

Abstract
Secreted Semaphorin3A (Sema3A) proteins are known to act as diffusible and repellant axonal guidance cues during nervous system development. A receptor complex consisting of a Neuropilin and a Plexin-A mediates their effects. Plexin-A signal transduction has remained poorly defined despite the documented involvement of collapsin response mediator protein and molecule interacting with CasL proteins (MICALs) as mediators of Plexin-A activation. Here, we defined a domain of Plexin-A1 required for Sema3A signaling in a reconstituted environment and then searched for proteins interacting with this domain. RanBPM is shown to physically interact with Plexin-A1, and the RanBPM/Plexin complex is regulated by MICAL expression. Overexpression of RanBPM cooperates with PlexinA1 to reduce non-neuronal cell spreading and strongly inhibit axonal outgrowth in vitro and in vivo. A truncated RanBPM protein blocks Sema3A responsiveness in non-neuronal and neuronal cells. Suppression of RanBPM expression reduces Sema3A responsiveness. Thus, RanBPM is a mediator of Sema3A signaling through Plexin-A. RanBPM has the potential to link Plexin-A receptors to retrograde transport and microtubule function in axonal guidance.
Introduction
Molecular cues guide axons to synaptic target areas during nervous system development. Semaphorins, netrins, ephrins, slits, and repulsive guidance molecule have all been identified as axon guidance molecules (Huber et al., 2003). The secreted class three semaphorins (Sema3s) are prototypic axon repellant proteins, originally defined as growth cone collapsing agents (Raper, 2000). It is now clear that Sema3 proteins function through receptor complexes formed from one of two Neuropilin (NRP) proteins and one of four Plexin-A (PlexA) proteins (He and Tessier-Lavigne, 1997; Kolodkin et al., 1997; Takahashi et al., 1999; Tamagnone et al., 1999). Neuropilins serve as the primary ligand binding sites and Plexins as the signal transducing components. Neuropilins also have roles as receptors for vascular endothelial growth factor isoforms, and Plexins can function as direct single-subunit receptors for some semaphorins (Gluzman-Poltorak et al., 2000; Gu et al., 2003; Suto et al., 2005). After binding to NRP/PlexA complexes, Sema3A induces collapse of the growth cone via a dramatic rearrangement of filamentous actin (F-actin) and endocytosis of the growth cone plasma membrane (Fan et al., 1993; Fournier et al., 2000; Jurney et al., 2002). Sema3A/NRP/PlexA signaling has also been shown to increase axoplasmic transport (Goshima et al., 1997, 1999; Li et al., 2004) and is mediated by local protein synthesis (Campbell and Holt, 2001; Brittis et al., 2002; van Horck et al., 2004).
Plexin signal transduction is less well defined than ligand specificity. PlexB receptors bind to transmembrane semaphorins and share some sequence homology with PlexA receptors. The Plexin-B cytoplasmic domain possesses a PDZ (postsynaptic density-95/Discs large/zona occludens-1)-binding tail that interacts with the Rho-guanine nucleotide exchange factor (GEF)/PDZ protein family including leukemia-associated Rho GEF (Driessens et al., 2002; Swiercz et al., 2002). In addition, the Plexin-B proteins can form a complex with R-Ras and Rnd1 to modulate G-protein activity in the cell (Oinuma et al., 2003). The Rho GTPases Rac1, Rnd1, and RhoD have been shown to signal downstream of PlexA1 (Jin and Strittmatter, 1997; Zanata et al., 2002; Turner et al., 2004). In addition, fyn, Cdk5, LIM-domain containing kinase, and glycogen synthase kinase-3β kinase activity is required for responses to Sema3A (Aizawa et al., 2001; Eickholt et al., 2002; Sasaki et al., 2002). The flavin monooxygenase, molecule interacting with CasL (MICAL), binds to Drosophila PlexA and is required for normal axon pathfinding. Blocking MICAL enzyme activity disrupts Sema3A signaling in flies and mammals (Terman et al., 2002; Pasterkamp et al., 2006). The collapsin response mediator protein (CRMP) family form a complex with PlexA proteins and also are required for growth cone responses to Sema3A (Goshima et al., 1995; Deo et al., 2004). The mechanism by which these proteins and signaling pathways are regulated by Plexin and Sema3 remain elusive
In defining the role of PlexA in Sema3A signaling, we developed a reconstituted non-neuronal cell assay. COS-7 cells expressing NRP1/PlexA1 proteins exhibit cell contraction in response to soluble Sema3A ligand. Furthermore, we found that PlexA1 is autoinhibited in the basal state by the Sema domain (Takahashi and Strittmatter, 2001). Deletion of the PlexA1 extracellular domain (PlexA1Δect) allows the transmembrane and cytoplasmic domains to exert constitutive activity in neuronal and non-neuronal cells leading to cell contraction or reduced axonal outgrowth (Takahashi and Strittmatter, 2001). In the current study, we analyzed the mechanism of constitutive activation of PlexA1Δect through deletion studies. After defining a segment of PlexA1 as being essential for cell contraction, this domain was used in a search for interacting proteins. We identified RanBPM as a protein that interacts with cytoplasmic PlexA. RanBPM participates in Sema3A-induced COS-7 cell contraction and axonal growth inhibition. Thus, PlexA signal transduction includes another partner with potential links to microtubule dynamics and retrograde transport and signaling.
