

Abstract
Here, we show that cultured Purkinje cells from inositol 1,4,5-trisphosphate receptor type 1 knock-out (IP3R1KO) mice exhibited abnormal dendritic morphology. Interestingly, despite the huge amount of IP3R1 expression in Purkinje cells, IP3R1 in granule cells, not in the Purkinje cells, was responsible for the shape of Purkinje cell dendrites. We also found that BDNF application rescued the dendritic abnormality of IP3R1KO Purkinje cells, and that the increase in BDNF expression in response to activation of AMPA receptor (AMPAR) and metabotropic glutamate receptor (mGluR) was impaired in IP3R1KO cerebellar granule cells. In addition, we observed abnormalities in the dendritic morphology of Purkinje cells and in the ultrastructure of parallel fiber–Purkinje cell (PF-PC) synapses in IP3R1KO mice in vivo. We concluded that activation of AMPAR and mGluR increases BDNF expression through IP3R1-mediated signaling in cerebellar granule cells, which contributes to the dendritic outgrowth of Purkinje cells intercellularly, possibly by modifying PF-PC synaptic efficacy.
Introduction
The proper growth and branching of dendrites profoundly affects the formation of neural networks and neural functions. The spatial patterns of dendritic morphology determine the number and patterns of synapses received by individual neurons and the efficacy with which synaptic information is transmitted to the soma. The molecular mechanism that regulates the development of the dendritic arbor has been intensively examined recently, and numerous lines of evidence indicate that dendritic growth is a dynamic process that is regulated by various environmental cues, including synaptic activity and molecular signals from adjacent cells (Whitford et al., 2002; Wong and Ghosh, 2002; Miller and Kaplan, 2003; Konur and Ghosh, 2005; Redmond and Ghosh, 2005).
Inositol 1,4,5-trisphosphate receptors (IP3Rs) constitute a family of Ca2+ channels that is responsible for Ca2+ mobilization from intracellular stores (Berridge et al., 2000), and three subtypes have been identified: IP3R1, IP3R2, and IP3R3 (Furuichi and Mikoshiba, 1995; Patel et al., 1999). IP3R1 is the predominant subtype in the CNS, especially in cerebellar Purkinje cells (Furuichi et al., 1993). IP3Rs have been shown to play crucial roles in several neuronal functions, including synaptic plasticity (Matsumoto et al., 1996; Inoue et al., 1998; Fujii et al., 2000; Nishiyama et al., 2000; Itoh et al., 2001), axonal extension (Takei et al., 1998), and nerve growth cone guidance (Xiang et al., 2002). However, in contrast to our knowledge of the role of IP3R-mediated signaling in axon growth and guidance, it remains unknown whether IP3R-mediated signaling contributes to shaping the dendrites of neurons.
In this study, we used IP3R1 knock-out (KO) mice to examine the role of IP3R1-mediated signaling in the dendritic outgrowth of Purkinje cells. First, we discovered that Purkinje cells from IP3R1KO mice exhibit abnormal dendritic morphology in culture, and by means of simple experimental techniques we demonstrated that the abnormal morphology of Purkinje cells was caused by a lack of IP3R1 in cerebellar granule cells rather than in Purkinje cells. We also observed a defect of the increase in BDNF production after AMPA receptor (AMPAR) and metabotropic glutamate receptor (mGluR) stimulation in IP3R1KO cerebellar granule cells, and that exogenous application of BDNF rescued the abnormal morphology of IP3R1KO Purkinje cells. In addition, we observed enlarged presynaptic terminals of parallel fiber-Purkinje cell (PF-PC) synapses and vesicle accumulation in IP3R1KO mice in vivo. Together, our findings strongly suggest that AMPAR and mGluR stimulation increases BDNF expression at least in part through IP3R1-mediated signaling in cerebellar granule cells, which intercellularly controls the dendritic outgrowth of Purkinje cells, possibly by altering PF-PC synaptic efficacy. Thus, our data provide an important clue to understanding the molecular basis of intercellular communication between neurons in shaping the dendritic morphology.
Materials and Methods
Cerebellar neuronal culture.
