

Abstract
The ability of KCNQ (Kv7) channels to form hetero-oligomers is of high physiological importance, because heteromers of KCNQ3 with KCNQ2 or KCNQ5 underlie the neuronal M-current, which modulates neuronal excitability. In KCNQ channels, we recently identified a C-terminal subunit interaction (si) domain that determines their subunit-specific assembly. Within this si domain, there are two motifs that comprise ∼30 amino acid residues each and that exhibit a high probability for coiled-coil formation. Transfer of the first or the second coiled-coil (TCC) domain from KCNQ3 into the KCNQ1 scaffold resulted in chimeras KCNQ1(TCC1)Q3 and KCNQ1(TCC2)Q3, both of which coimmunoprecipitated with KCNQ2. However, only KCNQ1(TCC2)Q3 enhanced KCNQ2 currents and surface expression or exerted a strong dominant-negative effect on KCNQ2. Deletion of TCC2 within KCNQ2 yielded functional homomeric channels but prevented the current augmentation measured after coexpression of KCNQ2 and KCNQ3. In contrast, deleting TCC1 within KCNQ2 did not give functional homomeric KCNQ2 or heteromeric KCNQ2/KCNQ3 channels. Mutations that disrupted the predicted coiled-coil structure of TCC1 in KCNQ2 or KCNQ3 abolished channel activity after expressing these constructs singly or in combination, whereas helix-breaking mutations in TCC2 of KCNQ2 gave functional homomeric channels but prevented the heteromerization with KCNQ3. In contrast, KCNQ3 carrying a coiled-coil disrupting mutation in TCC2 hetero-oligomerized with KCNQ2.
Our data suggest that the TCC1 domains of KCNQ2 and KCNQ3 are required to form functional homomeric as well as heteromeric channels, whereas both TCC2 domains facilitate an efficient transport of heteromeric KCNQ2/KCNQ3 channels to the plasma membrane.
Introduction
The KCNQ (Kv7) family of voltage-gated K+ channels comprises five members, Kv7.1–5 (Gutman et al., 2003) (referred to as KCNQ1–5 herein). Mutations in four of these channels cause human inherited diseases, highlighting their physiological importance (Jentsch, 2000). Heterozygous dominant-negative KCNQ1 mutations are associated with cardiac arrhythmias in the long-QT syndrome (Wang et al., 1996), whereas patients carrying loss-of-function mutations on both alleles additionally suffer from congenital deafness (Neyroud et al., 1997). A gain-of-function mutation was found in patients with an autosomal dominant atrial fibrillation (Chen et al., 2003). Mutations in either KCNQ2 or KCNQ3 lead to benign familial neonatal convulsions (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998), whereas mutations in KCNQ4 cause a form of dominant progressive hearing loss (Kubisch et al., 1999).
Voltage-gated K+ channels are tetramers of α-subunits, which surround a K+-selective pore. Two types of domains involved in K+ channel assembly have been described. In Shaker-related K+ channels, a conserved cytoplasmic N-terminal domain (T1), which preceeds the first transmembrane segment, is important for subfamily-specific channel assembly. In contrast, C-terminal sequences are required for assembly of functional rat eag, inward rectifier IRK1/Kir2.1 (Tinker et al., 1996), and KCNQ channels (Schmitt et al., 2000; Schwake et al., 2003).
Although all KCNQ α-subunits can form functional homotetramers, they can also assemble to heteromeric channels containing different KCNQ α-subunits or associate with KCNE β-subunits. Thus, KCNQ1 coassembles with KCNE1 and KCNE3 (Barhanin et al., 1996; Sanguinetti et al., 1996; Schroeder et al., 2000b) but is unable to form heteromers with other KCNQ subunits. In contrast, KCNQ3 can form functional heteromers with KCNQ2, KCNQ4, and KCNQ5 (Schroeder et al., 2000a).
Many details of KCNQ channel assembly remain incompletely understood. Previous work has indicated that homomeric assembly of KCNQ1 involves a C-terminal assembly (ca) domain (Schmitt et al., 2000). However, recent findings suggest that this region is required for normal cell surface trafficking of assembled KCNQ1 tetramers (Kanki et al., 2004). The subunit-specific heteromerization between KCNQ subunits is determined by a 115 amino acid-long subunit interaction (si) domain within the C terminus, because transfer of the si domain of KCNQ3 was sufficient to confer the broad KCNQ3 assembly properties to KCNQ1 (Schwake et al., 2003).
Recently, Jenke et al. (2003) reported that short C-terminal domains are required for tetrameric assembly in eag channels. These domains have a high probability for formation of coiled-coils, which are helical protein–protein interaction domains with a heptad amino acid repeat pattern (abcdefg)n, with the positions a and d preferably occupied by hydrophobic residues (Lupas, 1996). Using peptides derived from eag and erg, the authors proposed that these domains act as sites driving tetramerization and were therefore referred to as tetramerizing coiled-coil (TCC) sequences (Jenke et al., 2003). By sequence analysis, they have also identified possible coiled-coils in the C termini of other potassium channels, including KCNQs (Jenke et al., 2003). In this study, we set out to determine the individual role of these TCC domains on homomeric and heteromeric channel formation to further characterize the molecular determinants of KCNQ channel assembly.
Materials and Methods
cDNA constructs.
Starting from KCNQ cDNAs subcloned into expression vector pTLN, the KCNQ1/KCNQ3 chimeras, KCNQ2 and KCNQ3 mutants and deletion constructs (Fig. 1A), the HA-tagged KCNQ1, and pore mutant KCNQ1(G314S) were constructed by recombinant PCR and verified by sequencing. The HA-tagged KCNQ2 construct was as described previously (Schwake et al., 2000).
Expression in Xenopus laevis oocytes.
Individual stage V to VI oocytes were obtained from anesthetized frogs and isolated by collagenase treatment. Synthesis of cRNA was performed with the SP6 mMessage mMachine kit (Ambion, Austin, TX). KCNQ cRNA (10 ng) was injected into oocytes (also for coinjection experiments, which contained a 1:1 cRNA mixture). After injection, oocytes were kept at 17°C in MBS solution [88 mm NaCl, 2.4 mm NaHCO3, 1 mm KCl, 0.41 mm CaCl2, 0.33 mm Ca(NO3)2, 0.82 mm MgSO4, 10 mm HEPES, pH 7.6].
Electrophysiology.
Three to 5 d after injection, currents were measured in ND96 (96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, pH 7.4) at room temperature in two-electrode voltage-clamp recordings using a Turbotec 10C amplifier (NPI Electronics, Tamm, Germany) and pClamp8 software (Molecular Devices, Union City, CA). Voltage protocol for current recordings, unless stated differently, is as follows: from a holding potential of −80 mV, cells were pulsed for 2.5 s to voltages between −80 and +40 mV in steps of 20 mV.
Data analysis and statistics.
All data were obtained from at least three different batches of oocytes with at least six oocytes per batch and cRNA injection scheme. The total numbers of oocytes for the respective constructs are given in the figures. The current values from each oocyte were normalized to a batch average (constructs used for normalization are indicated in the respective figures) and thus combined from oocytes of different batches. Data are given as means ± SE. To probe for significance, we performed Student's t tests on pairs of data sets. All data presented are significant at least on the p < 0.05 level, unless stated differently.
Quantitative analyses of I/Imaxcurves andtetraethylammonium dose–response curves.