Materials and Methods
Expression plasmids.
The plasmids pSecTag2–PlexA1FL, pSecTag2–PlexA1ΔECT, or pcDNA3.1–PlexA2 were described previously (Takahashi and Strittmatter, 2001; Deo et al., 2004). To express truncation mutants of PlexA1 as myc-tagged protein, the coding region of amino acids 1238–1814, 1238–1657, 1238–1600, 1238–1550, 1238–1502, 1238–1412, or 1238–1294 were amplified by PCR for PlexA1-T1 through -T7 mutants, respectively. The amplified DNA fragment was subcloned into BamHI and EcoRI site in pSecTag2 vector (Invitrogen, San Diego, CA). To obtain deletion mutants of PlexA1, the coding region of 1412–1894, 1502–1894, or 1657–1894 amino acids were inserted at the corresponding site to C termini of PlexA1-T7, -T6, -T5, or -T2 mutant for PlexA1-DC1a, DC1b, DNC1, or DC2 mutant, respectively. The plasmid pEGFP–RanBPM was a kind gift from Dr. T. Nishimoto (Kyushu University, Fukuoka, Japan) (Nishitani et al., 2001). The coding region of full-length (FL), 1–280 (N), 207–492 (I), 280–729 (C), or 1–333 (N3) amino acids of RanBPM were subcloned into p3xFLAG–CMV7.1 vector (Sigma, St. Louis, MO) for transient expression in COS-7 cells. The coding region of RanBPM-N3 or green fluorescent protein (GFP) was subcloned into pHSVPrPUC vector for transient expression in chick DRG explant using herpes simplex virus (HSV) (Takahashi et al., 1999). The plasmid for mammalian Myc-MICAL1 was a generous gift from Dr. Alex Kolodkin (Johns Hopkins University, Baltimore, MD).
Yeast two-hybrid screening.
Two hybrid screening was performed using nonconserved region 1 (NC1) of PlexA1 as a bait. The coding region of NC1 (amino acids 1502–1657) was amplified by PCR and subcloned into pGBKT7 vector. The screening was performed with Matchmaker pretransformed human fetal brain library according to the manufacture (Clontech, Mountain View, CA).
COS-7 cell transfection, immunoprecipitation, and assessment of cellular morphology.
COS-7 cells were maintained in DMEM supplemented with 10% fetal bovine serum. Cells were transfected with expression vectors for myc–PlexinAs, FLAG–RanBPMs, or Myc–MICAL1 by Fugene6 (Roche Diagnostics, Indianapolis, IN). After 24 h of transfection, cells were lysed with buffer containing 50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT, 1% Triton X-100, Complete protease inhibitor (Roche Diagnostics), and centrifuged at 16,000 × g to obtain supernatant. Anti-myc antibody-conjugated beads (Santa Cruz Biotechnology, Santa Cruz, CA) were added to the supernatant and rotated overnight at 4°C. After washing the beads three times with wash buffer (50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT, 1% Triton X-100), the associated proteins were eluted with 1× SDS-PAGE sample buffer. The eluted proteins were detected by immunoblotting using anti-myc (Santa Cruz Biotechnology), anti-FLAG (Sigma), or anti-V5 (Invitrogen) antibody and anti-mouse secondary antibody.
For assessment of COS-7 morphology, transfected cells were fixed with 3.7% formaldehyde/PBS and permeabilized with 0.2% Triton X-100/PBS. To analyze cell size, the shapes of cells were visualized with rhodamine–phalloidin (Invitrogen) and FITC-conjugated anti-myc antibody. For Sema3A contraction assay, cells were transfected in six-well plates and replated after 12 h to 24-well plates. After 24 h of growth, conditioned media were added to each well at the indicated concentrations, and cells were incubated for 1 h. They were then washed three times with ice-cold HEPES-buffered HBSS with 0.1% BSA and fixed with 3.7% formaldehyde in 20 mm HEPES/150 mm NaCl. Cells were then visualized for bound activator protein (AP) using the nitroblue–tetrazolium–chloride/5-bromo-4-chlor-indolyl-phosphate alkaline phosphatase substrates. Cell images were captured with fluorescence or transmitted light microscope (Olympus, Melville, NY) equipped with a cooled CCD camera (Kodak, Rochester, NY) and cell area was quantified using Scion (Frederick, MD) image analysis software. Concentration of AP–Sema3A or AP–Sema3F was determined based on the AP activity of the conditioned media using the p-mitrophenyl phosphate assay (Vector Laboratories, Burlingame, CA).
DRG culture and assessment of axonal outgrowth.
DRGs were dissected from embryonic day 7 (E7) chicken embryos and dissociated with 0.25% trypsin. Dissociated neurons were then nucleofected with expression vector using the chick DRG neuron nucleofector kit and program G-13 or O-03 (Amaxa, Cologne, Germany) as per manufacturer’s protocol. Nucleofected DRG neurons were preplated on a plastic tissue culture dish for 90 min and then plated on glass slides coated with poly-d-lysine and laminin. After 24 h of incubation, cells were fixed with 3.7% formaldehyde, 20% sucrose/PBS and permeabilized with 0.2% Triton X-100/PBS. Cells were visualized with rhodamine–phalloidin (Invitrogen) for neurite outgrowth or used for apoptotic assay with In Situ Cell Death Detection kit, TMR-red (Roche Diagnostics).
