

Abstract
Although some studies showed the efficacy of phosphodiesterase (PDE) inhibitors as neuronal plasticity enhancers, little is known about the effectiveness of these drugs to improve plasticity in cases of mental retardation. Fetal alcohol syndrome (FAS) is the leading cause of mental retardation in the western world. Using a combination of electrophysiological and optical imaging techniques, we show here that vinpocetine, a PDE type I inhibitor, restores ocular dominance plasticity in the ferret model of fetal alcohol exposure. Our finding should contribute to a better understanding and treatment of cognitive deficits associated with mental disorders, such as FAS.
- alcohol
- ocular dominance plasticity
- visual development
- ferret
- monocular deprivation
- optical imaging
- neocortex development
- fetal alcohol syndrome
Introduction
The use of phosphodiesterase (PDE) inhibitors as cognitive enhancers is currently under intensive study (Barad et al., 1998; Vitolo et al., 2002). PDE inhibitors prevent breakdown of cAMP to 5′-AMP, maintaining activation of protein kinases and the transcription factor CREB (cAMP response element-binding protein), which may in turn improve memory processing. Vinpocetine (VINP), a PDE type 1 inhibitor, has been shown to facilitate long-term potentiation (Molnar and Gaal, 1992), enhance the structural dynamics of dendritic spines (Lendvai et al., 2003), improve memory retrieval (DeNoble, 1987), and enhance performance on cognitive tests in humans (Hindmarch et al., 1991). Although these studies showed the efficacy of VINP in enhancing plasticity in normal subjects, little is known about the effectiveness of PDE inhibitors to improve plasticity in cases of severe cognitive impairment.
Fetal alcohol syndrome (FAS) is the leading cause of mental retardation in the western world, and there is no adequate treatment for children with this syndrome. There is growing evidence that abnormalities of cortical plasticity contribute to the cognitive deficits that characterize FAS (Rema and Ebner, 1999; Medina et al., 2003). However, the mechanisms by which prenatal alcohol exposure disrupts neocortical plasticity remain elusive. Recently, we developed a ferret model of early alcohol exposure that provides a unique approach to unveil such mechanisms as well as to test different strategies to restore cortical plasticity. Animals exposed to alcohol during the third trimester equivalent of human gestation and monocularly deprived after a prolonged alcohol-free period are characterized by a permanent impairment of ocular dominance plasticity (Medina et al., 2003; Medina and Ramoa, 2005). Here, we try to restore ocular dominance plasticity in alcohol-exposed ferrets using the PDE type 1 inhibitor vinpocetine.
Materials and Methods
In vivo electrophysiology. Animals were premedicated by subcutaneous injection of a tranquilizer (acepromazine, 1 mg/kg), a muscarinic antagonist (methyl atropine bromide, 0.2 mg/kg) to reduce bronchial secretion, and dexamethasone sodium phosphate (0.5 mg/kg) to reduce inflammation. Animals were then anesthetized using sodium pentobarbital (35 mg/kg; Abbott Laboratories, North Chicago, IL) and placed in a stereotaxic frame. No procedures started until the animal was sufficiently anesthetized, as ascertained by the loss of withdrawal and cornea-blink reflexes. A tracheal cannulation was performed, and the animal was placed on a ventilator. Heart rate, expired CO2, and arterial blood oxygen saturation (SpO2) were monitored continuously and maintained at ∼4.0 and >90%, respectively. Body temperature was maintained at 38°C using a homeostatic blanket. The eyelids were opened, nictitating membranes were retracted using phenylephrine hydrochloride (2.5%), the pupils were dilated with atropine sulfate (1%), and contact lenses were placed on the corneas.
