

Abstract
Mitochondrial encephalomyopathies are common and devastating multisystem genetic disorders characterized by neuromuscular dysfunction and tissue degeneration. Point mutations in the human mitochondrial ATP6 gene are known to cause several related mitochondrial disorders: NARP (neuropathy, ataxia, and retinitis pigmentosa), MILS (maternally inherited Leigh's syndrome), and FBSN (familial bilateral striatal necrosis). We identified a pathogenic mutation in the Drosophila mitochondrial ATP6 gene that causes progressive, adult-onset neuromuscular dysfunction and myodegeneration. Our results demonstrate ultrastructural defects in the mitochondrial innermembrane, neural dysfunction, and a marked reduction in mitochondrial ATP synthase activity associated with this mutation. This Drosophila mutant recapitulates key features of the human neuromuscular disorders enabling detailed in vivo studies of these enigmatic diseases.
Introduction
Mitochondrial impairment results in a host of devastating conditions known generally as respiratory chain diseases (DiMauro and Schon, 2003). Furthermore, mitochondrial dysfunction has been conclusively implicated in nearly every chronic human neurodegenerative disease, including mitochondrial encephalomyopathies, amyotrophic lateral sclerosis, and Huntington's, Alzheimer's, and Parkinson's diseases (Orth and Schapira, 2001; Schon and Manfredi, 2003). Primary mutations in several distinct mitochondrial genes, as well as nuclear genes that encode proteins localized to the mitochondria, are known to cause a variety of related and devastating encephalomyopathies with complex clinical features, including neurological and muscular dysfunction that is often complicated by renal, endocrine, cardiac, and hepatic involvement (for review, see Schapira and Cock, 1999; Hart et al., 2002; DiMauro and Schon, 2003). There are over 150 mutations causing these related diseases and range from point mutations to large deletions. Because there are numerous mutagenic targets, these mitochondrial disorders have an extremely high prevalence similar to that of muscular dystrophy, ∼10–15 cases per 100,000 individuals (DiMauro and Schon, 2003).
Point mutations in the human mitochondrial ATP6 gene are known to cause neuropathy, ataxia, and retinitis pigmentosa (NARP), maternally inherited Leigh's syndrome (MILS) and familial bilateral striatal necrosis (FBSN) diseases. Also, disease is associated with a common 5kb mitochondrial genome deletion of ATP6 (Table 1). The ATP6 protein is an essential component of the mitochondrial F1F0-ATP synthase (Complex V) that functions as a hydrogen ion channel serving to couple ion transport with rotary ATP catalysis (Mitchell and Moyle, 1967; Walker, 1995; Boyer, 1997; Stock et al., 2000). Bioenergetic impairment and oxidative damage resulting from respiratory chain dysfunction are two prominent hypotheses for the pathogenesis of human mitochondrial encephalopathies. However, mechanistic details of pathogenesis associated with chronic mitochondrial impairment remain unclear, and as a result, therapeutic approaches remain limited.
Human diseases associated directly with ATP6 mutation
Autosomal dominant progressive external ophthalmoplegia (adPEO) results from mutation of three distinct nuclear loci: 4q34–35, 10q24 (Suomalainen et al., 1995), and 15q22–26 (Van Goethem et al., 2001). The 4q PEO locus was recently identified as the ANT1 gene, wherein four different mutation sites segregating in six PEO families were discovered (Kaukonen et al., 2000; Napoli et al., 2001; Komaki et al., 2002). Patients with PEO are predisposed to secondary mitochondrial mutations that appear to underlie, or at least contribute to, the pathophysiological basis for their degenerative disease (Servidei et al., 1991; Suomalainen et al., 1997). The basis for the mutator phenomenon associated with PEO disease is not understood.
We discovered a mitochondrial ATP6 mutation as a maternally inherited component in our Drosophila sesB1 (ANT1) mutant stock. To our knowledge this is the first pathogenic mitochondrial mutation isolated in an intact genetic animal system. Our studies of the Drosophila ATP61 and sesB1(ANT1) mutations revealed common phenotypes, including conditional paralysis, shortened lifespan, and degeneration of the neuromuscular system. The observed locomotor impairment, myodegeneration, neural dysfunction, and severe shortening of lifespan observed in the fly recapitulate key features of MILS disease in humans. Ultrastructural analysis of ATP61 revealed a novel mitochondrial morphological defect associated with ATP synthase impairment. ATP synthase activity was absent in ATP61 mutants. Surprisingly, respiration rates were unaltered in these mutants. These data demonstrate that impaired ATP synthase function associated with ATP6 dysfunction can cause severe encephalomyopathy in the absence of a defect in respiration rate in vivo.
Materials and Methods
Drosophila stocks and culture. Standard cornmeal molasses fly media was used, unless otherwise noted. sesB1 and DSR (Wolbachia-infected positive control) flies were obtained from Michael Ashburner (University of Cambridge, Cambridge, UK) and Michael Clark (University of Chicago, Chicago, IL), respectively. All other stocks were obtained from the Bloomington stock center. Canton S was our wild-type (WT) strain. Tetracycline treatment of flies involved maintaining cultures on standard media supplemented with 0.25 mg/ml tetracycline for two generations, as described previously (Min and Benzer, 1997b).
Lifespan and behavior analysis. Lifespan analysis was performed as described previously (Palladino et al., 2003). Mechanical sensitivity (“bang sensitivity”) was assayed by vortexing flies in a standard media vial for 20 s and measuring the length of paralysis, similar to a previously described protocol (Ganetzky and Wu, 1982).
Genetic mapping of MATBS. sesB1/sesB1 females mated with wild-type males produce exclusively bang-sensitive paralytic offspring, suggesting that sesB1 may be dominant for this phenotype. sesB1/Y males mated with wild-type females never produce paralytic offspring, suggesting that sesB1 may be recessive for this phenotype. This conundrum prompted us to mate affected sesB1/FM7a females with FM7a males, and sesB1/Y males mated with C(1)/Y females (compound first chromosome with two sesB+ chromosomes attached to one centromere). The former mating yielded genotypically FM7a/FM7a (sesB+/sesB+) animals with locomotor impairment. These animals were out-crossed to wild-type animals for two generations maintaining maternal inheritance while selecting against the FM7a chromosome to produce our MATBS (ATP61) stock. The latter yielded sesB1/Y animals completely lacking a maternal sesB1 (MATBS) contribution and were used to generate a sesB1, MAT+ strain (patriclinously cleaned).