For neurite outgrowth assays, dissociated DRG neurons were incubated with recombinant HSV on uncoated plastic tissue culture plates for 12–15 h or nucleofected with custom small interfering RNA (siRNA) designed against chick RanBPM (Qiagen, Hilden, Germany) or control (scrambled sequence with the same nucleotides) and preplated on uncoated plates overnight. Cells were then replated on poly-d-lysine and laminin-coated 96-well plates and grown 2 h. Conditioned media were added to each well, and cells were grown for an additional 5 h before being fixed as described above. Neurites were visualized with anti-βIII tubulin immunofluorescence, and outgrowth was analyzed using an Image Express system (Molecular Devices, Union City, CA).
The culture and HSV infection of chick DRG explants was described previously (Takahashi et al., 1998). Briefly, dissected DRG explants were infected with recombinant virus in growth media (10% fetal bovine serum, 1% penicillin, 1% streptomycin, 1% l-glutamine, 100 ng/ml NGF/F12 media) for 1–2 h. After washing DRG with fresh media, DRGs were placed into collagen matrix (12% F12 media, 45% OptiMEM, 18% DMEM, 1% penicillin, 1% streptomycin, 1% l-glutamine, 100 ng/ml NGF, 25% collagen) with human embryonic kidney (HEK) 293T cells. Clumps of HEK293T cells transiently expressing Sema3A were made by culturing the cells for 36 h as hanging drops. After the collagen matrix hardened, growth media were added and cultures were allowed to grow for 24 h. The cultures were fixed with 3.7% formaldehyde/PBS and permeabilized with 0.2% Triton X-100/PBS. GFP- or RanBPM-N3-expressing neurites were visualized with anti-GFP or anti-myc antibody and AP-conjugated secondary antibody. The average length of proximal or distal extension of neurite was measured (Deo et al., 2004).
Electroporation in ovo.
Surgical procedures for the micromanipulation of chick embryos and electroporation were adapted from published protocols (Momose et al., 1999; Nakamura and Funahashi, 2001). A window was cut into the shell of Hamburger and Hamilton (H&H) stage 15–18 chick eggs, and DNA solution containing 3 mg/ml of either pEGFP-C1 (Clontech) or pEGFP-RanBPM with 0.04% trypan blue was injected into the neural tube of the embryo. Gold-plated electrodes were placed on either side of the neural tube at the lumbar level, and a set of five 50 ms square pulses (60 V) was delivered using a BTX ECM-830 electroporation system. The window was taped shut and the eggs were returned to the incubator and allowed to develop until E7. Embryos were then dissected from the egg, fixed at 4°C for 24–48 h with 4% paraformaldehyde/PBS, and 100 μm transverse slices were made using a vibratome. Slices were permeabilized with 0.1% Triton X-100/PBS and immunostained with monoclonal anti-GFP (B-2; Santa Cruz Biotechnology) and polyclonal anti-neurofilament-200 (Sigma) and visualized with Alexa-488 anti-mouse or Alexa-568 anti-rabbit secondary antibodies (Invitrogen).
Results
In previous NRP/PlexA expression studies of COS-7 responsiveness to various Sema3 proteins, we found that PlexA1 and PlexA2, but not PlexA3, mediated Sema3A signals from NRP1 (Takahashi et al., 1999). We have extended these studies to include PlexA4 and Sema3F (Fig. 1) with the goal of comparing specificity in this reconstituted system to results reported from recent gene targeting in mice (Suto et al., 2005; Yaron et al., 2005). Sema3A/NRP1 can cause contraction of COS-7 cells coexpressing PlexA1, PlexA2, and PlexA4 at ligand concentrations >1 nm (Fig. 1A,B). The contraction of NRP1/PlexA3-expressing cells did not significantly differ from that of cells expressing NRP1 alone at all doses of AP–Sema3A tested. Several independent clones of PlexA3 were tested in this assay (data not shown). Surprisingly, NRP1/PlexA4-expressing cells were much more sensitive to Sema3A, because cell contraction was significantly increased at Sema3A concentrations of 0.05 and 0.26 nm (Fig. 1B). These findings match with the observation that PlexA4, to a much greater extent than PlexA3, mediates Sema3A action within DRG neurons (Suto et al., 2005). The activity of endogenous PlexA1 and PlexA2 is irrelevant for embryonic DRG neurons because these proteins are expressed at significantly lower levels in these cells (Murakami et al., 2001). The AP–Sema3A-conditioned media were able to collapse E7 chick DRG growth cones in a dose-dependent manner (Fig. 1C). The highest dose of AP–Sema3A (6.6 nm) used in the contraction assay was twice the amount sufficient to cause significant growth cone collapse (3.3 nm). These findings support the fidelity of reconstituted COS-7 cell contraction method for assessing PlexA function.