A craniotomy (3–4 mm in diameter) was performed to expose the binocular region of the left primary visual cortex where recordings were performed. Single-unit recordings were conducted using a glass-coated tungsten microelectrode with a 5 μm exposed tip lowered into the primary visual cortex at ∼20° to the vertical. To minimize sampling bias, single units used in this study were separated at least 100 μm along the electrode track. After the isolation of a single unit, its receptive field was mapped, and the optimal stimulus orientation, direction, and velocity were determined qualitatively with a moving bar of light (0.5° wide and 20° long) projected onto a tangent screen. Ocular dominance, spontaneous activity, and the number of spikes per stimulus were then quantitatively determined for each cell by presenting a computer-controlled bar of light to each eye. Each stimulus presentation consisted of the bar of light moving across the receptive field at the optimal orientation in one direction and back across in the opposite direction. A computer collected spikes during the 10 stimulus presentations using Spike 2 software (Cambridge Electronics Design, Cambridge, UK), and peristimulus histograms were generated. Spontaneous activity was determined by recording activity in the absence of stimulation. At the conclusion of the electrophysiology experiment, ferrets were killed with Euthasol (125 mg/kg; Delmarva Laboratories, Midlothian, VA).
To quantify cortical binocularity, an ocular dominance index was calculated for each cell using the following equation: LE/(LE + RE), where LE stands for response to stimulation of left eye and RE for right eye. Additionally, a contralateral bias index (CBI) was calculated for each animal. This index was defined as follows: {(P0.00–0.19 – P0.80–1.00) + [(P0.20–0.39 – P0.60–0.79)/2] + 100}/200, where PA–B denotes the percentage of cells with ocular dominance index values between A and B. A CBI of 0.0 indicates complete ipsilateral eye dominance, whereas a CBI of 1.0 indicates complete contralateral eye dominance.
Optical imaging of intrinsic signals. Optical imaging of intrinsic signals was performed with Imager 2001 VSD+ (Optical Imaging System, Germantown, NY) by using imaging methods slightly modified from those described previously (Medina et al., 2003; Krahe et al., 2005). Briefly, animals were premedicated, anesthetized, and ventilated with similar procedures described for single-unit recordings. Temperature, heart rate, expired CO2, and SpO2 were monitored continuously. A craniotomy was made over the left hemisphere to expose the dorsal area of the occipital cortex. The dura was reflected and the opening was filled with agar (2.5% in saline) and covered with a glass coverslip. First, an image of the vascular pattern of the cortical surface was obtained by illuminating the cortical surface with a green filter (∼550 nm). Then, images of intrinsic signals were obtained by using a red filter (∼700 nm). Visual stimulation consisting of high-contrast rectangular wave gratings (8.75° dark phase/1.25° light phase) was generated on a 21-inch monitor (Sony Trinitron; Sony, Tokyo, Japan) using SGT+ graphics board and STIM software (generously provided by K. Christian, Optical Imaging System, Germantown, NY). Gratings were presented monocularly at an angle of 0, 45, 90, or 135° and drifted (22.5°/s) in both directions along the axis orthogonal to the orientation of the grating. A single trial consisted of these four gratings and a blank screen presented to each eye for 9 s in a pseudorandom sequence, with data acquisition during the last 8 s. The acquisition computer controlled eye shutters (Optical Imaging System), and a total of 20 trials was performed for each eye. An ocular dominance image was obtained by subtracting the summed images obtained with the left eye from the summed images obtained with the right eye. In these images, dark areas responded best to stimulation of the right eye, and white areas responded best to stimulation of the left eye. To quantify the results, unfiltered ocular dominance images were first clipped at ±2 SD, and then a region of interest (ROI) outlining V1 was drawn manually by an investigator who was blind to the animal's treatment. The contralateral eye band, which marks the anterior border of V1 and lies caudal to V2 ipsilateral big domains (White et al., 1999), was used as an anterior reference to draw the ROI. The posterior reference was the caudal pole. The approximate location of the border between V1 and V2 was still distinguishable after 3 d of monocular deprivation (MD). However, longer periods of MD lead to saturating left eye dominance in V1/V2, making this distinction more difficult (Krahe et al., 2005). After defining the ROI, we created ocular dominance histograms based on the range of gray values (0–255) divided into five class intervals, where 0–50 and 204–255 correspond to classes containing the darkest and lightest pixels, respectively. A contralateral bias index for optical imaging (CBIOI) was defined as follows: {(P0–50 – P204–255) + [(P51–101 – P153–253)/2] + 100}/200, where PA–B denotes the percentage of pixels with gray values between A and B. A CBIOI close to 1.0 indicates a prevalence of darker pixels and right eye dominance. A CBIOI close to 0.0 indicates a prevalence of lighter pixels and left eye dominance.