Determining percentage of homoplasmy. Using subsaturating PCR conditions, we amplified the ATP6 gene from three independent mutant and control whole genomic DNA preparations and subcloned the ∼500 bp fragment into a TOPO T/A vector (Invitrogen, Carlsbad, CA). From each of the three independent ATP61-derived transformations, we isolated plasmid from ∼30 clones and determined whether they were mutant (Fok I-resistant) or wild type (Fok I-liable) by restriction digestion and visualization on an ethidium bromide-stained agarose gel. We similarly examined 30 clones, 10 each from three independent wild-type control experiments, to assess the frequency of Fok I-resistant clones in wild-type control flies. We never observed a Fok I-resistant clone from wild-type control flies.
Paraffin histology. Heads were removed, probosci were dissected away, and heads were fixed overnight in Carnoy's fixative as described previously (Palladino et al., 2002). Bodies were dissected so that only the thorax remained, which were fixed overnight in Carnoy's fixative. Sections (5 μm) were stained with hematoxylin and eosin (H&E) using standard methods.
Transmission electron microscopy. Animals were aged to day 10, and their brains were dissected and processed similar to methods described previously (Kawasaki et al., 1998). Briefly, fixation occurred overnight at 4°C in a buffered saline solution containing 2.5% paraformaldehyde and 1.5% gluteraldehyde (the primary fixative). Brains were postfixed in 1% osmium tetroxide solution, dehydrated in a graded ethanol series, and embedded in epon resin. Sections (65 nm) were obtained from a Reichert Ultracut ultramicrotome and stained with 4% uranyl acetate and 2.7% lead citrate. The tissue was imaged on a JEOL (Akishima, Japan) 100CX transmission electron microscope. Stereological protocols were followed to collect numerous images from the central brain (neuropil) and the brain periphery (rich in cell bodies) from day 10 mutants and wild-type controls. Randomly selected central brain micrographs were used to quantify the frequency of aberrant mitochondria. The evaluator, blinded to the genotype, was asked to count and characterize the morphology of mitochondria from six micrographs (711 μm2 area per genotype). A Student's t test was used to evaluate significance.
Three-dimensional electron imaging: high-voltage electron tomography. For tomography, 180- to 310-nm-thick sections were cut from the epon blocks, and colloidal gold particles (15 nm diameter) were applied to one side as alignment markers. Tomographic data sets were collected on two electron microscopes. Tilt series were recorded on an AEI EM7 high-voltage electron microscope operated at an accelerating voltage of 1000 kV. The images were recorded around two orthogonal tilt axes, each over an angular range of ±60° with a 2° tilt interval. Also, single-axis data sets were collected over an angular range of ±70° with a 1° tilt interval on a Tecnai F20 electron microscope (FEI Company, Hillsboro, OR) operated at 200 kV. Images were aligned as described previously (Penczek et al., 1995), and tomographic reconstructions were calculated by the weighted back-projection method (Radermacher, 1992); all procedures were implemented in the SPIDER image processing system (Frank et al., 1996). Three-dimensional models of membrane surfaces were generated by density threshold analysis, using Iris Explorer (NAG, Downers Grove, IL) after filtering the volume by anisotropic diffusion (Frangakis and Hegerl, 2001) using SPIDER and masking the mitochondrial outlines in Sterecon (Marko and Leith, 1996). In all cases, the z-dimension (section thickness) of the final models was increased by 20% to compensate for radiation-induced thinning of the plastic section (Deng et al., 1999).
ATP synthase assays. The micromolar quantity of ATP produced per microgram of protein was determined using a modified version of a previously described assay (Manfredi, 2001). Mitochondria were isolated from ∼2 g of adult flies. Flies were homogenized with a glass Dounce homogenizer, three passes with plunger A, in 50 ml of mitochondrial isolation buffer (320 mm sucrose, 1 mm EDTA K-salt, 10 mm Tris, pH 7.4). The homogenate was centrifuged at 1500 × g for 10 min at 4°C. The supernatant was homogenized, three passes with plunger B, and centrifuged at 17,000 × g for 10 min at 4°C. The mitochondrial pellet was resuspended in ATP synthesis buffer A (150 mm KCl, 25 mm Tris-HCl, 2 mm EDTA, 0.1% BSA, 10 mm KH2PO4, 0.1 M MgCl, pH 7.4). Each sample is split into two tubes, and each tube contains the following concentrations of reagents (in mm): 0.15 di(adenosine)pentaphosphate, 0.1 ADP, 1 malate, and 1 pyruvate. Oligomycin at 10 μg/ml was added to only one of the two tubes. An ATP standard curve was generated (0.0, 0.01, 0.1, 1.0, and 10.0 μm). ATP synthase measurements were taken in a black 96-well plate after the addition of buffer B (0.8 mm Luciferin, 20 μg/ml Luciferase, 0.5 m Tris-acetate, pH 7.75), and the luminescence was immediately recorded using a Victor2 plate reader (PerkinElmer, Wellesley, MA). The light emitted was integrated for 1 s every 30 s for a total of 3 min. The data from this experiment were normalized to the total protein in the sample. The homogenate was brought to a final concentration of 0.1 m NaOH and sonicated for 10 s (setting 5 Fisher sonic dismembrator model 100; Fisher Scientific, Houston, TX). The concentration of protein was determined using a colorimetric protein assay reagent (BioRad, Hercules, CA) and a BSA protein standard curve. There was no significant difference in ATP synthase activity between untreated ATP61 and oligomycin-treated wild type, demonstrating that the ATP6 protein encoded by the mutant is not simply oligomycin insensitive.
Population respiration rates. We directly measured respiration of ∼5 g of flies (∼5000 flies) as the emergence of CO2 in a fixed volume respirometer. The respirometer was a sealed acrylic 3.570 L box containing a dual wavelength infrared-absorbance analyzer to monitor CO2 concentration between 0 and 10,000 ppm (model 2820; Bacharach, New Kensington, PA) and 30 glass shell vials each containing 8 ml of yeast-free food and ∼150 flies. Cheesecloth was used to confine flies to within the vials while allowing free air exchange. Flies aged 2–10 d were placed in the respirometer, which was purged with 60% N2, 40% O2 (20 min) to remove CO2 and establish a standard initial O2 concentration. The intake and exhaust ports were sealed, and the CO2 levels were recorded every 2 min for 5 h. We performed this experiment, sans flies, to determine the integrity of the container and identify any other sources of CO2: no detectable CO2 was produced in all experiments without flies over the course of 5 h. The mass of the flies was determined immediately after the experiment, and respiration data are presented as CO2 emergence per unit mass (parts per million per gram of fly) as a function of time. The effective respirometer chamber volume (total volume less volume of vials, food, meter, and flies) is ∼2.760 L. Experiments were performed alternating wild-type and age-matched mutant flies (n = 4).