Sema3A/PlexA specificity in COS-7 contraction assay. A, COS-7 cells were transfected with plasmids encoding the indicated PlexinA1–4 and NRP1 proteins and then exposed for 60 min at 37° to 1 nm AP–Sema3A or 1 nm AP–Sema3F, as indicated. After fixation, receptor-expressing cells were visualized by staining for bound AP. Cell expressing NRP1 and PlexA1, A2, and A4 contracted in response to AP–Sema3A but not AP–Sema3F. PlexA3 failed to mediate AP–Sema3A contraction. B, The cell area of AP-bound COS-7 cells treated with fivefold increasing concentrations of AP–Sema3A (0.05, 0.26, 1.32, or 6.60 nm) or 1 nm AP–Sema3F was measured, and the percentage of contracted cells (area ≤ 1600 μm2) is reported (mean ± SEM from 6–10 separate determinations). NRP1/PlexA1- and NRMP1/PlexA2-expressing cells contracted in response to AP–Sema3A at concentrations above 1 nm, and 0.05 nm was sufficient to cause contraction in NRP1/PlexA4 cells. The dotted line indicates the combined mean contraction with Sema3F. C, Quantification of the percentage of collapsed DRG growth cones (mean ± SEM) in response to increasing doses of the AP–Sema3A-conditioned media used in A and B (0, 3.3, 17.0, 34.0 nm). A significant increase in growth cone collapse was seen with 3.3 nm AP–Sema3A.
We next determined the cytoplasmic domains of PlexA1 required for Sema3A signal transduction (Fig. 2). Previously, we documented that deletion of the entire extracellular portion of PlexA1 (PlexA1Δect) resulted in a constitutively active protein (Takahashi and Strittmatter, 2001). The intracellular portion of PlexA1 consists of two domains that are highly conserved among all Plexin family members (C1 and C2) and two relatively nonconserved domains (NC1 and NC2). We generated a series of deletion mutants of PlexA1Δect that lacked either one or more of these domains (Fig. 2B,D) and tested their ability to cause COS-7 cell contraction. Our results indicate that NC1 is required for signaling because truncation (mutant T4) or deletion (mutant T5, T6, ΔNC1) of this domain blocked cell contraction (Fig. 2C,E). Mutagenesis of other intracellular domains reduced cell contraction when compared with normal PlexA1Δect, but only NC1 mutants abolished contraction altogether.

A short segment of PlexinA1 is the primary effector domain. A, Typical morphology of COS-7 cells expressing PlexA1 mutant. Overexpressed PlexA1 mutants were visualized with anti-myc antibody and FITC-conjugated secondary antibody (green), and F-actin was visualized using rhodamine/phalloidin (red). Expression of the constitutively active ΔECT mutant and the ΔC1b mutant caused cell contraction (left and right, respectively). In contrast, expression of ΔNC1 did not reduce cell area (middle). B, D, Schematic representation of PlexinA1 truncation mutants. The cytoplasmic domain of PlexinA1 can be divided into two large conserved regions that share homology with R-ras GAP. Transmembrane domain (TM; yellow), conserved region 1 (C1; orange), conserved region 2 (C2; pink), nonconserved regions 1 and 2 (NC1–2; purple). C, E, Quantification of contraction (cell area < 1600 μm2) of COS-7 cells expressing the indicated mutant proteins. Data are mean ± SEM from three to six experiments.
Interestingly, PlexA1 proteins lacking the NC1 domain are still able to physically interact with two known PlexA mediators, CRMP and MICAL (data not shown). Therefore, a yeast two-hybrid screen was performed using the NC1 domain as bait to identify novel proteins required for signaling. The screen isolated the following clones from a library of 5,000,000 million clones as potential PlexA1–NC1-interacting proteins: RanBPM (Ran-small GTPase binding protein; three independents/four clones), CKAP1 (tubulin folding cofactor B), MAP1A (microtubule-associated protein 1A), SKIP (sphingosine kinase type 1 interacting protein), JAB1 (Jun activation domain binding protein), Murr1 (copper metabolism protein), DnaJB1 (DnaJ/Hsp40 homolog, B1). It is notable that neither MICAL nor CRMP were isolated in this screen. Also, several of these clones encode microtubule-regulating proteins, implicating this region of Plexin in tubulin-dependent processes.
Multiple independent clones for RanBPM were isolated in this screen, so we focused our efforts on this protein. All clones were partial sequences of RanBPM, but all contained parts of the N-terminal region an a portion of SPRY domain (data not shown). RanBPM was originally identified as a Ran GTPase binding protein (Nakamura et al., 1998) and is widely expressed in adult tissues. There are several conserved domains including a proline- and glutamine-rich N-terminal segment thought to mediate formation of protein complexes (Nishitani et al., 2001), an SPRY (SP1a and ryanodine receptor) domain that is a protein interaction motif shared with the ryanodine receptor, and regions that share homology with Lis1 [Lissencephaly type-1-like homology and C-terminal to LisH (LisH-CTLH)], a dynein-regulating protein critical in neuronal migration (Wang et al., 2002). RanBPM has been demonstrated to localize primarily to the cytoplasmic face of the plasma membrane and to interact with integrin β subunits and the hepatocyte growth factor receptor Met to modulate cellular morphology and motility (Wang et al., 2002, 2004; Denti et al., 2004).
The interaction of RanBPM and PlexA family members in eukaryotic cells was confirmed by coimmunoprecipitation from transfected HEK293T cell lysates (Fig. 3A,B). All four class A plexins were able to coimmunoprecipitate (co-IP) with RanBPM, suggesting a common signaling pathway. In addition, RanBPM coimmunoprecipitated with Myc–MICAL1 (Fig. 3C), another molecule known to mediate PlexA signaling (Terman et al., 2002), suggesting a complex of molecules may collaborate at the cytoplasmic region of Plexins. Domain deletion analysis of RanBPM (Fig. 3D) mapped the Plexin-interacting domain to a portion of the N-terminal segment (Fig. 3E), consistent with the original RanBPM clones identified in the yeast two-hybrid screen. Therefore, the Lis1 domain, the proline-rich domain, and a portion of the SPRY domain remain available for interaction with other partners that might alter cell shape.