Results
Ferrets were injected with alcohol (3.5 mg/kg, 25% in saline, i.p.) every other day from postnatal day 10 (P10) until P30, which approximately mimics a binge-drinking episode once a week during the third trimester of gestation in humans. This alcohol exposure leads to ∼250 mg/dl of blood alcohol concentration after the first 5 h and is undetectable after 34 h (Medina et al., 2003). Control littermates received saline solution or did not receive any treatment. At P40–P50, ferrets were injected with VINP (20 mg/kg, i.p., daily) or vehicle solution (Veh) for 4 d. One day after the beginning of VINP treatment, animals had the lid of the right eye sutured closed. At the fourth day of VINP treatment, third day of MD, quantitative single-unit electrophysiology or optical imaging of intrinsic signals were performed to assess changes in ocular dominance in the left hemisphere. To quantify ocular dominance assessed by single-unit recordings, an index was calculated for each cell using the following equation: LE/(LE + RE), where LE stands for response to stimulation of left eye and RE for right eye. An ocular dominance index of 1.0 indicates that a neuron is responsive only to the left eye, and an ocular dominance index of 0.0 indicates responses only to the right eye.

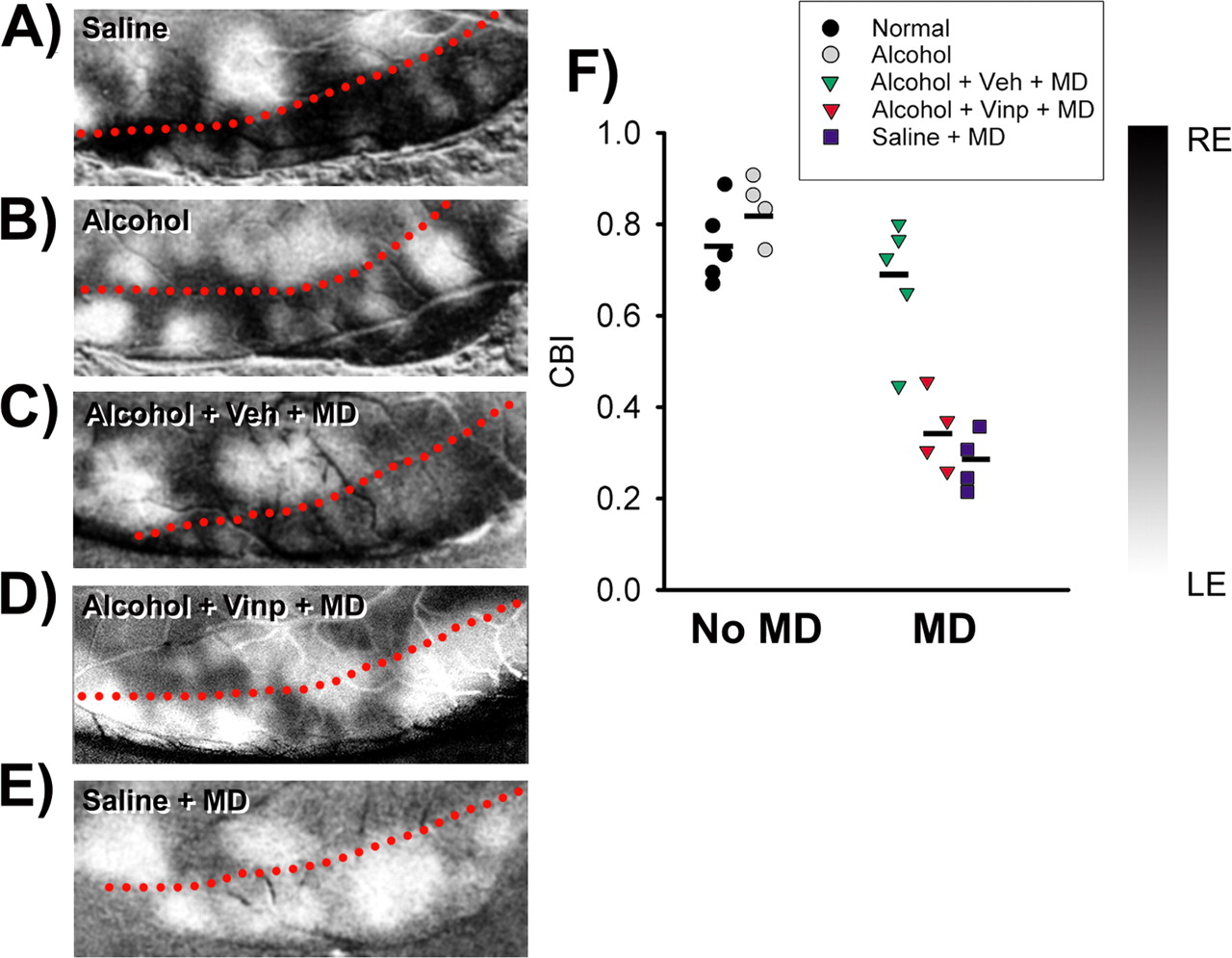
Single-unit electrophysiology shows that VINP restores plasticity in alcohol-exposed animals. Ocular dominance profiles of non-deprived normal (A, 186 cells in 8 ferrets) and alcohol-exposed (B, 195 cells in 8 ferrets) animals show a large number of binocular cells. Most cells remain activated by the deprived (right) eye in Alcohol + Veh + MD animals (C, 129 cells in 5 ferrets) after 3 d of MD. In contrast, the same period of MD resulted in a striking ocular dominance shift toward