To determine the parameters for the voltage dependence of activation, I/Imax curves were fitted by a Boltzmann function as follows:  in which Bmin and Bmax are the minimal or maximal I/Imax values, respectively, R is the molar gas constant, zq is the slope factor (equivalent charge), F is the Faraday constant, T is the absolute temperature in K, V is the transmembrane potential, and V1/2 is the potential of half-maximal activation.
in which Bmin and Bmax are the minimal or maximal I/Imax values, respectively, R is the molar gas constant, zq is the slope factor (equivalent charge), F is the Faraday constant, T is the absolute temperature in K, V is the transmembrane potential, and V1/2 is the potential of half-maximal activation.
For probing tetraethylammonium (TEA) sensitivity, 2-s-long voltage pulses were applied from −80 to −40 mV in the presence of different TEA concentrations, and the total activation amplitude at the end of the test pulse was measured. Amplitudes were normalized for each oocyte to the value measured in the absence of TEA. Data are means ± SE of normalized amplitudes. The resulting dose–response curves were fitted with the following equation:  in which Imax is the maximal activation amplitude, c is the TEA concentration, IC50 is the concentration for the half-maximal inhibition, and nH is the Hill coefficient.
in which Imax is the maximal activation amplitude, c is the TEA concentration, IC50 is the concentration for the half-maximal inhibition, and nH is the Hill coefficient.
Surface expression experiments.
Analysis of surface expression of HA-tagged KCNQ constructs was as described previously (Zerangue et al., 1999; Schwake et al., 2000). Briefly, oocytes were placed for 30 min in blocking solution (BS) (ND96 plus 1% BSA), incubated for 1 h with rat monoclonal anti-HA antibody 3F10 (Roche Diagnostics, Mannheim, Germany) diluted in BS (1 μg/ml), washed three times, and incubated with HRP-conjugated secondary antibody (goat anti-rat FAB fragments; Jackson ImmunoResearch, West Grove, PA) in BS, followed by three washes each, first in BS and then in ND96. All incubation and washing steps were performed on ice. Surface expression was quantified by placing individual oocytes in 50 μl of SuperSignal ELISA Femto Maximum Sensitivity Substrate solution (Pierce, Rockford, IL), and luminescence was measured in a TD 20/20 luminometer (Turner Designs, Sunnyvale, CA).
Coimmunoprecipitation.
For coimmunoprecipitation experiments, we used Cos7 cells that exhibited sufficiently high efficiency for transfection with KCNQ channel cDNAs. Cells were cultured in DMEM with 4.5 g/L glucose (PAA Laboratories, Cölbe, Germany) containing 10% fetal bovine serum, penicillin, and streptomycin at 37°C in 5% CO2. The cells were transiently transfected using FuGENE 6 (Roche Products). When KCNQ2-FLAG was contransfected with KCNQ1, KCNQ3, and various KCNQ1/KCNQ3 chimeras, four times more KCNQ2-FLAG DNA was used. Forty-eight hours after transfection, homogenization buffer [ice-cold PBS (137 mm NaCl, 2.7 mm KCl, 7.4 mm Na2HPO4, 1.5 mm KH2PO4, pH 7.4) containing 1× Complete (Roche Products)] was added to the plates, and cells were gathered with a scraper. Cells were lysed and homogenized by sonication. Cell lysates were centrifugated at 1000 × g for 10 min to remove cellular debris, and supernatants were collected. Membranes were pelleted at 100,000 × g (0.5 h; 4°C) and resuspended in lysis buffer (120 mm NaCl, 5 mm DTT, 1 mm EGTA, 0.5% NP-40, 10% glycerol, 1× Complete, 50 mm Tris-HCl, pH 8.0). Protein concentration was determined using the BCA protein assay system (Pierce). The samples were adjusted with lysis buffer to obtain equal protein concentrations. Proteins were immunoprecipitated with the 3F10 anti-HA monoclonal antibody (Roche Products) for 4.5 h and protein-G Sepharose (Roche Products) for an additional 0.5 h at 4°C. After five washes with lysis buffer, immunoprecipitates were released at 55°C in SDS sample buffer for 12 min and separated on 10% SDS polyacrylamide gels. Immunodetection used primary mouse anti-FLAG monoclonal M2 (Sigma, St. Louis, MO) and secondary HRP-conjugated goat anti-mouse (Jackson ImmunoResearch) antibodies. Reacting proteins were detected by using the ECL detection system (Amersham Biosciences, Arlington Heights, IL). Signals were recorded by a luminescent image analyzer (Fujifilm image reader, LAS1000; Fujifilm, Tokyo, Japan).
Western blot analysis.
The oocytes used for TEA-sensitivity measurements were subsequently pooled and stored at −20°C. After homogenization of the pooled oocytes in an ice-cold solution containing 250 mm sucrose, 0.5 mm EDTA, 5 mm Tris-HCl, pH 7.4, and a protease inhibitor mix (Complete; Roche Products), the yolk platelets were removed by three low-speed centrifugations. The resulting supernatant was mixed with SDS-Laemmli sample buffer, and the protein equivalent to one oocyte was analyzed by SDS-PAGE (10% polyacrylamide). The separated proteins were transferred to polyvinylidene difluoride membrane. Blots were blocked with TBS (150 mm NaCl, 25 mm Tris, pH 7.4) containing 5% milk powder and 0.1% Nonidet P-40. Primary [KCNQ2(N-19); 1:500; Santa Cruz Biotechnology, Santa Cruz, CA] and secondary (horseradish peroxidase-conjugated rabbit ant-goat IgG; 1:5000) antibodies were diluted in TBS blocking solution. Washes were with TBS with 0.1% Nonidet P-40. Reacting proteins were detected by using the ECL detection system (Amersham Biosciences). Signals were recorded by a luminescent image analyzer (Fujifilm image reader, LAS1000).
Results
Both TCC domains of KCNQ3 are sufficient to determine the subunit specificity of KCNQ channel assembly
As shown previously (Schwake et al., 2003), the broad heterooligomerization properties from KCNQ3 can be transferred to KCNQ1 by replacing a C-terminal segment of KCNQ1 (amino acids 530–620) by the corresponding stretch of KCNQ3 (amino acids 535–650). This strategy resulted in a construct termed KCNQ1-sidQ3, in which the si domain was swapped between KCNQ3 and KCNQ1. To determine the role of the two TCC domains (Jenke et al., 2003) within the KCNQ si domain (Fig. 1A) on channel assembly, we first replaced both TCC domains within the si domain of KCNQ1 by the corresponding segments from KCNQ3 (Fig. 1A). The resultant KCNQ1(TCC1+TCC2)Q3 construct was compared with KCNQ1-sidQ3 in its ability to interact with KCNQ2 by performing two-electrode voltage-clamp experiments on Xenopus oocytes (Fig. 2). Currents measured after coexpression of KCNQ1-sidQ3 with KCNQ2 (Fig. 2B) or KCNQ1(TCC1+TCC2)Q3 with KCNQ2 (Fig. 2C) were very similar in time course and voltage dependence (Fig. 2D). Both KCNQ1(TCC1+TCC2)Q3 and KCNQ1-sidQ3 increased currents by a factor of ∼8–10 compared with KCNQ2 expressed alone (Fig. 2D,E).

Sequence alignment and helical wheel projection. A, Alignment of C-terminal sequences of KCNQ1, KCNQ2, and KCNQ3 according to Schroeder et al. (2000a). The si domains from KCNQ1, KCNQ2, and KCNQ3 are indicated by light gray boxes, and the TCC domains within the si domains are shown as dark gray boxes, respectively. Conserved amino acids within the TCC domains are shown in white. The conserved leucine residues within the TCC domains, which were substituted for prolines, are marked with asterisks. The X indicates the last amino acid in the truncated KCNQ2-TCC1-X construct, whereas Y and the Ω indicate the first amino acids of the TCC2-CT-KCNQ2 and dCT-KCNQ2 construct, respectively. To generate the KCNQ2(NT-TCC2) construct, we used the truncated KCNQ2-TCC1-X construct and inserted, after the start methionine, the TCC2 fragment of KCNQ2 shown in italics. B, Schematic helical wheel projection for coiled-coils. Heptad positions are labeled a–g. Residues of the first two helical turns are boxed (positions a and d) or circled (positions b, c, e, f, and g).