Interaction between PlexAs and RanBPM. A, FLAG-tagged RanBPM was coexpressed with Myc-tagged PlexA1, A2, A3, or A4 in COS-7 cells, and anti-FLAG immunoprecipitations were performed on cell lysates. All four PlexA isoforms were able to co-IP with FLAG–RanBPM. B, Myc-tagged full-length PlexA1 or constitutively active PlexA1Δect and FLAG-tagged RanBPM were transiently coexpressed in COS-7 cells. Cell lysates were subjected to immunoprecipitation using anti-Myc antibody. FLAG–RanBPM was able to co-IP with both Plexins. C, FLAG–RanBPM and Myc–MICAL1 were coexpressed in COS-7 cells, and anti-Myc immunoprecipitations were performed on cell lysates. FLAG–RanBPM co-IPed with Myc–MICAL. D, Schematic representation of RanBPM and deletion mutants. E, Myc-tagged PlexA1 and RanBPM mutants were transiently coexpressed in COS-7 cells. Cell lysates were subjected to immunoprecipitation using anti-myc antibody, and RanBPM was detected by immunoblot. N-terminal region of RanBPM coprecipitates with PlexinA1. IB, Immunoblot; WB, Western blot.
As a first step toward assessing the functional significance of the RanBPM interaction with the NC1 domain of PlexA1, we coexpressed the proteins in COS-7 cells and analyzed morphology. Although neither protein alone altered cell spreading, coexpression of the two proteins led to pronounced ligand-independent cell contraction, similar to that produced by Sema3A stimulation of NRP1/PlexA1 cells or by expression of constitutively activated PlexA1Δect (Fig. 4A). Similarly, coexpression of RanBPM with PlexA4 significantly increased cell contraction compared with PlexA4 or RanBPM expressed alone. The PlexA1/RanBPM cell contraction is not a toxic effect because terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining revealed no increase in apoptosis and there was no change in trypan blue exclusion from double-transfected cells (data not shown).
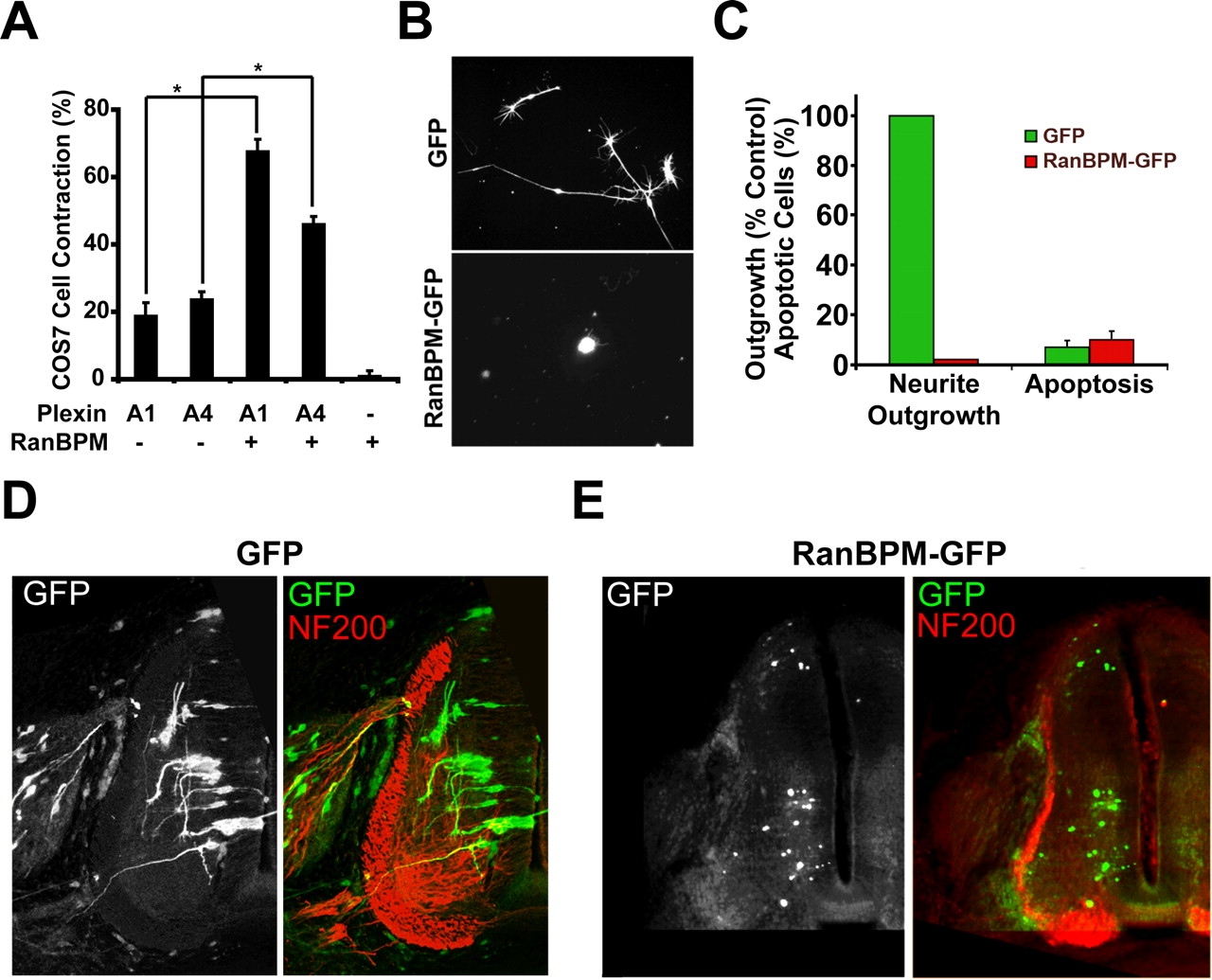
Overexpression of RanBPM and PlexA1 stimulates inhibitory signaling. A, FLAG–RanBPM, Myc–PlexA1, or Myc–PlexA4 were transiently expressed in COS-7 cells. Cells were visualized with anti-FLAG and anti-Myc immunocytochemistry, and cellular area was measured. Expression of PlexA1, PlexA4, or RanBPM alone did not significantly alter the percentage of contracted cells (area ≤ 1600 μm2). However, expression of RanBPM together with PlexA1 or PlexA4 significantly increased the ratio of contracted COS-7 cells (∗p < 0.005, Student’s t test) Data are mean ± SEM. B, Chick E7 DRG neurons were nucleofected with expression plasmids for GFP or RanBPM–GFP. GFP fluorescence reveals neurite outgrowth from transfected cells. All cells in these images are β-III tubulin-immunoreactive neurons (data not shown). C, Quantification of neurite outgrowth or apoptosis from DRG neurons nucleofected with GFP or RanBPM–GFP (both GFP and β-III tubulin positive). Overexpression of RanBPM–GFP completely blocked the formation of neurites. This was likely not because of a toxic effect of RanBPM, because TUNEL staining revealed no difference in apoptosis in GFP- or RanBPM–GFP-positive cells. Data are mean ± SEM. D, The RanBPM–GFP expression plasmid was electroporated into the neural tube of H&H stage 15–18 chick embryos. At E7, embryos were fixed and sectioned transversely and visualized by GFP (green) and neurofilament (NF200; red) immunofluorescence. There was robust outgrowth from control GFP neurons. GFP fluorescence demonstrated the presence of many RanBPM-expressing cells in the spinal cord but no neurite outgrowth. This is one of 12 embryos with similar results.