Dissociated cerebellar neuron cultures were prepared from postnatal day 1 (P1) to P2 wild-type (WT) and IP3R1KO mice according to a method for rat Purkinje cell culture established previously (Furuya et al., 1998) with some modifications. Briefly, cerebellar cells were suspended in medium containing 10% fetal bovine serum at a density of 7.5 × 106 cells/ml. Forty microliters of the suspension were spotted (spots are ∼4–5 mm in diameter, resulting in ∼1.5–2.5 × 106 cells/cm2) onto plastic coverslips (13.5 mm in diameter; Sumilon; Sumitomo Bakelite, Tokyo, Japan) coated with 200 μg/ml poly-l-lysine and placed in a humidified CO2 incubator (5.0% CO2 at 37°C). After 12 h, serum-free medium was added to each well (final serum concentration, 1.0%). The medium was composed of Eagle's minimal essential medium supplemented with 1.0 mg/ml bovine albumin (Sigma, St. Louis, MO), insulin–transferrin–serenium (Invitrogen, Carlsbad, CA), 0.1 nm l-tyroxine (T4) (Sigma), 1.0 μg/ml aprotinin (Sigma), 0.25% glucose, 100 μm l-serine, 2.0 mm glutamine, 2.0 mg/ml Na2CO3, 100 U/ml penicillin, and 135 μg/ml streptomycin. To determine the effect of exogenous BDNF on dendritic outgrowth, 10 ng/ml BDNF (Sigma) was added to culture from 9 d in vitro (div) for 5 d.
For overlay assay, 40 μl of cerebellar cell suspension in medium containing 10% bovine serum (7.0 × 106cells/ml) from either P7 WT or IP3R1KO mice were spotted on the plastic coverslips. After 24 h, 10 μl of cerebellar cells (7.0 × 106 cells/ml) from P1 IP3R1KO mice were spotted over the P7 cerebellar cells. After 24 h, serum-free medium was added to each well. The final percentage of serum was 1.0%.
Cerebellar glial cell culture was prepared from P1–P2 WT or IP3R1KO mice cerebellum. Briefly, the cerebellar cells were cultured in the 25 ml noncoated flask. On the next day, nonadherent cells were washed away, and adherent cells were cultured further. After cells were confluent, cells were removed with 1.0% trypsin-EDTA in HBSS and replated on noncoated flask. The cells obtained by this method were confirmed to consist of glia using anti-GFAP antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Glial cells were mixed with cerebellar cells at a ratio of 3:1, and the mixed cells were spotted on plastic coverslips. After 12 h, serum-free medium was added to make final serum concentration to be 1.0%.
Immunoblotting.
For Western blotting of granule cells, cerebellar cells taken from P7 WT or IP3R1 KO mice were cultured in the media (final, 1.0% serum) containing 20 μm AraC (50 μl of 7 × 106 cells/ml) for 5–6 d. In this condition, no Purkinje cells and glial cells survived. Cells were lysed with 150 μl of sample buffer, and the cell lysates were boiled and separated by 7.5% SDS-PAGE and transferred to a polyvinyldene difluoride membrane. The membrane was treated with the blocking solution (0.05% Tween 20/PBS) containing 5.0% skim milk and probed with the indicated primary antibodies. The antibodies were used at the following concentrations: rabbit polyclonal anti-mGluR5 antibody (1.0 μg/ml; Upstate Biotechnology, Lake Placid, NY), mouse monoclonal anti-calreticulin antibody (1.0 μg/ml; Affinity BioReagents, Golden, CO), mouse monoclonal anti-β-actin antibody (1.0 μg/ml; Sigma), rabbit polyclonal anti-pan-IP3R antibody (Hattori et al., 2004) (× 200), and rabbit polyclonal anti-IP3R1 antibody (1.0 μg/ml). After incubation with horseradish peroxidase-labeled secondary antibodies (GE Healthcare, Arlington Heights, IL), the immobilized specific antigen was visualized with ECL plus detection kit (GE Healthcare).
Immunocytochemistry.
