al., 1991), a process that is essential for learning and memory (Martin et al., 2000; Milekic and Alberini, 2002). Furthermore, the impairment of the increase in CBF evoked by brain activation produces an imbalance between substrate delivery through CBF and energy requirements which, if protracted in time, may deprive the brain of energy substrates and lead to brain dysfunction (Farkas et al., 2002). Another deleterious vascular effect of Aβ1-40 is a disruption of cerebrovascular autoregulation (Niwa et al., 2002b), a mechanism in part mediated by vasoactive factors produced in endothelial cells, which prevents fall of CBF during hypotension (Huang et al., 1996; Jones et al., 1999; Chillon and Baumbach, 2002). Failure of autoregulation leads to ischemic damage of cortical white matter (Matsushita et al., 1994), a finding that may be responsible for the white matter lesions observed in patients with AD (Brun and Englund, 1986; Van Dijk et al., 2004). These observations, collectively, suggest that Aβ peptides, in addition to their well known deleterious effects on neurons (Selkoe and Schenk, 2003), can also disrupt the function of cerebral blood vessels. Because Tg2576 recapitulates the pathobiology of Aβ peptides overproduction in humans, the findings suggest that the alterations in CBF induced by Aβ1-40 are an early event in the mechanisms of the diseases that precede the deleterious effects resulting from direct actions of Aβ peptides on neurons. Such cerebrovascular dysfunction increases the vulnerability of the brain and is likely to contribute to the mechanisms of AD.
Footnotes
This work was supported by grants from the National Institutes of Health (NIH) (NS38252 and HL18974 to C.I.; AG15453 to G.A.C.). C.I. is the recipient of a Javits Award from NIH-National Institute of Neurological Disorders and Stroke.
Correspondence should be addressed to Dr. C. Iadecola, Division of Neurobiology, Weill Medical College of Cornell University, 411 East 69th Street, KB410, New York, NY 10021. E-mail: coi2001{at}med.cornell.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251769-09$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}