Exchange of both TCC sequences within the si domain. A, Schematic illustration of the chimeras KCNQ1-sidQ3 and KCNQ1(TCC1+TCC2)Q3. B, C, Representative current recordings from Xenopus oocytes injected with cRNAs of KCNQ2 plus KCNQ1-sidQ3 (B) and KCNQ2 plus KCNQ1(TCC1+TCC2)Q3 (C). D, Current–voltage (I/V) curves of KCNQ2 (n = 20), KCNQ2 plus KCNQ1 (n = 24), KCNQ2 plus KCNQ1(TCC1+TCC2)Q3 (n = 55), and KCNQ2 plus KCNQ1-sidQ3 (n = 45). Steady-state current levels measured at the end of a 2-s-long voltage pulse are shown. E, Bar graph of current levels obtained at the end of a 2-s-long voltage pulse to +40 mV. Currents from different oocyte batches were normalized to the mean KCNQ2 plus KCNQ1-sidQ3 current at +40 mV.
Next, we constructed chimeras in which only the first or the second TCC domain were exchanged, resulting in constructs KCNQ1(TCC1)Q3 and KCNQ1(TCC2)Q3, respectively (Fig. 3A). When KCNQ1(TCC1)Q3 was coexpressed with KCNQ2, currents were not significantly enhanced compared with KCNQ2 alone (Fig. 3B,D,F), whereas KCNQ1(TCC2)Q3/KCNQ2 coexpression led to an approximately eightfold increase, similar to a KCNQ1-sidQ3/KCNQ2 expression scheme (Fig. 3C,E,G).

Exchange of individual TCC sequences within the si domain. A, Schematic illustration of the chimeras KCNQ1(TCC1)Q3 and KCNQ1(TCC2)Q3. B, C, Representative current recordings from oocytes injected with cRNAs of KCNQ2 plus KCNQ1(TCC1)Q3 (B) and KCNQ2 plus KCNQ1(TCC2)Q3 (C). D and E, Current–voltage relationships of KCNQ2 (n = 20), KCNQ2 plus KCNQ1 (n = 24), KCNQ2 plus KCNQ1(TCC1)Q3 (n = 38), and KCNQ2 plus KCNQ1-sidQ3 (n = 45) (D) and of KCNQ2 (n = 20), KCNQ2 plus KCNQ1 (n = 24), KCNQ2 plus KCNQ1(TCC2)Q3 (n = 43), and KCNQ2 plus KCNQ1-sidQ3 (n = 45) (E). Steady-state current levels measured at the end of a 2-s-long voltage pulse are shown. Currents from different oocyte batches were normalized to the level of KCNQ2 plus KCNQ1-sidQ3 currents at +40 mV. F, G, Bar diagrams of mean current levels obtained at +40 mV from currents depicted in D and E, respectively. Analysis of dominant-negative effects (H) and surface expression (I) by exchange of individual TCC sequences within the si domain. H, Effect of coexpressing pore mutants KCNQ1-G314S (n = 24), KCNQ1(TCC1)Q3-G314S (n = 44), KCNQ1(TCC2)Q3-G314S (n = 55), KCNQ1(TCC1+TCC2)Q3-G314S (n = 31), and KCNQ3-G318S (n = 20) on KCNQ2 (n = 20) currents. Average current levels at the end of a test pulse to +40 mV from oocytes (co-)injected with cRNAs as indicated are shown. I, Surface expression determined from luminescence measurements of oocytes coinjected with HA-tagged KCNQ2 and nontagged KCNQ2 (n = 35), KCNQ1(TCC1)Q3 (n = 39), KCNQ1(TCC2)Q3 (n = 39), or KCNQ3 (n = 42). Data were normalized to the value for the KCNQ2(HA) plus KCNQ3 coinjection. Oocytes expressing nontagged KCNQ2 (n = 40) served as a background control. Values are means ± SE. J, Coassembly of the HA-tagged KCNQ1(TCC1)Q3 and KCNQ1(TCC2)Q3 constructs with FLAG-tagged KCNQ2. Protein complexes obtained from Cos-7 cells expressing various KCNQ subunits were immunoprecipitated with anti-HA antibody, separated by SDS-PAGE, and detected by Western blot analysis using an anti-FLAG antibody. The arrowhead denotes the molecular weight of FLAG-tagged KCNQ2 proteins, and the asterisk represents the weight of the heavy chain of the anti-HA antibody. Left, Molecular weight marker. K, Summary of three independent coimmunoprecipitation experiments. The corresponding Western blots were analyzed by densitometric analysis. The band intensities were normalized to the Q3HA/Q2FLAG value in each experiment and combined.
To further analyze this interaction, we constructed KCNQ1(TCC1)Q3, KCNQ1(TCC2)Q3, and KCNQ1(TCC1+TCC2)Q3 mutants that contained the dominant-negative pore mutation KCNQ1-G314S (Wollnik et al., 1997) and coexpressed these mutants [termed KCNQ1(TCC1)Q3-G314S, KCNQ1(TCC2)Q3-G314S, and KCNQ1(TCC1+TCC2)Q3-G314S] with KCNQ2. Additionally, we coexpressed the pore mutants KCNQ1-G314S (which should not interact) and KCNQ3-G318S (which should interact most efficiently) with KCNQ2 to control the dominant-negative experiments.
Whereas KCNQ1-G314S had no effect on KCNQ2 currents, which excludes a dominant-negative effect, KCNQ1(TCC1+TCC2)Q3-G314S exerted a strong dominant-negative effect by suppressing currents to the level observed for KCNQ3-G318S coexpression (Fig. 3H). In this context, KCNQ1(TCC2)Q3-G314S reduced KCNQ2 currents by 70% (p < 0.000005), whereas coexpression of KCNQ1(TCC1)Q3-G314S led to only 40% current reduction (p < 0.000005). Currents from KCNQ1(TCC2)Q3-G314S plus KCNQ2 and KCNQ1 (TCC1)Q3-G314S plus KCNQ2 coexpression were significantly different (p < 0.0005).
Thus, KCNQ1(TCC1+TCC2)Q3-G314S and KCNQ1(TCC2)Q3-G314S exerted a dominant-negative effect in a 1:1 coinjection scheme with KCNQ2. However, the reduction of KCNQ2 currents by the KCNQ1(TCC1)Q3-G314S chimera indicates a weaker effect, although an interaction between these subunits could be shown in coimmunoprecipitation experiments (see below).
Because the strong current increase after KCNQ2/KCNQ3 and KCNQ2/KCNQ1-sidQ3 coexpression results primarily from an increased surface expression (Schwake et al., 2000, 2003), we tested whether such an effect also explains the enhancement of KCNQ1(TCC2)Q3/KCNQ2 currents. Figure 3I shows that the KCNQ1(TCC2)Q3 chimera was as efficient as KCNQ3 in stimulating the surface expression of epitope-tagged KCNQ2. In contrast, coexpression of the KCNQ1(TCC1)Q3 chimera resulted in a surface expression of epitope-tagged KCNQ2 of about the same magnitude as in a 1:1 coinjection scheme with WT KCNQ2.