Individual metabolic measurements. Flies eclosed at room temperature and were placed in a 25°C incubator with a 12/12 h photoperiod for at least 48 h to acclimate to standard metabolic conditions. Resting metabolic rates were measured on flies that were 5–6 d postemergent using methods described previously (Van Voorhies et al., 2003, 2004). Briefly, respiration was measured as the CO2 produced by individual flies maintained in a 2.2 ml glass sealed chamber flushed with CO2-free, water-saturated (100% relative humidity) air. Gas samples were removed from the chamber with a Hamilton SampleLock syringe (Hamilton, Reno, NV) and directly injected into a 150 ml/min (±1%) standard temperature pressure dry, CO2-free carrier air stream. Flow of the carrier air stream was controlled with a mass flow meter (Sierra Instruments, Monterrey, CA) and was scrubbed of water vapor with a magnesium perchlorate filter before entering into a Li-Cor 6251 carbon dioxide gas analyzer (Li-Cor, Lincoln, NE). The Li-Cor 6251 analyzer has a sensitivity of <0.1 ppm and an accuracy of <1 ppm. The amount of CO2 produced by each fly was calculated using DATACAN software (Sable Systems International, Henderson, NV). Each fly examined was briefly frozen at –80°C, and its mass was determined using a Sartorius (Goettingen, Germany) M2P microbalance. The CO2 gas analysis system was zeroed daily against CO2 free air and calibrated against a 51 ppm certified gas standard (Air Products, Long Beach, CA.). Previous studies have established that CO2 production provides a sensitive and accurate proxy for oxygen consumption in Drosophila melanogaster (Van Voorhies et al., 2004). Results expressed in microwatts/fly are as follows: 19.6 ± 0.7 ATP61 females, 20.8 ± 0.9 wild-type females, 15.7 ± 0.4 ATPalphaDTS1R1 (1R1)-positive control females, 15.1 ± 0.5 ATP61 males, 13.3 ± 0.5 wild-type males, and 10.2 ± 0.2 ATPalphaDTS1R1 (1R1)-positive control males (SE provided). Our studies revealed severely reduced respiration in ATPalphaDTS1R1 flies that served as a positive control for these studies. ATPalphaDTS1R1 are heterozygous for ATPalpha, the Drosophila gene encoding the Na+/K+ ATPase α subunit (Palladino et al., 2003).
Results
Maternally inherited mutation causes conditional paralysis
Mutations affecting ANT1 in Drosophila were originally identified as a stress-sensitive strain of animals (sesB) that are conditionally paralytic in response to mechanical stress (Homyk and Sheppard, 1977). It was later discovered that sesB1 is an adult viable hypomorphic allele of the gene encoding ANT1 and that null alleles are lethal (Zhang et al., 1999). Previous studies have shown that conditional paralytic mutants in Drosophila are enriched for those that cause neurodegeneration (Palladino et al., 2002). During the course of studying sesB1(ANT1), a separate maternally inherited genetic component was discovered that enhances the conditional paralysis of sesB1(ANT1) and itself causes conditional paralysis. This maternal component was genetically isolated and named for the maternally derived phenotype MATBS (maternal isolate, bang sensitive; see Materials and Methods for details). MATBS animals transmit conditional paralysis and reduced lifespan (with 100% penetrance) to every strain tested but only when maternal inheritance is maintained. The maternal effect is stable; MATBS strains have been maintained for numerous generations without phenotypic erosion.
Previous studies have shown that a pathogenic variant of the bacterium Wolbachia, a naturally occurring insect symbiont, could infect Drosophila and cause a shortening of lifespan (Min and Benzer, 1997b). To test whether a pathogenic variant of Wolbachia could be the source of the MATBS maternally inherited phenotypes, Wolbachia-specific primers and an established PCR detection protocol were used as an assay for the presence of these bacteria (Min and Benzer, 1997b). Wolbachia were detected in a positive control strain known to harbor the bacteria but were not present in our mutant strain (data not shown). Furthermore, tetracycline treatment for two generations has been shown to completely rid flies of microbial parasites, including Wolbachia, and this treatment did not modify the phenotypes associated with MATBS (Min and Benzer, 1997b). The strict maternal inheritance and antibiotic resistance of the MATBS phenotypes led us to hypothesize that there was a mitochondrial mutation in this strain of flies.
MATBS animals have a mitochondrial mutation in their ATP6 gene
The mitochondrial genome was amplified in overlapping ∼3 kb fragments using PCR and directly sequenced from our MATBS and two wild-type control strains. tRNA and rRNA sequences revealed no changes. One nonpolymorphic change in the coding regions was discovered that was unique to the MATBS strain. Interestingly, this G to A transition appeared homogeneous in the sequence chromatogram and results in a glycine to glutamate codon change at position 116 in the ATP6 gene (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Alignment of the ATP6 amino acid sequences revealed that the glycine residue is invariant in insects and mammals. We have renamed MATBS as ATP61. The ATP6 gene encodes an essential subunit of the mitochondrial F1F0-ATP synthase (Complex V), which is localized to the inner mitochondrial membrane. Specifically, ATP6 is known to function as a hydrogen ion channel that couples ion transport with rotary ATP catalysis (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). ATP6 is a known target for several human disease mutations that result in complex neuromuscular diseases, such as NARP, MILS, and FBSN syndromes (Table 1).

Lifespan analysis of mitochondrial mutants. sesB1 (ANT; blue) and ATP61 (red) mutant flies have a 31 and 67% reduction in median lifespan from wild-type control animals (WT; green), respectively. Flies bearing both sesB1 (ANT) and ATP61 mutations (purple) have an additional reduction in lifespan: 76% reduction in median lifespan from control. All comparisons are based on six populations of each genotype. All mutant median lifespans are significantly reduced from wild type (Student's t test; p < 0.001). Error bars represent SEM.