We also assessed the effect of RanBPM overexpression on axonal growth (Fig. 4). When full-length RanBPM was overexpressed in NGF-dependent DRG neurons by nucleofection, there was drastically reduced neurite outgrowth (Fig. 4B,C). This cannot be explained by a toxic effect because the total number of neurons in the culture and the percentage of apoptotic neurons were unchanged by RanBPM overexpression (Fig. 4C). When RanBPM was overexpressed in the neural tube of intact chick embryos by in ovo electroporation, axon outgrowth from spinal cord neurons was suppressed to nil levels (Fig. 4D), showing strong outgrowth regulation by RanBPM in vivo and in vitro. These observations suggest that high levels of RanBPM have an inhibitory effect on basal neurite outgrowth and that this regulation is not Plexin specific.
Whereas gain of RanBPM function in neurons suppressed outgrowth, reduced RanBPM function had the opposite effect. A fragment of RanBPM (RanBPM-N3) that contains the PlexA1-interacting domain and SPRY domain (Fig. 5A) but lacks the microtubule-regulating LisH domain functioned as a dominant blocker of PlexA1 signaling (Fig. 5). In COS-7 cells expressing NRP1/PlexA1, responsiveness to Sema3A was attenuated by expression of RanBPM-N3 (Fig. 5B). To test the effect of RanBPM-N3 in neurons, DRG explants and Sema3A-expressing HEK293T cells were cocultured in a collagen matrix and HSV vectors were used to overexpress GFP or RanBPM-N3. Although the axons of GFP-expressing DRG explants strongly avoided a Sema3A source in collagen gels, this axon repulsion was significantly reduced in explants expressing the RanBPM-N3 fragment (Fig. 5C,D).

RanBPM-N3 disrupts Sema3A signaling. A, Schematic illustration of the RanBPM-N3 protein fragment. RanBPM-N3 is truncated at amino acid 320 of RanBPM. B, COS-7 cells were transfected with a bicistronic PlexA1/NRP-1 expression plasmid and/or the RanBPM-N3 expression plasmid. After 48 h, the cells were exposed to either AP–Sema3A or AP–Sema3F at 1 nm for 60 min at 37°. The percentage of contracted cells binding AP ligand was measured. Sema3A treatment resulted in significantly more contracted PlexA1/NRP1 cells than Sema3F; however, this difference was abolished in cells coexpressing RanBPM-N3 with PlexA1/NRP1. Data are mean + SEM from three to six experiments (∗p < 0.01, Student’s t test). C, Chick E7 DRG explants were cocultured in collagen gels adjacent to HEK293T aggregates expressing Sema3A or not. Before culture, the explants were infected with EGFP-expressing control virus or truncated RanBPM-N3-expressing virus, and the transfected protein was visualized by immunohistology. D, Quantification. The extent of immunoreactive axonal growth in the quadrant proximal (p) versus distal (d) to the cell aggregate is reported as a ratio. Sema3A-expressing cells repel control neurites to a significantly greater extent than RanBPM-N3-expressing neurons (∗p < 0.01, Student’s t test). a.u., Arbitrary units. Data are mean ± SEM from three to six experiments.