To confirm the role of the TCC domains in channel assembly on the protein level, we performed coimmunoprecipitation experiments after coexpressing HA-tagged KCNQ1, KCNQ1(TCC1)Q3, KCNQ1(TCC2)Q3, or KCNQ3 with FLAG-tagged KCNQ2.
Figure 3J shows that FLAG-tagged KCNQ2 coprecipitated with HA-tagged KCNQ1(TCC1)Q3, KCNQ1(TCC2)Q3, and KCNQ3. In contrast, there was only a very faint signal when coimmunoprecipitation was attempted with HA-tagged KCNQ1, which was also observed in controls in which HA-tagged KCNQ1 was not included. Figure 3K shows results from a densitometric analysis of Western blots from three such experiments, which demonstrate that FLAG-tagged KCNQ2 coprecipitated with HA-tagged KCNQ1(TCC1)Q3 or KCNQ1(TCC2)Q3 as well as with HA-tagged KCNQ3.
These four lines of evidence suggested that both TCC domains from KCNQ3 can mediate interactions between KCNQ3 and KCNQ2. The transfer of the TCC1 domain from KCNQ3 to KCNQ1 already facilitates subunit interaction, which, however, does not lead to current enhancement. In addition, the transfer of the TCC2 domain is responsible for the efficient forward transport of heteromeric KCNQ2/KCNQ3 channels to the plasma membrane.
TCC2 of KCNQ2 is needed for heteromeric KCNQ2/KCNQ3 but not for homomeric KCNQ2 channel assembly
Given the importance of the TCC2 domain from KCNQ3 for an efficient plasma membrane expression of heteromeric KCNQ2/KCNQ3 channels, we also investigated a possible involvement of the TCC2 domain of KCNQ2 in this process. Therefore, we replaced TCC2 of KCNQ2 by the corresponding TCC2 of KCNQ1 and tested the resulting chimera KCNQ2(TCC2)Q1 for its effect on KCNQ3 currents. KCNQ2(TCC2)Q1 yielded functional K+ channels (Fig. 4A,C). Coexpression of KCNQ2(TCC2)Q1 with KCNQ3 led to a significant, albeit only an approximately twofold current increase (Fig. 4B,C). This result contrasts with the strong augmentation of KCNQ2 currents by KCNQ1(TCC2)Q3 (Fig. 3C,E,G) or KCNQ3 (Fig. 4C), suggesting that the interaction between KCNQ2(TCC2)Q1 and KCNQ3 is impaired, resulting in inefficient plasma membrane expression of KCNQ2/KCNQ3 heteromers.
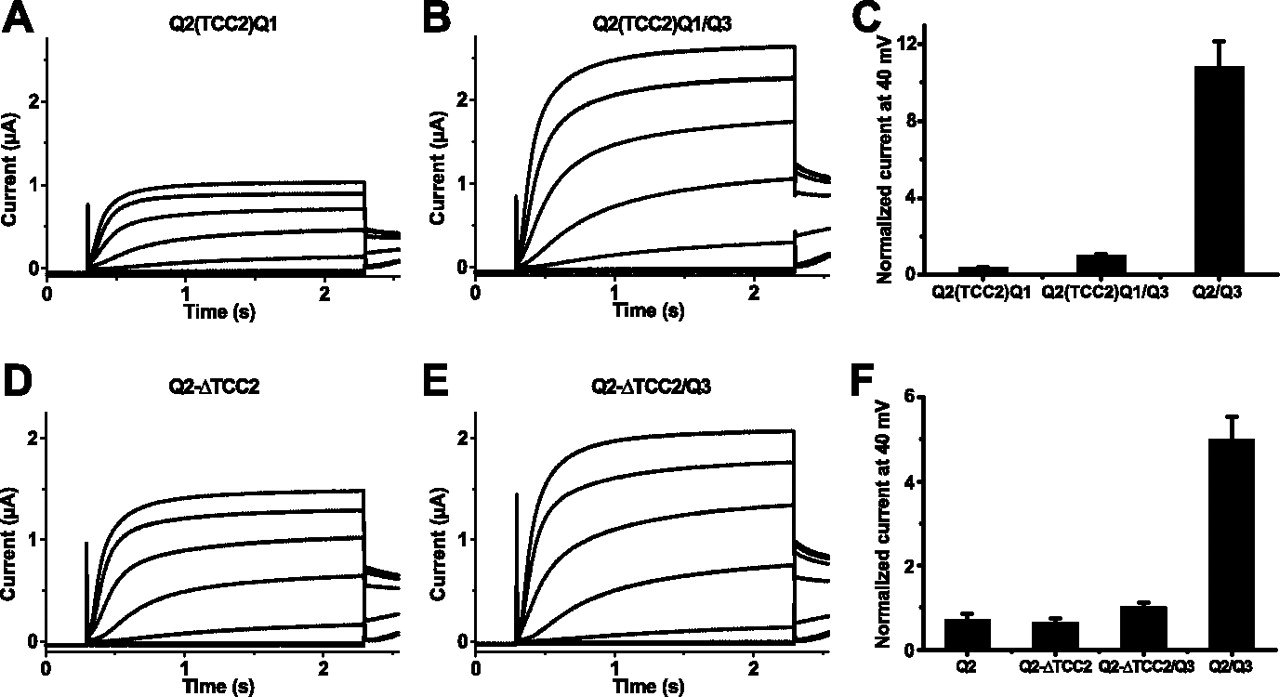
Effect of the KCNQ2(TCC2)Q1 chimera and of KCNQ2 constructs with individually deleted TCC domains. A, B, Representative current recordings from oocytes (co-)injected with cRNA(s) of KCNQ2(TCC2)Q1 (A) and KCNQ2(TCC2)Q1 plus KCNQ3 (B). C, Bar diagram of current levels recorded at the end of a 2-s-long test pulse to +40 mV from oocytes expressing KCNQ2(TCC2)Q1 (n = 32), KCNQ2(TCC2)Q1 plus KCNQ3 (n = 42), and KCNQ2 plus KCNQ3 (n = 42). Currents were normalized to the value obtained from (KCNQ2 plus KCNQ3)-expressing oocytes. D, E, Representative current recordings from oocytes (co-)injected with cRNA(s) of KCNQ2-ΔTCC2 (D) and KCNQ2-ΔTCC2 plus KCNQ3 (E). F, Bar diagram of current levels from oocytes (co-)expressing KCNQ2 (n = 19), the deletion construct KCNQ2-ΔTCC2 (n = 18), KCNQ2-ΔTCC2 plus KCNQ3 (n = 25), or KCNQ2 plus KCNQ3 (n = 22). Currents were normalized to the value obtained from oocytes coexpressing KCNQ2 plus KCNQ3.
To test this hypothesis and to further investigate the individual roles of TCC1 and TCC2 within the si domain in channel assembly and trafficking, we analyzed KCNQ2 deletion constructs, in which either the complete si domain (KCNQ2-Δsid), TCC1 (KCNQ2-ΔTCC1), or TCC2 (KCNQ2-ΔTCC2) were removed, respectively. Expression of KCNQ2-Δsid and KCNQ2-ΔTCC1 did not yield K+ currents (data not shown), suggesting that TCC1 of KCNQ2 is also essential for forming functional homomeric channels. In contrast, currents obtained from the deletion construct KCNQ2-ΔTCC2 (Fig. 4D) were very similar to those of wild-type KCNQ2 channels (Fig. 5B). Hence, the formation of KCNQ2 homomers does not depend on TCC2. Coexpression of either the KCNQ2-Δsid or the KCNQ2-ΔTCC1 with KCNQ3 again did not yield K+ currents (data not shown). Additionally, no significant increase in currents was observed after coexpression of KCNQ2-ΔTCC2 with KCNQ3 (Fig. 4E,F).