Human ATP6 mutations that affect several well characterized residues cause numerous related neuromuscular diseases. Interestingly, each of these mutations is aphenotypic when present at low heteroplasmic levels but results in disease when at high levels. For example, one of the best-studied ATP6 mutations, which results in an L156R substitution, is generally aphenotypic when below ∼70% heteroplasmy but causes NARP disease when ∼70–90% heteroplasmic (Tatuch et al., 1992). NARP is characterized by adult-onset progressive wasting, neurogenic muscle weakness, and neuropathy. However, when the same mutation is present with greater than ∼90% heteroplasmy, a much more severe disease, MILS, typically results (Schon et al., 2001). MILS is a devastating multisystem childhood syndrome with an extremely poor prognosis. Like many mitochondrial encephalomyopathies, NARP and MILS are complex diseases characterized by a variety of complications resulting from renal, digestive, and cardiac system impairment. The chromatographic data suggest that the ATP61 transition is nearly homoplasmic (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Although heterozygosity can often be seen in chromatographic data, such findings are not strictly quantitative and cannot be used to accurately measure heteroplasmy. To more precisely quantify the frequency of the ATP61 mutation, 90 ATP61-derived clones from three independent experiments were examined and revealed a 98 ± 2% mutant sequence. A similar analysis of 30 wild-type-derived clones from three independent experiments revealed that 100 ± 0% of the clones contained the predicted wild-type sequence. These data demonstrate that the ATP61 mutation is nearly, but not completely, homoplasmic in the fly.
ATP61 animals have shortened lifespans and tissue degeneration
It is intriguing that the ATP61 mutation was discovered in the sesB1 (ANT1) mutant genetic background and that each mutation results in a qualitatively similar conditional paralysis phenotype. Lifespan analyses revealed that both sesB1 (ANT1) and ATP61 animals have a significant reduction in lifespan from wild-type controls (Fig. 1). Despite the similar paralysis phenotype, each mutant strain's lifespan impairment was distinct: ATP61 and sesB1 median adult lifespan was 12 and 25 d, respectively, whereas control animals' median lifespan was ∼50 d. The lifespan of the ATP61; sesB1 double mutants revealed an additional impairment of adult viability beyond each of the individual mitochondrial mutants (Fig. 1). It is important to note that ATP61 does not appear to compensate for the sesB1 mutation and improve viability.
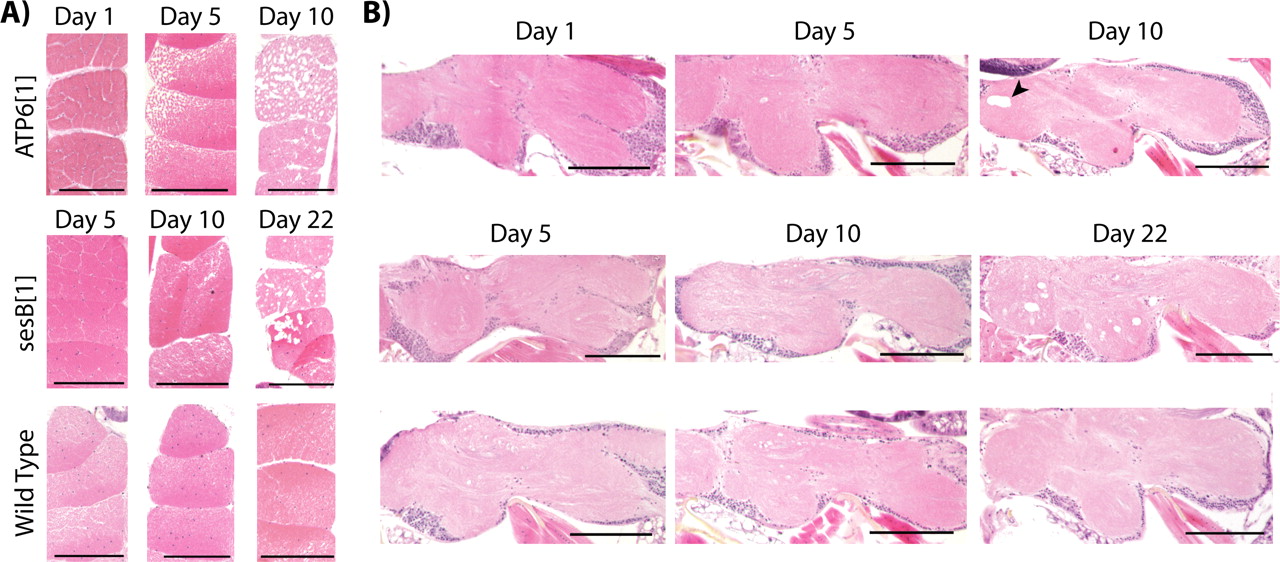
Mitochondrial dysfunction is known to manifest as neurological and muscular impairment in animals because of the intense metabolic demands of myofibrils and neurons. Furthermore, several Drosophila mutants with shortened lifespan have been characterized and have been found to result in neurodegeneration (Kretzschmar et al., 1997; Min and Benzer, 1997a, 1999; Rogina et al., 1997; Palladino et al., 2002, 2003). Thus, to test whether neuropathology may be causing the reduction in the ATP61 median lifespan, mutant brains were examined for neurodegeneration using standard paraffin histology and H&E staining procedures. There were no apparent histologic abnormalities in aged ATP61 mutants (Fig. 2). However, severe neuropathology was observed in aged ANT (sesB1) and ATP61; sesB1 double mutant brains (Fig. 2). In all genotypes examined, young animals were free of pathology, demonstrating the neurodegeneration is strictly progressive (data not shown). Although ATP61 mutants do not have obvious neurodegeneration, the mutation dramatically accelerates the onset of severe neuropathology in sesB1 (ANT1) mutants from days 23 to 12 (Fig. 2B,C). Day 12 sesB1 (ANT1) mutants did not exhibit neurodegeneration and were indistinguishable with age-matched ATP61 mutants and aged wild-type flies (data not shown). NARP and MILS diseases are associated with profound neuromuscular impairment, prompting examination of muscle integrity in ATP61 mutants. Histology was performed to examine the dorsal flight muscles for the presence of pathology in young and aged animals using standard paraffin histology and (H&E) staining procedures, which revealed muscle degeneration in ATP61 mutants that was strictly progressive (Fig. 3A). Young animals revealed no evidence of myopathology in ATP61, sesB1 (ANT1), or wild-type controls. In contrast, ATP61 mutants revealed striking pathology at day 10, a time consistent with the onset of poor viability (Figs. 1, 3A). Day 5 ATP61 mutants have less severe myopathology. In contrast, sesB1 (ANT1) mutants revealed only minimal evidence of myopathology by day 10; however, this mutant strain had marked myopathology by day 22 (Fig. 3A). Examination of the thoracic ganglion, which comprises another significant portion of the fly's CNS, revealed age-dependent neurodegeneration in sesB1 (ANT1) mutants (Fig. 3B). ATP61 mutant histopathology revealed some evidence of thoracic ganglion neurodegeneration, namely individual large vacuolar structures that were not observed in age matched wild-type animals (Fig. 3B). As seen in the brain, the ATP61 mutant results in neural dysfunction and accelerates sesB1 degeneration of the thoracic ganglion (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). It is intriguing that sesB1 (ANT1) mutation results in severe neuromuscular pathology and ATP61 mutation results predominantly in muscle pathology, yet both mutants are predicted to disrupt mitochondrial ATP output and cause a similar locomotor impairment.