We next tested viral vector-mediated overexpression of dominant-negative RanBPM on neurite outgrowth in dissociated DRG neurons. Overexpression of the N3 fragment led to an ∼25% increase in basal NGF-dependent neurite outgrowth (Fig. 6B). This observation is constant with the idea that full-length RanBPM exerts a negative effect on axon growth. Because of the difference in basal outgrowth, we present the data as the percentage of the mean outgrowth from HEK293T-conditioned media-treated control for each virus group. Sema3A treatment led to a 60% decrease in outgrowth compared with HEK293T-conditioned media in GFP-overexpressing cells; however, this effect was completely blocked in RanBPM-N3-overexpressing cells (Fig. 6C). Therefore, overexpression of HSV–RanBPM-N3 led to decreased sensitivity to Sema3A compared with GFP controls. EphrinA5 has also been shown to have an inhibitory effect on neurite outgrowth in NGF-dependent DRG neurons (Munoz et al., 2005). Expression of the N3 fragment did not alter the ability of ephrinA5 to attenuate axon growth, thus demonstrating that this effect was specific to Sema3A/Plexin signaling.

RanBPM-N3 blocks Sema3A inhibition. A, Dissociated chick DRG neurons were infected with HSV–GFP or HSV–RanBPM-N3 (HSV-N3) and grown in the presence of control, Sema3A-, or ephrinA5-conditioned media. Cells were then fixed and visualized with anti-βIII tubulin immunofluorescence. B, Comparison of neurite outgrowth between control (GFP) and RanBPM-N3 (N3)-overexpressing cells. Mean neurite outgrowth was measured and is expressed as a percentage of the mean outgrowth of GFP controls. Data are mean ± SEM from six independent experiments (∗p < 0.01, Student’s t test). C, Quantification of neurite outgrowth of cells from A (mean + SEM). Cells infected with HSV–GFP showed decreased outgrowth in the presence of Sema3A and ephrinA5 compared with control. GFP-N3 overexpression led to no difference between control and Sema3A-treated cells but still showed responsiveness to ephrinA5 (∗p ≤ 0.01, Student’s t test).
Finally, the effect of knocking down endogenous expression of RanBPM was tested in neuronal responses to Sema3A. Synthetic siRNA oligonucleotides targeting the chick RanBPM sequence were nucleofected into DRG neurons, and neurite outgrowth in the presence of Sema3A or ephrinA5 was tested. Whereas outgrowth was reduced to 40% by Sema3A in cultures treated with control siRNA, there was no difference between Sema3A or HEK293T-conditioned media in neurons treated with siRNAs directed against RanBPM (Fig. 7A,B). These data corroborate those observed with overexpression of the dominant-negative N3 fragment. Knocking down RanBPM expression did cause a 27% increase in basal outgrowth of NGF-sensitive DRG neurons (Fig. 7C), similar to that observed with HSV–RanBPM-N3 (Fig. 6B). However, disruption of RanBPM expression did not alter responsiveness to ephrinA5, confirming that the differences seen between control and siRNA groups are specific for PlexA signaling and not a result of a nonspecific increase in basal outgrowth. These results also show that RanBPM is required for proper PlexA function but may not be used by other axon guidance receptors. The siRNA-mediated reduction of RanBPM expression was confirmed by reverse transcription-PCR (RT-PCR) (Fig. 7D).

Knock-down of RanBPM expression by siRNA blocks Sema3A responses. A, Dissociated chick DRG neurons were treated with control siRNA or siRNA molecules directed against chick RanBPM and grown in the presence of control, Sema3A-, or ephrinA5-conditioned media. Cells were visualized with anti-βIII tubulin immunofluorescence. B, Quantification of neurite outgrowth (mean + SEM) of cells in A was measured. RanBPM siRNA blocked Sema3A-dependent decreases in outgrowth, but had no effect on ephrinA5 treatment (∗p < 0.01, Student’s t test). Data are presented as percentage of the mean outgrowth from control conditioned media-treated group. C, Quantification of the basal neurite outgrowth from RanBPM siRNA treated DRG neurons from A. RanBPM siRNA led to a ∼27% increase in basal neurite outgrowth. Data are expressed as a percentage of the mean outgrowth of control siRNA (mean ± SEM; ∗p < 0.01, Student’s t test). D, The level of endogenous RanBPM mRNA in extracts from chick E7 DRG exposed to RanBPM directed siRNA or to control siRNA was measured by RT-PCR by varying the number of cycles. After 25 cycles, a 600 bp PCR product is seen in control (c), but not siRNA-treated (si), extracts, demonstrating a knock-down of RanBPM expression.