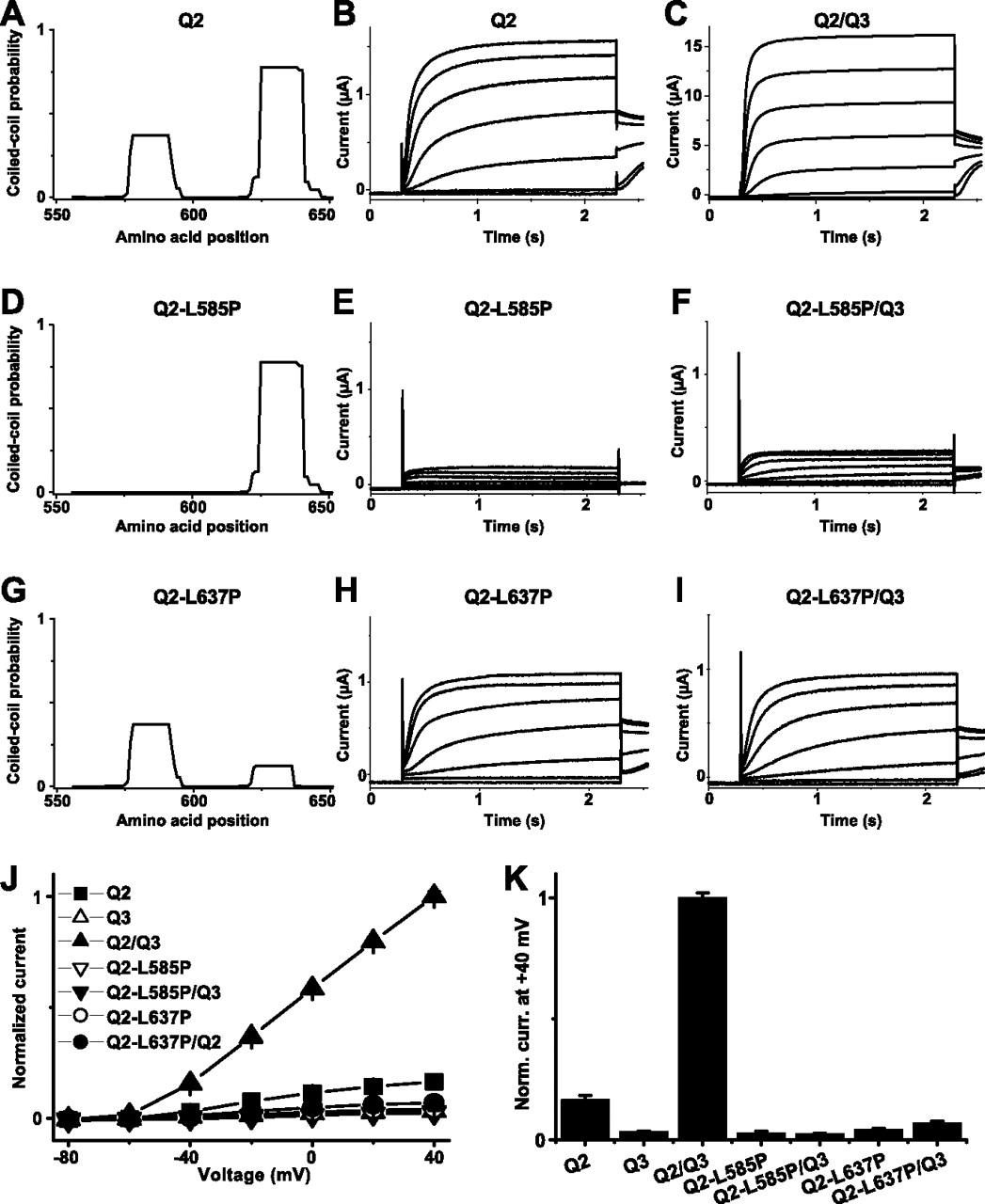
Effects of disrupting the coiled-coil probability within the TCC domains of KCNQ2. Coiled-coil probabilities within the si domain of KCNQ2 wild-type (A), KCNQ2-L585P (D), and KCNQ2-L637P (G), determined with the program Coils (version 2.2). Representative current recordings from oocytes injected with cRNAs of KCNQ2, KCNQ2-L585P, and KCNQ2-L637P are shown in B, E, and H and from coexpressions of these constructs with KCNQ3 in C, F, and I, respectively. J shows I/V curves, and K shows a bar diagram of current levels recorded at the end of a 2-s-long test pulse to +40 mV for KCNQ2 (n = 36), KCNQ3 (n = 42), KCNQ2 plus KCNQ3 (n = 45), KCNQ2-L585P (n = 38), KCNQ2-L585P plus KCNQ3 (n = 40), KCNQ2-L637P (n = 37), and KCNQ2-L637P plus KCNQ3 (n = 37) (co-)injection schemes. Data in J and K were normalized to KCNQ2 plus KCNQ3 currents.
Together, we conclude that TCC1 is necessary for assembly of both homomeric and heteromeric channels, whereas TCC2 determines the formation of heteromers but is dispensable for homomeric channel assembly.
Effects of disrupting the predicted coiled-coil structure of individual TCC domains of KCNQ2 and KCNQ3
To test the importance of coiled-coil-specific structural features on homomerization and heteromerization properties of KCNQ2 and KCNQ3, we inserted helix-breaking amino acids within the TCC domains of KCNQ2 and KCNQ3 (Fig. 1A) and probed for effects on the formation of functional homotetramers and heterotetramers. The program Coils version 2.2 [http://www.ch.embnet.org (Lupas et al., 1991)] was used to predict the coiled-coil probability within the si domains of either wild-type KCNQ2 (Fig. 5A) or KCNQ3 (Fig. 6A). Whereas the coiled-coil probabilities for TCC2 were rather high (0.75 for KCNQ2 and 0.6 for KCNQ3), it was only ∼0.4 for TCC1 of KCNQ2 and even lower for TCC1 of KCNQ3 (<0.1). Disruption of the coiled-coil structure of TCC1 of KCNQ2 by the L585P mutation (Fig. 5D) reduced currents nearly to background levels (Fig. 5E,J,K). Coexpressing KCNQ2-L585P with KCNQ3 did not enhance KCNQ3 currents (Fig. 5, compare F and K).

Effects of disrupting the coiled-coil probability within the TCC domains of KCNQ3. Coiled-coil probabilities within the si domain of KCNQ3 wild-type (A), KCNQ3-L564P (D), and KCNQ3-L636P (G), determined with the program Coils (version 2.2). Representative current recordings from oocytes injected with cRNAs of KCNQ3, KCNQ3-L564P, and KCNQ3-L636P are shown in B, E, and H and from coexpressions of these constructs with KCNQ2 in C, F, and I, respectively. J shows I/V curves, and K shows a bar diagram of current levels recorded at the end of a 2-s-long test pulse to +40 mV for KCNQ2 (n = 36), KCNQ3 (n = 42), KCNQ2 plus KCNQ3 (n = 45), KCNQ3-L564P (n = 24), KCNQ3-L564P plus KCNQ2 (n = 28), KCNQ3-L636P (n = 31), and KCNQ3-L636P plus KCNQ2 (n = 47) (co-)injection schemes, as indicated. Data in J and K were normalized to current levels obtained from oocytes expressing KCNQ2 plus KCNQ3.