Neuropathology in Drosophila mitochondrial mutants. H&E-stained midbrain frontal sections from ATP61 day 14 (A), ATP61; sesB1 (ANT1) day 12 (B), sesB1 (ANT1) day 23 (C), and aged wild-type control (day 22) (D) animals. Both ATP61; sesB1 (ANT1) and sesB1(ANT1) mutants show marked vacuolar pathology throughout the central brain and optic lobes. This analysis did not reveal neuropathology in aged ATP61 or wild-type animals. In young animals of all genotypes, there is no evidence of neural pathology (data not shown). n ≥15 animals per genotype. Scale bar, 100 μm.
Locomotor impairment in mitochondrial mutants
One hallmark of mitochondrial encephalomyopathies is the progressive onset of symptoms, including locomotor impairment. Both sesB1 (ANT1) and ATP61 mutants have reduced locomotor function and conditional paralysis brought on by mechanical stress. We characterized the time required for recovery from physical stress in both mitochondrial mutants. Wild-type flies are relatively unaffected by this stress and recover within ∼2–3 s at all time points examined. There is a small but significant decrement with age in wild-type animals observed using this assay, as seen by comparing day 2 and day 24 animals (Fig. 4) (p < 0.05). In contrast, both sesB1 (ANT1) and ATP61 mutants revealed a markedly progressive paralysis phenotype. Young mutants, day 2 and 4, are not more affected by mechanical stress than wild type (p > 0.05), although there is greater variability in recovery time in the mutants than age-matched wild-type animals. However, by day 6, both mitochondrial mutants demonstrate significant locomotor impairment (Fig. 4). The trend continues for sesB1 (ANT1) mutants at day 24, in which paralysis typically lasts ∼3 min. Interestingly, ATP61; sesB1 double mutants revealed enhanced locomotor impairment with an earlier onset than either of the separate mitochondrial mutants.

Progressive neuromuscular pathology in Drosophila mitochondrial mutants. Cross sections of dorsal flight muscles (A) and longitudinal sections of the thoracic ganglion (B) were examined. A, ATP61 mutants (top) revealed severe progressive degeneration of myofibrils in day 5 and day 10 animals that was never observed in wild-type controls (bottom). ATP61 mutants (top) revealed occasional large vacuoles (arrowhead) on day 10 that were not observed in wild-type animals. sesB1 (ANT1) mutants (middle) revealed intermediate myodegeneration on day 10 and severe degeneration on day 22. B, sesB1 (ANT1) mutants (middle) show vacuolar pathology of the thoracic ganglion by day 22 compared with age-matched wild-type controls (bottom). Animals were killed, fixed in 2.5% gluteraldehyde, and paraffin embedded, and serial 5 μm sections of the thorax were obtained and stained with H&E. In young animals of all genotypes, there is no evidence of neuromuscular pathology. n ≥ 8 animals per genotype per time point. Scale bars, 100 μm.
Transmission electron microscopy analysis reveals mitochondrial pathology
The F1F0 ATP synthase (Complex V) uses the mitochondrial electrochemical potential in the form of a hydrogen ion gradient to drive rotary catalysis of ATP. Mutations affecting ATP6, the hydrogen ion channel through which dissipation of this gradient is coupled to ATP production, are thought to have a profound affect on mitochondria function (Mitchell and Moyle, 1967; Walker, 1995; Boyer, 1997; Stock et al., 2000). Mitochondrial dysfunction is known to manifest as neurological and muscular impairment in animals because of the intense metabolic demands of these tissues. The ATP61 mutation causes severe progressive muscle degeneration but not marked neurodegeneration. It does, however, enhance the neurodegeneration phenotype of sesB1 (ANT1), demonstrating that the underlying dysfunction is present in this tissue. To examine the consequence of this dysfunction without risk of the results being confounded by tissue degeneration, thin sections of the cortical and central brain regions of day 10 ATP61 and age-matched control animals were examined using transmission electron microscopy (TEM). The analysis revealed mitochondria displaying highly abnormal internal compartmentalization that was not typical in age-matched wild-type flies (Fig. 5). The numerous small, round cristae (∼50–80 nm in diameter) give the mitochondria a honeycomb appearance. This was not an isolated or rare finding: the vast majority of mitochondria observed in numerous midbrain micrographs from independent ATP61 animals had abnormal internal membrane morphology (n > 40 micrographs from four independent animals). TEM micrographs were acquired using stereological protocols allowing the frequency of this morphological abnormality to be quantified in a blinded analysis of randomly selected midbrain micrographs covering 711 μm2 tissue from ATP61 and wild-type control animals (Fig. 5C). The frequency of abnormally compartmentalized mitochondria was significantly higher in mutant brains and, correspondingly, the frequency of normal mitochondrial was significantly reduced (p < 0.01). Although mitochondria are morphologically abnormal, the density (number of mitochondria per unit area) of mitochondria in ATP61 (31 ± 3 per 100 μm2) is not significantly different from that of wild-type control animals (35 ± 2 per 100 μm2; Student's t test; p = 0.45). Previous ultrastructural examination of mitochondria has revealed the dynamic nature of mitochondrion structure (Hackenbrock, 1966). More recently, tomographic modeling has confirmed the functional importance of the mitochondrial internal membrane, which can become compartmentalized in conditions associated with mitochondrial impairment (Frey and Mannella, 2000).

Progressive locomotor impairment in mitochondrial mutants. Locomotor impairment is quantified as the recovery time from stress-induced paralysis. Mechanical stress/hyperstimulation induces sustained paralysis in sesB1 (ANT1; blue) and ATP61 (red) mutant strains, which increase significantly with age. The ATP61; sesB1 double mutants (purple) show increased severity relative to each individual mutant strain and wild-type flies and exhibit a faster progression of the locomotor impairment. Wild-type animals are extremely tolerant to mechanical hyperstimulation and generally recover within 1 or 2 s with little increase over time (green). The asterisk indicates significant difference from age-matched wild-type controls (Student's t test; p < 0.01). Error bars represent SEM. Three populations of 15 animals/genotype were tested. ATP61 and ATP61; sesB1 double mutants were not tested beyond day 8 because of morbidity in the populations (Fig. 1).