the experienced (left) eye in Alcohol + VINP + MD (D, 161 cells in 5 ferrets) and Saline + MD (E, 164 cells in 5 ferrets) animals. Error bars indicate SE. F, CBI (see Materials and Methods) values are plotted for each group. Each symbol represents a single animal (number of animals as in A–E), and the horizontal bars represent means. CBI values in non-deprived animals reflect contralateral eye dominance. In the Alcohol + Veh + MD group, MD does not result in significant changes of CBI values. In contrast, Alcohol + VINP + MD animals present CBI values that are significant lower than those of non-deprived and Alcohol + Veh + MD animals (one-way ANOVA, p < 0.001; Tukey's test, p < 0.001 for all comparisons) but similar to those of Saline + MD animals. G, The cumulative number of cells (as percentage) plotted as a function of the ocular dominance index for all groups. Curves skewed to the right indicate an ocular dominance shift. The distribution of ocular dominance indexes in the Alcohol + VINP + MD (n = 161 cells) group is similar to that of the Saline + MD (n = 164 cells) group [Kolmogorov–Smirnov (KS) test, p = 0.7] and significantly different from those of the Alcohol (n = 195 cells; p < 0.01), Normal (n = 186 cells; p < 0.01), and Alcohol + Veh + MD (n = 129 cells; p < 0.01) groups. H, VINP treatment does not alter ocular dominance index distribution in non-deprived normal (n = 51 cells from 2 animals) and alcohol-treated (n = 53 cells from 2 animals) groups (compare with non-deprived animals shown in F). Moreover, VINP treatment does not alter the ocular dominance shift after MD. The curve in the Normal + VINP + MD (n = 46 cells from 2 animals) group is significantly different from those of the Normal + VINP (KS test, p < 0.01) and the Alcohol + VINP (KS test, p < 0.01) groups.