In contrast, reducing the predicted coiled-coil probability of TCC2 from KCNQ2 by the L637P mutation (Fig. 5G) gave functional channels. Their currents were somewhat smaller than those of wild-type KCNQ2 (Fig. 5H,K). However, the KCNQ2-L637P mutant failed to enhance currents when being coexpressed with KCNQ3. These observations further strengthen the conclusion that an intact TCC1 structure is required for both homomeric and heteromeric channel formation, whereas structural integrity of TCC2 is essential for heteromerization.
Similar results were observed with equivalent mutations within the TCC domains of KCNQ3. Although the predicted coiled-coil probability within TCC1 of KCNQ3 is very low (Fig. 6A), complete disruption of the TCC1 coiled-coil probability by mutation L564P again abolished currents (Fig. 6E, compare with KCNQ3 wild-type currents in B). Furthermore, no functional interaction of the KCNQ3-L564P mutant with KCNQ2 was seen. Currents from this coexpression scheme reached KCNQ2 wild-type current amplitudes (Fig. 6F,K). When the coiled-coil probability of TCC2 from KCNQ3 was mostly disrupted by mutation L636P (Fig. 6G), again functional K+ channel currents were obtained (Fig. 6H) in accordance with results from KCNQ2. However, coexpression of the KCNQ3-L636P mutant with KCNQ2 still led to a robust enhancement of currents, which were comparable in amplitude with KCNQ2/KCNQ3 heteromeric channels. This indicates that in contrast to the TCC2 domain of KCNQ2, less rigorous structural requirements apply for the KCNQ3 TCC2 domain during KCNQ2/KCNQ3 heteromer formation.
The fact that the KCNQ1(TCC2)Q3 chimera was able to form heteromeric channels with KCNQ2, whereas KCNQ1 was not, raised the possibility that specific residues within the TCC2 domain mediate the subunit-specific assembly. We therefore generated further chimeras in which either only single helix turns within TCC2 were exchanged or the hydrophobic residues along the positions a and d alone or together with the residues at positions b, c, e, f, and g (Fig. 1B) were exchanged between KCNQ1 and KCNQ3 (see scheme of the investigated constructs in Table 1). None of the resultant subchimeras of KCNQ1(TCC2)Q3 led to a robust enhancement of currents in coexpression with KCNQ2 as seen for the KCNQ2/KCNQ1(TCC2)Q3 coexpression. Thus, we were unable to identify individual residues that are absolutely required for the formation of KCNQ2/KCNQ3 heteromers. These results, together with those from proline substitutions that disrupt the α-helical structure, suggest structural flexibility of the KCNQ3 TCC2 domain.
KCNQ1/KCNQ3 chimeras with single-helix turn exchanges or exchanges of certain hydrophobic or residues within the TCC2 region
During our survey of the KCNQ2 and KCNQ3 protein sequences using the coils program [http://www.ch.embnet.org (Lupas et al., 1991)], we also identified a region with a high coiled-coil probability within the extracellular turret domain (segment termed “TD”) of KCNQ3 (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). After exchange of this domain by the corresponding sequence of KCNQ1, a large increase in current amplitudes of the resulting chimera [KCNQ3(TD;Q1)] compared with KCNQ3 was observed. It was shown that this is primary a result of a boosted expression, most likely because an N-glycosylation site is present in the TD domain from KCNQ1 (Schenzer et al., 2005). Interestingly, analysis with the coils program revealed that the KCNQ3(TD;Q1) chimera does not exhibit a significant coiled-coil probability within the extracellular turret domain (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Therefore, we tested the homomeric and heteromeric assembly properties of the KCNQ3(TD;Q1) chimera in coexpression experiments with dominant-negative constructs of KCNQ1, KCNQ2, and KCNQ3. Whereas KCNQ1-G314S reduced KCNQ3(TD;Q1) currents by only 5%, KCNQ2-G279S and KCNQ3-G318S exerted strong dominant-negative effects and suppressed currents to background levels (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material) [all current values were significantly reduced, except for the KCNQ1-G314S/KCNQ3(TD;Q1) coexpression, with p < 0.005 from Student's t test]. These results suggest that the turret domain is not involved in the assembly of homomeric and heteromeric KCNQ channels.
Analysis of the interaction of the two TCC domains within one KCNQ subunit
We next addressed the question of whether the two adjacent TCC domains within the KCNQ2 si domain could interact with each other, as might be expected for tetramerizing coiled-coil sequences. Therefore, the KCNQ2 sequence was split between the TCC domains (Figs. 1A, 7A), resulting in constructs KCNQ2-TCC1-X (comprising the KCNQ2 transmembrane domains including TCC1) and TCC2-CT-KCNQ2 (a soluble C-terminal fragment including TCC2), which were tested for functional channel formation after coexpression in oocytes. KCNQ2 was chosen for these experiments because of its robust expression. Neither the expression of truncated polypeptides KCNQ2-TCC1-X or TCC2-CT-KCNQ2 alone nor coexpression of KCNQ2-TCC1-X with TCC2-CT-KCNQ2 yielded measurable K+ currents (data not shown).

Effect of different KCNQ2 channel fragments on KCNQ3 currents. A, Schematic illustration of the KCNQ2 constructs. The N terminus and the transmembrane region of KCNQ2 are shown as light gray boxes, and the C-terminus is shown as dark gray boxes. The TCC domains are indicated in black. B, Comparison of current levels after 2 s at +40 mV recorded from oocytes (co-)injected with KCNQ3 (n = 55), KCNQ2 plus KCNQ3 (n = 27), KCNQ2-TCC1-X plus KCNQ3 (n = 28), KCNQ2-TCC1-X plus TCC2-CT-KCNQ2 plus KCNQ3 (n = 41), TCC2-CT-KCNQ2 plus KCNQ3 (n = 74), and dCT-KCNQ2 plus KCNQ3 (n = 26) cRNA(s), respectively. Data were normalized to TCC2-CT-KCNQ2 plus KCNQ3 currents. C, Representative current recording from an oocyte coinjected with cRNAs of KCNQ2-TCC1-X and KCNQ3 measured 5 d after injection. D, Surface expression determined from luminescence measurements of oocytes expressing either the HA-tagged KCNQ3 (n = 47) alone or coinjected with TCC2-CT-KCNQ2 (n = 47) and dCT-KCNQ2 (n = 47). Data were normalized to the value for the KCNQ3(HA) plus TCC2-CT-KCNQ2 coinjection. Oocytes expressing nontagged KCNQ3 (n = 44) served as a background control. E, TEA sensitivity of KCNQ2, KCNQ3, KCNQ2 plus KCNQ3, TCC2-CT-KCNQ2 plus KCNQ3, and dCT-KCNQ2 plus KCNQ3 (n = 10 for each data set). F, Western blot analysis probing expression of KCNQ2 and N-terminal KCNQ2 fragments performed on pooled oocytes that had been used for measurements of TEA sensitivity as shown in E. Antibody KCNQ2(N-19) directed against an N-terminal epitope of KCNQ2 was used for detection. The arrowhead denotes the molecular weight of KCNQ2 proteins, and the asterisk represents the weight of the truncated KCNQ2-TCC1-X proteins. Right, Molecular weight marker. G, Comparison of current levels after 2 s at +40 mV recorded from oocytes (co-)injected with KCNQ3 (n = 23), KCNQ2 (n = 38), KCNQ2 plus KCNQ3 (n = 37), KCNQ2(NT-TCC2) plus KCNQ3 (n = 44), and KCNQ2(NT-TCC2;ΔTCC1) plus KCNQ3 (n = 29) cRNA(s), respectively. Data were normalized to the mean of KCNQ2(NT-TCC2) plus KCNQ3 currents. All values are means ± SE
Next, we tested for effects of the fragments KCNQ2-TCC1-X and TCC2-CT-KCNQ2 in coexpression with KCNQ3. Currents measured after KCNQ2-TCC1-X plus KCNQ3 coexpression could be differentiated from those of KCNQ3 (Fig. 7C), but no enhancement compared with KCNQ3 current levels was detected (Fig. 7B). In contrast, functional channels were obtained in a KCNQ2-TCC1-X plus TCC2-CT-KCNQ2 plus KCNQ3 coexpression scheme, reaching ∼50% of the KCNQ2/KCNQ3 current level. Strikingly, coexpression of KCNQ3 with only the cytosolic fragment TCC2-CT-KCNQ2 led to an approximately fourfold increase in currents compared with KCNQ3 alone (Fig. 7B). To test whether this effect depends on the presence of TCC2, we generated an additional C-terminal fragment (termed dCT-KCNQ2), which comprises only the distal KCNQ2 C terminus following TCC2 (Figs. 1A, 7A). Coexpression of this construct did not enhance KCNQ3 currents (Fig. 7B), suggesting that the TCC2 domain of KCNQ2 augments KCNQ3 currents even when present within a soluble protein fragment.