TEM tomography analysis of ATP61
The appearance of small, round cristae in mitochondria of ATP6 mutants is reminiscent of a morphological abnormality associated with a human mitochondrial myopathy, Senger's syndrome (Huizing, 1998; Jordens et al., 2002). In the latter case, three-dimensional imaging of the mitochondria, using electron tomography of thick sections, demonstrated that the round cristae are, in fact, vesicles that are not attached to the inner boundary membrane by the usual crista junctions (Frey and Mannella, 2000). To elucidate the structure of the mitochondrial inner membrane in the Drosophila mutant, we generated tomographic reconstructions of the aberrant ATP61 mitochondria. The tomographic analysis verified the roughly spherical shape of the cristae in the honeycomb regions of these mitochondria but also revealed a more complex three-dimensional structure than was evident by standard TEM analysis. Specifically, the round inner membrane compartments are not individual, unconnected vesicles (as in the case of Senger's syndrome) but rather are part of a highly interconnected membrane network (Fig. 6). The tightly packed spherical compartments are connected by narrow tubular junctions to each other and to the inner boundary membrane (i.e., the region that parallels the outer membrane) (Fig. 6C,D,G). An even more intriguing feature of the tomographic reconstructions is that the roughly spherical compartments are clearly contiguous with the lamellar regions of the inner membrane (Fig. 6E,F). We were unable to find a single unambiguous example of a truly vesicular (unattached) inner membrane compartment within the tomogram, suggesting that the round compartments represent local dilations in the lamellae that remain physically coupled with the rest of the inner-membrane space. At numerous places the lamellar membranes were observed to be fenestrated (evident in Fig. 6E,F). See supplemental movies 1 and 2 (available at www.jneurosci.org as supplemental material) for two independent tomograms of ATP61 mitochondria, movie 3 (available at www.jneurosci.org as supplemental material) for an animation of the full membrane model of Figure 6, C and D, and movie 4 (available at www.jneurosci.org as supplemental material) for a tomogram of wild-type mitochondrion.
Mitochondrial function in ATP61 mutants
Mitochondrial encephalomyopathies are collectively an extremely prevalent and devastating class of diseases that can result from a variety of mitochondrial or nuclear mutations. Mitochondrial encephalomyopathies associated directly with ATP6 mutation, namely NARP, MILS, and FBSN, can result from numerous mutagenic events (Table 1) with the resulting disease onset and severity highly dependent on the percentage of homoplasmy of the mutation. The pathological basis of these diseases is uncertain, but it has been hypothesized that bioenergetic impairment and oxidative stress resulting from respiratory chain dysfunction contribute to the disease condition. To test whether the ATP61 mutation altered mitochondrial ATP synthase activity in vivo, luciferin-luciferase luminescence assays (Manfredi, 2001) were used to biochemically measure the oligomycin-dependent component of ATP generation from ATP61 mitochondria (Fig. 7A). Wild-type control mitochondria had robust ATP synthase activity (23.3 μmol/min/mg), whereas ATP6 mutant mitochondria never revealed significant ATP synthase activity in this assay (Fig. 7B) (p < 0.01). We similarly examined ATP synthase activity from wild-type flies acutely treated with dinitrophenol (DNP). DNP-treated flies produced 6.9 μmol/min/mg ATP synthase activity, a significant reduction from untreated wild-type flies (p < 0.05). These data demonstrate that ATP synthase function in ATP61 mutants is markedly reduced and that the resulting bioenergetic impairment likely contributes to disease pathogenesis.

Abnormal mitochondrial morphology in ATP61 mutants. TEM reveals abnormal internal mitochondria membrane structure in ATP61 mutants. Within ATP61 brain tissues, mitochondria have a highly abnormal internal membrane morphology characterized by numerous vesicular cristae resulting in mitochondria with a honeycomb appearance (B) that were only rarely observed in age-matched wild-type animals (A). Aside from the apparent mitochondrial pathology, brain morphology is essentially normal. Scale bars, 500 nm. C, Quantification of the mitochondria vacuolar morphology revealed an extremely high frequency in ATP61 brains (62% of total mitochondria), whereas this morphology was infrequent in wild-type control animals (<5%). Less than 25% of ATP61 mitochondria appeared grossly normal compared with > 85% in wild-type brains. At times, there was insufficient tissue in the micrograph plane to be certain as to whether the mitochondria were of normal morphology (ambiguous). Statistical comparisons using a Student's t test between mutant and wild type (*p < 0.01) are shown. The frequency of ambiguous mitochondria was not different between mutants and controls (p = 0.38). Error bars represent SEM.
The abnormal morphology of ATP61 mitochondria and the reduction in mitochondrial ATP synthase activity prompted us to investigate whether an altered rate of respiration in these animals may also be contributing to the disease pathogenesis. The rate of respiration of ATP61 and age-matched wild-type control populations was measured in a fixed volume respirometer. Surprisingly, both genotypes produced an indistinguishable linear response (Fig. 7C). The respiration rate was derived from the slope of the best-fit line and was not different between the ATP61 mutants and controls (p = 0.68; n = 4). Although these data strongly argue that respiration rate is not affected in the mutant strain, an independent and extremely sensitive single fly assay was used to verify this finding (Van Voorhies et al., 2003, 2004). The resting metabolic rate of individual ATP61 and age-matched wild-type control flies was measured and, in accord with population measurements, there was no significant difference in metabolic rate between these strains (Fig. 7D)(p > 0.33). There was a significant decrease in the respiration rate from a positive control ATPalphaDTS1R1 (1R1) strain (p < 0.001). These data suggest that our assay of respiration rate is sufficiently sensitive to detect differences in metabolic rate and that there is no apparent dysfunction in respiration rate contributing to the neuromuscular condition in ATP61 mutant flies.
Discussion
Mitochondrial encephalomyopathies are a collection of devastating medical conditions that are poorly understood and for which therapeutic treatments remain extremely limited. Our understanding of these mysterious diseases is constrained by our limited ability to genetically manipulate mitochondria, difficulties associated with studying progressive conditions, and a lack of tractable in vivo models that recapitulate these diseases. This is especially true of conditions associated directly with mutation of the mitochondrial genome, such as NARP, MILS, and FBSN. We identified marked neuromuscular degeneration associated with mutations of the nuclear gene sesB that encodes the mitochondrial ANT1 protein. We also isolated a nearly homoplasmic mitochondrial mutation of the mitochondrial ATP6 gene that results in a marked shortening of lifespan, locomotor impairment, and progressive myodegeneration. These studies demonstrate that normal respiration rates are maintained despite a defect in mitochondrial ATP synthase activity. To our knowledge, this ATP61 Drosophila mutant is the first isolation of an endogenous mitochondrial mutation that recapitulates key pathological features of human mitochondrial disease. This tractable genetic model of MILS disease will allow detailed studies to further elucidate the pathogenic basis of this disease and related mitochondrial encephalomyopathies.