Visual responses of striate cortical cells to a moving bar of light. VINP treatment preserved robust responses to visual stimulation. A shows mean maximal responses (in spikes per second) of cortical neurons to stimulation at the optimal orientation. Similar values were obtained for all groups. Additionally, VINP treatment did not change spontaneous activity (B). Error bars indicate SD.

Optical imaging of intrinsic signals shows that VINP restores plasticity in alcohol-exposed animals. A–E, Representative cases of ocular dominance maps from each group. The dashed red line represents the approximate location of the border between V1 (bottom) and V2 (top). The dark and light regions represent are as dominated by the RE and LE, respectively. Note that, after MD, the Alcohol + Veh + MD animal still presents a large area of V1 responding to the deprived (right) eye (C). In contrast, after the same period of MD, the Alcohol + VINP + MD animal (D) presents a strong dominance of the experienced (left) eye in V1 similar to the Saline + MD animal (E). F, CBIOI (see Materials and Methods) values for individual animals. Horizontal bars represent means. Alcohol + VINP + MD animals presented significantly lower CBIOI values than non-deprived and Alcohol + Veh + MD animals (one-way ANOVA, p < 0.001; Tukey's test, p < 0.001 for both comparisons). The Alcohol + VINP + MD group did not differ from the Saline + MD group.
Ocular dominance profiles observed in untreated and saline-exposed animals (both non-deprived) were similar and pooled together (hereafter defined as normal group). Figure 1 shows that ocular dominance profiles of normal and alcohol-exposed animals (non-deprived) were very similar (Fig. 1, compare A, B). Remarkably, whereas MD had little effect on cortical ocular dominance in alcohol-exposed animals treated with vehicle (Fig. 1C, Alcohol + Veh + MD), animals treated with VINP (Alcohol + VINP + MD) exhibited a striking shift toward the experienced eye (Fig. 1D). This ocular dominance shift was similar to that observed in monocularly deprived saline-exposed animals (Fig. 1E, Saline + MD). To further quantify ocular dominance, we calculated a CBI for each animal (see Materials and Methods). A CBI of 0.0 indicates that the ipsilateral (left) eye dominated the responses in every neuron tested, whereas a CBI of 1.0 indicates that the contralateral (right) eye dominated all responses. Mean CBI values (±SEM) were significantly lower in Alcohol + VINP + MD (0.3 ± 0.04; n = 5 ferrets) and Saline + MD (0.2 ± 0.03; n = 5) animals than in Alcohol + Veh + MD (0.49 ± 0.04; n = 5) and non-deprived (Normal, 0.58 ± 0.03, n = 8; Alcohol, 0.62 ± 0.03, n = 8) animals (Fig. 1F). The cumulative distributions of the ocular dominance index values for all cells analyzed in each group confirm these results (Fig. 1G). Alcohol + VINP + MD and Saline + MD animals exhibited right-shifted curves when compared with non-deprived and Alcohol + Veh + MD animals. Moreover, VINP treatment neither altered ocular dominance distribution in non-deprived animals nor affected the expected ocular dominance shift after monocular deprivation in normal animals (Fig. 1H).
Restoration of ocular dominance plasticity in VINP-treated animals did not result in abnormal visually driven activity. Quantitative assessment of single-unit responses revealed that VINP treatment preserved visual responsiveness and normal spontaneous activity (Fig. 2A,B).