To test whether this current increase depends on an increased surface expression, we coexpressed the TCC2-CT-KCNQ2 or dCT-KCNQ2 constructs together with an extracellular epitope-tagged KCNQ3 construct. Figure 7D shows that the C-terminal fragment carrying the second TCC domain (TCC2-CT-KCNQ2) stimulated the surface expression of epitope-tagged KCNQ3, whereas the fragment lacking TCC2 (dCT-KCNQ2) had no effect.
At this point, we wanted to demonstrate the exact nature of channel pores being created by coexpressing KCNQ3 and KCNQ2 fragments and therefore intended to use their TEA sensitivity for identification, because IC50 values vary >100-fold between KCNQ2 and KCNQ3. In accordance with Hadley et al., (2000), we found that the TEA sensitivity of KCNQ2 is high, that of KCNQ3 is low, and that of KCNQ2/KCNQ3 is intermediate (Fig. 7E). As expected, the TEA sensitivity of channels formed after either KCNQ3/KCNQ2-TCC1-X/TCC2-CT-KCNQ2 or KCNQ3/KCNQ2-TCC1-X coexpression was comparable with the intermediate sensitivity of KCNQ2/KCNQ3 channels (data not shown). Remarkably, the TEA sensitivity of channels formed by KCNQ3 and the cytoplasmatic KCNQ2 fragment (TCC2-CT-KCNQ2) was also comparable with that of KCNQ2/KCNQ3 heteromers (Fig. 7E). In contrast, the dCT-KCNQ2 construct did not influence the TEA sensitivity of KCNQ3 (Fig. 7E). To make sure that no KCNQ2 pore-containing subunits were present in this coexpression situation, oocytes used for the TEA-sensitivity measurements were analyzed by Western blotting using an antibody that recognizes the N terminus of KCNQ2. The resulting Figure 7F shows that no contaminating KCNQ2 pore-forming subunits were present in oocytes expressing KCNQ3 plus TCC2-CT-KCNQ2. Thus, although the exact nature of the channel pores formed after KCNQ3 plus TCC2-CT-KCNQ2 coexpression cannot be inferred from TEA sensitivity, the results suggest that the increase in currents is most likely attributable to a more efficient plasma membrane targeting of KCNQ3 channels when TCC2-CT-KCNQ2 is present. Furthermore, it appears that the second TCC domain of KCNQ2 might influence the pore properties of KCNQ3.
Position-independent effect of the TCC2 domain
Because the current-enhancing effect of the KCNQ2 TCC2 domain could essentially be observed with split KCNQ2 channels or the cytosolic TCC2-CT-KCNQ2 fragment alone, we asked whether TCC2 could exert a current-stimulating effect when placed in a different position within the KCNQ2 backbone. The KCNQ2 TCC2 fragment was fused to the N terminus of a KCNQ2 channel that was truncated after TCC1 [termed KCNQ2(NT-TCC2)] (Figs. 1A, 7A). This construct did not yield measurable K+ currents when expressed alone but increased KCNQ3 currents in coexpression experiments nearly as efficiently as KCNQ2 wild-type (Fig. 7G). Next, we deleted the TCC1 domain within the KCNQ2(NT-TCC2) construct and expressed the resultant chimera KCNQ2(NT-TCC2;ΔTCC1) in oocytes. No functional K+ channels were observed, and the current-stimulating effect on KCNQ3 was absent (Fig. 7G). In line with the results obtained from the coexpression of KCNQ3 with the C-terminal fragment TCC2-CT-KCNQ2, these experiments indicate that the current-stimulating effect of the KCNQ2 TCC2 domain does not depend on its location behind TCC1 in the primary sequence.
Discussion
KCNQ channels can either form homotetramers or heteromers composed of different α-subunits. Previous work has already demonstrated that the subunit-specific assembly of KCNQ subunits is mediated by the C-terminal si domain (Maljevic et al., 2003; Schwake et al., 2003). Structural motifs with a high probability to form TCC sequences have been identified within the si domain of KCNQ C termini (Jenke et al., 2003). Unlike most other cases, in which the TCC domains are organized as a single stretch of periodically repeated hydrophobic residues, the TCC motif of KCNQ channels is bipartite with a variable linker region in between (Jenke et al., 2003). The aim of this study was to elucidate the role of these TCC domains in KCNQ channel assembly.
The present work demonstrates that insertion of TCC2 from KCNQ3 into KCNQ1 is sufficient to transfer assembly properties. The KCNQ1(TCC2)Q3 chimera interacted nearly as efficiently with KCNQ2 as wild-type KCNQ3, as inferred from the following: (1) the strong current enhancement after KCNQ1(TCC2)Q3/KCNQ2 coexpression; (2) a dominant-negative effect of the KCNQ1(TCC2)Q3-G314S pore mutant on KCNQ2 currents; (3) stimulation of surface expression of epitope-tagged KCNQ2; and (4) coimmunoprecipitation of the KCNQ1(TCC2)Q3 chimera with KCNQ2. Moreover, TCC2 of KCNQ2 is required for heteromerization with KCNQ3, because the KCNQ2(TCC2)Q1 chimera and a KCNQ2 construct with a deletion of TCC2, although both functional as homotetramers, did not increase currents after coexpression with KCNQ3. These experiments indicate that the TCC2 domains from KCNQ2 and KCNQ3 are needed for an efficient transport of the heteromeric channel complex to the plasma membrane. Several reasons are conceivable. Either an efficient forward trafficking motif is formed or exposed by the interaction of the two TCC2 domains, or an endoplasmatic reticulum retention motif is masked by this interaction. Both alternatives would explain that only assembled KCNQ2/KCNQ3 heteromers are efficiently transported to the plasma membrane.
In contrast, the function of the first TCC domain of KCNQ2 and KCNQ3 remains less well understood. The KCNQ1(TCC1)Q3 chimera did not significantly stimulate KCNQ2 currents and surface expression, and no current suppression consistent with a dominant-negative effect was observed. However, in contrast to KCNQ1, it could be coimmunoprecipitated with KCNQ2. This indicates a specific interaction with KCNQ2 mediated by the TCC1 domain of KCNQ3. This is supported by functional channels obtained for the coexpression of a truncated KCNQ2 channel (KCNQ2-TCC1-X) with KCNQ3. Additionally, for the deletion construct KCNQ2-ΔTCC1, no K+ currents could be observed and no current enhancement after coexpression with KCNQ3 was seen. These results suggest that the TCC1 domain is necessary for the formation and/or function of homomeric KCNQ2 and heteromeric KCNQ2/KCNQ3 channels. Thus far, it cannot be distinguished, whether deletion of TCC1 impairs the assembly to homomers and heteromers, or whether channels that are not able to gate open might be formed. However, the formation of functional heteromeric channels by coexpression of the truncated KCNQ2-TCC1-X construct together with KCNQ3 favors the idea that the TCC1 domains of KCNQ2 and KCNQ3 are needed for assembly.