Mitochondrial morphology and neuromuscular pathology
Examination of brain mitochondria in ATP61 mutants revealed a striking morphological defect associated with the neural dysfunction in these mutants. The pathology resulted in mitochondria with a honeycomb appearance that was presumably caused by vesicularization of the inner mitochondrial membrane. More detailed TEM tomography revealed that the roughly spherical inner membrane compartments were highly interconnected and contiguous with lamellar regions of the mitochondrial inner membrane. These findings are especially intriguing because similar (albeit not identical) morphological abnormalities are seen in a human myopathy (i.e., vesicular cristae in Senger's syndrome) (Huizing, 1998) and an avian model system (canary myocardial mitochondria) (Slautterback, 1965; Frey and Mannella, 2000). The ultrastructural similarities among these three diverse systems suggest a conserved underlying pathophysiology and argue that our model system will provide valuable insight into the molecular basis of human mitochondrial diseases. In the case of Senger's syndrome, there is evidence that the disorder is causally linked to a deficiency of ANT1 protein (Jordens et al., 2002). Such a defect could account for both a severe bioenergetic deficit in these patients and altered physical properties of the inner membrane. For example, an inhibitor-induced conformational change in ANT causes aberrant morphology of the inner membrane of isolated liver mitochondria (Scherer and Klingenberg, 1974). Likewise, a mutation in the subunit e of yeast ATP synthase that inhibits dimerization of the protein complex results in abnormal cristae morphology (Paumard et al., 2002). Whether the ATP61 mutation affects intermolecular interactions of the ATP synthase and whether this might account for the observed abnormality in inner membrane morphology of Drosophila mitochondria will need to be examined.

Tomography TEM analysis of ATP61 mitochondrial innermembranes. Day 10 ATP61 central brain mitochondria were examined using electron tomography. A, Projection image of an untilted thick section (∼0.2 mm) of a typical mitochondrion shows a honeycomb appearance similar to that in Figure 6 B; numerous colloidal gold particles used for alignment are evident in the field. B, A representative slice, 2 nm thick, from the center of the tomogram reconstructed from 122 tilted views of the mitochondrion in A. C–G, Volume-rendered models of the tomogram reveal complex inner membrane compartmentalization. For clarity, the model is split into top (1–36; C) and bottom (37–72; D) halves that each reveal an extensively interconnected network of inner membrane compartments. The round compartments are contiguous at numerous sites with lamellae and the intermembrane space. Removing the outer and peripheral inner membranes from the model and clipping it along various planes clearly reveals the transitions between the round, vesicle-like regions and the curving lamellar regions within the inner mitochondrial membrane (E, F). The interconnectedness of the compartments is revealed by regular forking membranous structures seen in the row of vesicle-like cristae. G. Note that the model is oriented in plates E–G such that “up” in these side views corresponds to the top surface shown in C. Also see supplemental movies (available at www.jneurosci.org as supplemental material) of the entire tomogram.
The rate of mitochondria with aberrant morphology was extremely high (>60% of the mitochondria in ATP6 mutants), suggesting widespread dysfunction throughout the brains of these animals. Significant neurodegeneration was not prominent in ATP61; however, neural dysfunction was evident and dramatically increased the onset of neuropathology in sesB1 animals. Severe myopathy was observed in ATP61 mutants coincident with the onset of morbidity in this strain, suggesting a causal relationship. It is curious that sesB1 (ANT) dysfunction results in neuromuscular pathology and ATP61 results primarily in myopathy. It is possible that the severe myopathy in ATP61 results in lethality before the onset of neurodegeneration and that the ultrastructural pathology observed is an early indicator of neuropathogenesis. Although the prevalence of the mitochondrial pathology is extremely high, it does not precisely match the near homoplasmy of the ATP61 mutation (98%). It is possible that the morphological defect represents a more advanced stage of organellar dysfunction and that a small subset of affected (mutant) mitochondria will maintain essentially normal morphology. Alternatively, if one assumes the ATP61 mutation is fully recessive, only mitochondria in which all of the mitochondrial genomes are mutated would exhibit abnormal morphology. Mitochondria are dynamic polyploid organelles that regularly undergo morphological changes as well as fusion and fission. It is extremely difficult to determine the extent of the polyploidy of mitochondria: estimates range from ∼5–25 genomes per organelle (Satoh and Kuroiwa, 1991; Iborra et al., 2004; Ashley et al., 2005). If the ATP61 mutation was fully recessive, one would estimate ∼60–90% of the mitochondria would exhibit the morphological defect (0.98n × 100), which is in good accord with experimental data. It is intriguing that mitochondria with a general honeycomb appearance were observed in age-matched wild-type brains, albeit at a very low rate. This suggests the dysfunction may result from a normal biological function occurring at an accelerated rate. Possibly the inner membrane vesicularization is the result of excessive or poorly controlled mitochondrial fission, fusion, maturation, or a pathology associated with senescence that prematurely manifests with ATP6 dysfunction.

Mitochondrial function is dramatically reduced in ATP61 mutants. A, ATP synthase activity was measured as the oligomycin-sensitive component of ATP production using a luminescent assay. B, ATP synthase activity per microgram of total mitochondrial protein is significantly lower in ATP61 and DNP-treated wild-type animals (acute 100 mm) compared with wild-type controls (error bars represent SEM; n > 14). C, Metabolic rate was determined for each genotype population as the emergence of CO2 in parts per million in a fixed-volume respiration chamber. Respiration rate for ATP61 mutants (22.1 ppm/g/min) was no different from wild-type controls (22.6 ppm/g/min; p = 0.68). Error (SEM) was comparable in both strains; error bars shown are wild-type error (n = 4). D, Resting metabolic rate was additionally measured from individual ATP61 mutants and age-matched wild-type controls. There was no difference in rate of respiration between the mutant and wild-type controls (p > 0.3 males; p > 0.7 females). We also examined a mutant strain with a consistently reduced respiration rate as a positive control. Comparisons of microwatts per fly resulted in the same statistical conclusions as the microwatts per milligram of data presented here. SE is given; n > 15 per genotype per gender. Statistical significance was determined by Student's t test (*p < 0.05, **p < 0.01, and ***p < 0.001).