To further assess the effects of VINP treatment on ocular dominance plasticity, we conducted optical imaging of intrinsic signals in alcohol-exposed ferrets (Fig. 3). Dark areas in ocular dominance maps are regions that respond preferentially to stimulation of the right eye, whereas white areas respond preferentially to the left eye. After MD, Alcohol + Veh + MD animals still show a large area of V1 responsive to the deprived eye (Fig. 3C), similar to non-deprived animals (Fig. 3A,B). In contrast, Alcohol + VINP + MD and Saline + MD animals show a strong dominance of the experienced (left) eye in V1 after MD (Fig. 3D,E). To obtain a quantitative estimate of ocular dominance, we created a contralateral bias index for optical imaging (CBIOI) (see Materials and Methods). A CBIOI close to 1.0 indicates right eye dominance (prevalence of darker pixels), whereas a CBIOI close to 0.0 indicates left eye dominance (prevalence of lighter pixels). Similarly to what was observed in single-unit recordings, mean CBIOI values (±SEM) were significantly lower in Alcohol + VINP + MD (0.35 ± 0.04; n = 4 ferrets) and Saline + MD animals (0.28 ± 0.03; n = 4) than in Alcohol + Veh + MD (0.68 ± 0.06; n = 5) and non-deprived animals (Normal, 0.75 ± 0.04, n = 5; Alcohol, 0.83 ± 0.03, n = 4).
Discussion
Collectively, our results show that inhibition of PDE1 restores plasticity in ferrets exposed early to alcohol. This finding has a potential significance from a clinical standpoint and suggests that defective phosphorylation of intracellular signaling pathways underlie the impairment of cortical plasticity in FAS. In addition to PDE1 inhibition, VINP treatment may also affect voltage-dependent sodium channels (Molnar and Erdo, 1995). However, recent findings suggest that VINP affects more potently a subset of sodium channels resistant to tetrodotoxin (Zhou et al., 2003). Nevertheless, if VINP substantially affects sodium channel function in the neocortex, one may expect a reduction of cortical activity. However, in our study, we showed that VINP treatment did not affect visual responsiveness as well as spontaneous activity in visual cortex. Moreover, studies in humans showed lack of side effects of VINP at clinical doses (Hindmarch et al., 1991).
What are the possible mechanisms underlying the restoration of ocular dominance plasticity by PDE1 inhibition? VINP treatment is likely to increase cAMP signaling, which in turn would enhance the activity of a large number of factors downstream from the NMDA receptor, any of which may be downregulated in FAS. In fact, it has been suggested that an alteration in the cAMP cascade may play an important role in the deleterious effects of early alcohol exposure on brain development (Maas et al., 2005). A possible explanation for an alteration of cAMP levels after early alcohol exposure is related to the upregulation of calmodulin (CaM) that has been demonstrated to occur after chronic alcohol exposure (Pant et al., 1985; Hamoudi et al., 1995). Increase in CaM can result in an enhancement of PDE1 activity and a consequent decrease on cAMP (Kakkar et al., 1999). In fact, it has been shown recently that an increase in calcineurin levels, which can enhance PDE1 activity as well, impairs ocular dominance plasticity (Yang et al., 2005). Additional studies will be needed to verify whether CaM and cAMP levels are indeed altered in our model. However, we cannot discard the possibility that PDE inhibition may simply compensate for problems outside the cAMP signaling system. In conclusion, our findings provide for the first time evidence that PDE1 inhibitors may be useful to enhance neuronal plasticity in severe cases of mental retardation such as in FAS.
Footnotes
This work was supported by National Institute on Alcohol Abuse and Alcoholism Grant 013023-05A1 to A.E.M. This study is dedicated to the memory of Dr. Ary S. Ramoa.
Correspondence should be addressed to Alexandre E. Medina, Department of Anatomy and Neurobiology, Box 980709, Virginia Commonwealth University, 1101 East Marshall Street, Room 12-042, Richmond, VA 23298-0709. E-mail: amedina{at}vcu.edu.
DOI:10.1523/JNEUROSCI.4177-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/261057-04$15.00/0
↵† Deceased, July 1, 2005.





{kind=link}
{kind=link}
{kind=link}