Discussion
The present study identifies RanBPM as a PlexA-binding protein required for Sema3A signal transduction in chick DRG neurons. RanBPM was found to interact with the intracellular NC1 domain of PlexA1 that is essential for the regulation of cell morphology. A complex consisting of RanBPM and PlexA1 was copurified from transfected cells, and overexpression of RanBPM in the presence of PlexA1 induces cell contraction. RanBPM also interacts with PlexA4, the primary Sema3A signal transducing coreceptor in DRG sensory neurons, and coexpression of these molecules results in cell contraction similar to PlexA1. RanBPM negatively regulates axonal outgrowth, a phenomenon believed to be independent from its role in PlexA signaling. Moreover, overexpression of truncated RanBPM or the knock-down of endogenous RanBPM protein levels promote neurite outgrowth and attenuate Sema3A responsiveness. Therefore, RanBPM joins MICAL and CRMP as a component of Sema3A signal transduction (Terman et al., 2002; Deo et al., 2004). A full understanding of signaling downstream of PlexA will require an integrated assessment of all three mediator proteins.
RanBPM was originally identified in a protein-interaction screen searching for novel binding partners of Ran GTPase (Nakamura et al., 1998). Ran GTPase plays a role in nuclear/cytoplasmic transport and in microtubule organization at the centrosome (Fahrenkrog et al., 2004). Thus, the regulation of Ran itself may or may not be the primary target of RanBPM in axonal growth cones. The feasibility of this hypothesis is supported by the presence of Ran GTPase being concentrated in discrete domains of the axonal growth cone (our unpublished data). The Ran GTPase-regulated nuclear transporters, importin-α and -β, have been shown to deliver retrograde signals from the axon to the nucleus in response to injury in sensory neurons (Hanz et al., 2003). There is also evidence that importins are localized to hippocampal synapses and play a role in synaptic plasticity (Thompson et al., 2004). An intriguing possibility is that RanBPM acts to link Plexin signaling to importin function and retrograde signaling to the nucleus. It is established that Sema3A increases retrograde axoplasmic transport (Goshima et al., 1997, 1999), so it is likely that long-range signaling occurs. Such a hypothesis is consistent with our data because RanBPM perturbation effected long-term axonal outgrowth and guidance events and not the immediate growth cone collapse normally associated with Sema3A responses (data not shown).
Interestingly, nerve injury initiates the local translation of importin-β in the axon before retrograde signaling (Hanz et al., 2003), and Sema3A-facilitated axon transport correlates with the activation of translation initiation (Li et al., 2004). Therefore, it is conceivable that Sema3A-mediated responses are dependent on local translation of long-distance signaling molecules (Hanz and Fainzilber, 2004) in addition to proteins for local use as described previously (Campbell and Holt, 2001; Brittis et al., 2002; van Horck et al., 2004).
RanBPM also possesses domains that resemble Lis1 and could therefore have a function in dynein-mediated transport of organelles underlying axonal growth versus inhibition. Initial subcellular localization studies demonstrated a close association of RanBPM with the centrosome (Nakamura et al., 1998), although this localization has not been confirmed (Nishitani et al., 2001). Regulation of microtubule structure from the centrosome is known to contribute to neuronal plasticity and axon determination (de Anda et al., 2005), so this is another potential mechanism of RanBPM action.
Although the majority of the RanBPM localizes to the cytosol, a significant fraction can be found near the inner surface of the plasma membrane (Denti et al., 2004; Cheng et al., 2005). It is this pool that is likely to be most relevant for Sema3A/NRP1/PlexA signaling. RanBPM has been shown to interact with other plasma membrane proteins, suggesting a large role in mediating cell growth and signaling (Wang et al., 2002; Denti et al., 2004; Cheng et al., 2005; Hafizi et al., 2005). Because both MICAL and RanBPM have been implicated in integrin-based cellular responses to the extracellular matrix (Suzuki et al., 2002; Denti et al., 2004), both proteins provide potential sites for cross-talk between Sema3-dependent and matrix-dependent regulation of axonal growth. Because RanBPM is also capable of interacting with Met and p75NTR (Wang et al., 2002; Bai et al., 2003), it could provide a focal point for growth cone modulation by multiple extracellular cues.
It has been shown that the Rho-like GTPase Rnd1 binds to PlexA1 within the NC1 domain and mutation of residues 1598, 1599, and 1600 (mutant A1P2) abolish this interaction (Zanata et al., 2002). The investigators found that coexpression of wild-type PlexA1 and Rnd1 led to COS-7 cell contraction and this was blocked by A1P2 expression. Although it is possible the initial cell morphology results from PlexA1ΔectΔNC1-expressing COS-7 cells were attributable in part to the disruption of Rnd1 binding, our data using dominant-negative RanBPM in COS-7 cells and neurons and targeting RanBPM expression in neurons demonstrate the specificity of PlexA–RanBPM signaling. Zanata et al. (2002) do not demonstrate a direct interaction between PlexA1 and Rnd1; thus, it is also possible such an interaction is mediated by RanBPM.
It is now clear that the Plexin signaling pathway does not rely on a single transducing molecule functioning in a simple linear cascade to activate downstream effectors. Instead, a macromolecular complex consisting of several proteins appears to be assembled at the cytoplasmic surface of Plexin. Whether these proteins initiate separate signaling pathways or work in concert remains to be determined and will be the topic of future studies.
Footnotes
-
This work was supported by a research grant from the National Institutes of Health (NIH) to S.M.S. and by an individual predoctoral training grant from the NIH to E.F.S.
-
↵*H.H. and E.F.S. contributed equally to this work.
- Correspondence should be addressed to Stephen M. Strittmatter, Department of Neurology, Yale University School of Medicine, P.O. Box 208018, New Haven, CT 06510. Email: stephen.strittmatter{at}yale.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}