adPEO and ANT dysfunction
The discovery of a nearly homoplasmic mitochondrial mutation suggests that ANT dysfunction resulting from sesB1 acts as a mutator causing mitochondrial DNA instability and that this dysfunction also provides selective pressure for the inheritance of the mutated mitochondrial DNA in germ cells. In human adPEO disease, muscle biopsies from those inheriting ANT mutations reveal mitochondrial deletions by Southern blot analysis demonstrating the mutator effect and resulting DNA instability (Zeviani et al., 1989; Kaukonen et al., 2000). It is not known whether adPEO disease is solely the result of the primary ANT dysfunction or whether it is attributable to the secondary mutations sustained to mitochondrial DNA. Flies with an ANT point mutation (sesB1) also have progressive myopathy, as do flies with the ATP61 point mutation (lacking the primary ANT dysfunction). These findings do not completely resolve, which is causing the myopathy in adPEO patients; however, our data demonstrate that ATP6 mutation alone causes myopathy independent of ANT dysfunction. Our data additionally suggest that point mutations result from ANT dysfunction and that these, unlike large-scale deletions, are heritable and likely selected for in the germ line.
Generating a nearly homoplasmic mitochondrial DNA (mtDNA) point mutation in a Drosophila strain would require selective pressure. This could result from any of the following conditions: (1) the ATP6 mutation improved the overall fitness of the animal (which appears unlikely given our lifespan, locomotor impairment, and histological data), (2) continued selection in the laboratory for the most affected animals in the strain, or (3) selective inheritance such as a meiotic drive system operating in the female germ line of the animals. The latter is supported by experimental data demonstrating selective maintenance of the variant ATP6 (T8993G) mtDNA (Vergani et al., 1999) and selective inheritance during oogenesis (Blok et al., 1997). Interestingly, in MILS disease, a more severe variant of NARP manifests when the mutant heteroplasmy is above ∼90%. Yet, these diseases are not extremely rare, as might be predicted. It is an enigma how such high heteroplasmy levels are achieved when the mutations that cause these diseases are predicted to have adverse effects on the cells bearing mitochondria with a high mutation load. The discovery and long-term maintenance of the nearly homoplasmic ATP61 mutation in vivo and the findings of Blok et al. (1997) support the hypothesis that meiotic drive contributes to the prevalence of these diseases through selection within the female germ cells for mitochondrial mutations.
Disease pathogenesis
Mitochondrial encephalomyopathies are widely recognized as an important and devastating class of diseases; however, the mechanism of pathogenesis remains controversial. Studies have reported severe oxygen consumption defects (Trounce et al., 1994; Mattiazzi et al., 2004), whereas other studies of the same T8993G mutation did not observe such a defect (Nijtmans et al., 2001). Additionally, studies of ATP synthase activity from cellular models nearly homoplasmic for the T8993G mutation revealed a ∼50% reduction in activity, whereas an Escherichia coli model bearing the analogous mutation abolished all activity (Hartzog and Cain, 1993). More recent studies of the T8993G mutation in cell cybrid models revealed a ∼95% reduction in ATP synthesis and described such a large reduction as a common feature associated with several respiratory chain disease mutations, with the notable exception of the T8993C mutation that revealed only a modest decrease (Pallotti et al., 2004). Nearly homoplasmic ATP61 mutant Drosophila clearly revealed severe neuromuscular impairment associated with marked decrease in ATP synthase activity similar to that reported by cellular models of human mutations (Hartzog and Cain, 1993). Respiration rates are maintained at normal rates in ATP61 mutants similar to what was observed by Nijtmans et al. (2001). These data demonstrate that bioenergetic crisis is integral to the pathogenesis of ATP6 dysfunction. Additionally, the finding that mitochondrial respiration rates are unaffected in vivo suggests that the mitochondria may be functionally uncoupled, which may be integral to the pathogenesis in these mutants. Additional studies will be needed to test whether normal rates of respiration indicate normal reactive oxygen species generation to determine whether oxidative stress may also be contributing to the pathogenesis.
In humans, ATP6 dysfunction results in three different diseases with overlapping clinical manifestations; however, differences between these diseases and even between patients diagnosed with the same disease are evident. Thus, the genotype/phenotype relationship remains ambiguous. It is not evident why numerous mutations all affecting the same biochemical process, presumably resulting in bioenergetic impairment in the affected tissues, result in a veritable plethora of clinical manifestations that are not fully explained by the heteroplasmy of the mutations (Schon et al., 1997; DiMauro and Andreu, 2000). Innovative techniques have been used to model mitochondrial diseases, such as cybrid cellular models derived from affected patient cells (Pallotti et al., 2004; Mattiazzi et al., 2004) and mouse models with mitochondrial DNA that was introduced via embryonic stem cells (Sligh et al., 2000). However, these model systems are difficult to study, and a complete understanding of disease pathogenesis has not emerged. This sentiment was exemplified in a review on mitochondrial encephalomyopathies: “thus far insurmountable problem of obtaining animal models for mtDNA mutations has prevented detailed studies of pathogenesis” (DiMauro and Andreu, 2000). The Drosophila strain reported here will enable additional such detailed studies of pathogenesis associated with ATP6 impairment in vivo.
Footnotes
This work was supported by National Institutes of Health (NIH)–National Institute of Neurological Disorders and Stroke Grant T32NS07391-07 (A.M.C.), Pittsburgh Institute for Neurodegenerative Diseases research award (M.J.P.), The Wadsworth Center's Resource for Visualization of Biological Complexity, NIH National Biotechnological Resource Grant RR01219 from the National Center for Research Resources (Department of Health and Human Services, Public Health Service), and the University of Pittsburgh Department of Pharmacology and School of Medicine. We thank Dr. Christian Renken for assistance with tomographic data collection, Matt Levy and the University of Pittsburgh core facilities for assistance with sequencing, the Pittsburgh Center for the Environmental Basis of Human Disease (CEBHD) for support, Dr. Simon Watkins and the Center for Biologic Imaging for access to microscopes and histology equipment, Ana Bursick for sharing her microscopy expertise, Dr. Valerian Kagan for helpful advice, Drs. Michael Ashburner and Michael Clark and the Bloomington Stock Center for fly strains, Dr. Ian Reynolds for his ongoing support and numerous helpful discussions, and Dr. Bruce Freeman for comments on this manuscript.
Correspondence should be addressed to Michael J. Palladino, Pittsburgh Institute for Neurodegenerative Diseases and Department of Pharmacology, University of Pittsburgh School of Medicine, 200 Lothrop, Biomedical Science Tower E1355, Pittsburgh, PA 15261. E-mail: mjp44{at}pitt.edu.
DOI:10.1523/JNEUROSCI.4162-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260810-11$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}