To further investigate the importance of TCC-specific structural features on homomerization and heteromerization properties of KCNQ2 and KCNQ3, we probed for the effects of helix-breaking amino acids within the TCC domains. Leu-to-Pro substitutions in TCC1 of KCNQ2 and KCNQ3 disrupted homomeric and heteromeric channel formation. This implies that the structural integrity of the TCC1 domain is a key requirement for both homomeric and heteromeric KCNQ channel assembly. In contrast, Leu-to-Pro substitutions within TCC2 yielded functional homomeric channels with properties similar to wild-type.
Whereas coexpression of the KCNQ3-L636P mutant with KCNQ2 led to current augmentation as seen with KCNQ2/KCNQ3 coexpression, coinjection of KCNQ3 with the KCNQ2-L637P mutant did not increase currents. These results imply that the structural integrity of TCC2 of KCNQ2 is necessary for KCNQ2/KCNQ3 heteromerization, whereas the putative coiled-coil structure of TCC2 of KCNQ3 is less important in this process. Because the KCNQ3-L636P mutant together with KCNQ2 still led to a robust current enhancement, the structural requirements of the KCNQ3 TCC2 domain for heteromerization with KCNQ2 are less stringent than that for TCC2 of KCNQ2.
This conclusion is in line with results obtained from our attempts to identify TCC domain-specific features within TCC2 of KCNQ3, which might underlie heteromeric channel assembly. Because coiled-coil structures are characterized by a heptad amino acid repeat (with positions designated a–g), one would assume that the swapping of amino acids at equivalent positions between KCNQ1 and KCNQ3 subunits (especially the hydrophobic residues at position a and d) would suffice to exchange subunit-specific assembly properties between KCNQ3 to KCNQ1. However, all subchimeras failed to stimulate KCNQ2 currents in coexpression experiments. These results, together with the analysis of the KCNQ3-L636P mutant, support the idea that structural features beyond the probability to form coiled-coil structures underlie the important role of the TCC2 domain from KCNQ3 in assembly and/or cell surface trafficking of heteromeric channels.
Given the pivotal role of coiled-coil domains in protein–protein interactions, we wanted to test whether the first and second TCC domain within one KCNQ subunit interacted with each other by taking advantage of a “split channel” approach, in which functional channels are formed from complementary fragments. Such a strategy was successful in case of voltage-gated chloride channels of the CLC family. When CLC-1 was truncated after the first of the two cytoplasmatic cystathionine β-synthase (CBS) domains, the resulting channel was not functional. Function could be restored after coexpression of the C-terminal part comprising the second CBS domain (Schmidt-Rose and Jentsch, 1997; Estevez et al., 2004). In contrast, we were unable to reconstitute functional KCNQ2 channels from fragments KCNQ2-TCC1-X and TCC2-CT-KCNQ2, not even when TCC2-CT-KCNQ2 cRNA was coinjected in large molecular excess. This suggests that the first and second TCC domains might not significantly interact within one subunit.
Surprisingly, coexpression of the TCC2-CT-KCNQ2 fragment with KCNQ3 led to a large current augmentation that was strictly dependent on the presence of TCC2. This increase in current was primarily a result of an increase in surface expression of KCNQ3, suggesting that the second TCC domains from KCNQ2 and KCNQ3 facilitate an efficient transfer of heteromeric KCNQ2/KCNQ3 channels to the cell surface. Importantly, the KCNQ2(NT-TCC2) construct carrying TCC2 at the N terminus also increased currents in coexpression with KCNQ3. This further suggests that the TCC2 domain of KCNQ2 does not need to be physically linked in sequence after TCC1. The intracellular presence of TCC2, either as soluble protein fragment or tethered to the membrane-embedded parts of the channel protein, suffices to enhance currents in a heteromeric expression scheme. Therefore, we hypothesize that the specific requirement for TCC2 from KCNQ2 may be to either mask a retention motif or that it helps to expose a plasma membrane-targeting motif present on KCNQ3.
Another surprising finding is the modulation of the TEA sensitivity of KCNQ3 by the TCC2 domain from KCNQ2. According to Hadley et al., (2000), the high TEA sensitivity of KCNQ2 depends on a tyrosine in the pore loop. In KCNQ3, which displays a lower TEA sensitivity, this tyrosine is replaced by threonine. However, although KCNQ1 and KCNQ4 lack this tyrosine, both are more TEA sensitive than KCNQ3 (Hadley et al., 2000), suggesting that the TEA block depends on additional structures. Our results favor this view and suggest that the TCC2 domain of KCNQ2 might modulate pore properties of KCNQ3.
It is well established, that the N-terminal T1 domain of Kv channels plays an important role in subunit tetramerization and subfamily-specific assembly (Li et al., 1992; Shen et al., 1993; Bixby et al., 1999). In addition, there is evidence that this cytoplasmatic domain can influence pore properties (Cushman et al., 2000; Minor et al., 2000; Kurata et al., 2002). Our TEA inhibition data indicate that this could also apply for the TCC2 domain of KCNQ2. However, the reason for this effect is obscure and opens up interesting perspectives for future investigations.
Our results on KCNQ2/KCNQ3 heteromerization agree with the established concepts of KCNQ1 subunit assembly (Schmitt et al., 2000; Kanki et al., 2004). Whereas Schmitt et al., (2000) suggested that amino acids 590–620 (essentially comprising TCC2) in KCNQ1 serve as a tetramerization domain, Kanki et al., (2004) claimed that this region is important for cell surface trafficking of assembled KCNQ1 channels. However, both reports did not investigate the role of TCC1 on KCNQ1 tetramerization, and additional experiments are needed to address this question. KCNQ1 might be unique in this context, because it does not interact with other KCNQ α subunits, so that KCNQ1 TCC structures might have to meet particular requirements.
Footnotes
-
This work was supported by Deutsche Forschungsgemeinschaft Grants SCHW 866/3 (M.S.) and FR 1276/1 (T.F.), the Max-Planck-Gesellschaft zur Förderung der Wissenschaften, and a fellowship from the Novartis Foundation for Therapeutic Research (M.S.). T.F. gratefully acknowledges current support from the German Federal Ministry of Education and Research program “Unternehmen Region–Innovations Offensive Neue Länder, Zentren für Innovationskompetenz” and the Technical University of Ilmenau/Thuringia, Germany. We thank Katharina Stiebeling for excellent technical assistance, and we are grateful to Ernst Bamberg for personal and scientific support.
- Correspondence should be addressed to either of the following: Michael Schwake, Institut für Biochemie, Christian-Albrechts-Universität zu Kiel, Olshausenstrasse 40, D-24098 Kiel, Germany, Email: mschwake{at}biochem.uni-kiel.de; or Thomas Friedrich, Center for Innovative Competence MacroNano, Technical University of Ilmenau, Center for Micro- and Nanotechnologies, Gustav-Kirchhoff-Strasse 7, D-98693 Ilmenau, Germany, Thomas.Friedrich{at}mpibp-frankfurt.mpg.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}