Cultured cells were washed with PBS and fixed with 4.0% paraformaldehyde in PBS for 15 min at room temperature (RT). After being washed with PBS, cells were permeabilized with 0.2% Triton in PBS for 10 min at RT and blocked with 3.0% skim milk in PBS for 60 min at RT. The coverslips were incubated with primary antibodies for 2 h at RT or overnight at 4°C. After being washed with PBS three times for a total of 15 min, the coverslips were incubated with fluorescence-conjugated secondary antibodies (Alexa 488 or Alexa 596) (Invitrogen, Eugene, OR) for 60 min at RT. After being washed three times with PBS for 15 min, the coverslips were mounted with Vectashield (Vector Laboratories, Burlingame, CA) and observed under an Olympus BX50 microscope (Olympus, Tokyo, Japan). Antibodies used were as follows: rat anti-IP3R1 monoclonal antibody (18A10) (Maeda et al., 1988), mouse anti-calbindin monoclonal antibody (1:400; Swant, Bellinzona, Switzerland), rabbit anti-NSE (neuron-specific enolase) polyclonal antibodies (1:200; Polysciences, Warrington, PA), and mouse anti-GFAP monoclonal antibody (1.0 μg/ml; Santa Cruz Biotechnology).
To measure maximum length of a primary dendrite, numbers of branch points on the primary dendrite, and dendritic area, images were stored and analyzed using IPLab (Scanalytics, Fairfax, VA) or a custom-made software (TI Workbench, written by T.I.). Branch points were counted along a longest dendrite of each Purkinje cell. Dendritic area was measured by connecting the tips of dendrites including soma with straight line.
Ca2+ imaging.
Cerebellar cells plated on poly-l-lysine-coated plastic disk were placed in a 3.5 cm glass-bottom dish (Matsunami, Tokyo, Japan) and loaded with 5.0 μm fura-2 AM (Dojin Chemical, Kumamoto, Japan) for 30 min at room temperature in HEPES-buffered salt solution (HBSS) (in mm): 115 NaCl, 5.4 KCl, 1 MgCl2, 2 CaCl2, 20 HEPES, 10 glucose, pH 7.42. Fura-2 fluorescence images were analyzed using an inverted microscope (IX70; Olympus) and TI workbench with excitation filters at 340 ± 10 and 380 ± 10 nm, a beam splitter at 400 nm, and a bandpass emission filter at 510–550 nm. HBSS was used for the recording solution unless otherwise mentioned.
Reverse transcription-PCR.
Cerebellar granule cells (3.5 × 105) from P7 WT or IP3R1KO mice in 12-well dishes cultured for 6 d in the presence of 20 μm AraC were incubated with 1.0 μm TTX for 2 h to reduce basal neuronal activity. Then, the neurons were stimulated with 50 μm (RS)-3,5-dihydroxyphenylglycine (DHPG) and 10 μm AMPA. After 4 h, the neurons were washed with PBS once and dissolved in 1.0 ml of Trizol reagent (Invitrogen), and total RNA was extracted according to the instructions of the manufacturer. First-strand cDNA was produced from the total RNA using reverse transcriptase Superscript II (Invitrogen) and oligonucleotide (dT) primers. The cDNAs were amplified with specific primers: BDNF, sense 5′-TACTTCGGTTGCATGAAGGCGGCG-3′, antisense 5′-TCCAAAGGCACTTGACTGCTGAGC-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), sense 5′-ATGGTGAAGGTCGGTGTGAACCG-3′, antisense 5′-AAACATGGGGGCATCGGC-AGAA-3′. After an initial cycle of 2 min at 94°C, the reaction was cycled for 30 s at 94°C, 30 s at 55°C, and 30 s at 72°C, 25 times for GAPDH and 35 times for BDNF. For analysis, PCR products were separated by electrophoresis in 1.5% agarose gel and stained with ethidium bromide, and the band intensities were quantified with TI workbench. The expression level of GAPDH mRNA was used as an internal control.
Visualizing Purkinje cell shape in acute brain slices.
Cerebellar slices (250 μm thick) were prepared according to a standard method (Kuruma et al., 2003) using P17–P20 WT and IP3R1KO mice. Purkinje cells in the slices were patch clamped at the soma with internal solution containing 500 μm fluorescein (Sigma). After waiting 15 min for the dye perfusion into the dendrite, the shape of single Purkinje cells was recorded with a two-photon confocal microscope with a 60× water immersion lens (numerical aperture, 0.90; Olympus). Consecutive 50–70 focal planes separated by 1 μm were taken, and two-dimensional images of Purkinje cells were reconstructed by picking brightest pixel along each z-axis after correcting tilts of the dendritic plane by fitting to a horizontal plane in the three-dimensional space. Data analysis was performed with TI Workbench.
Ultrastructure study.
Anesthetized mice were intracardially fixed with 2.0% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m phosphate buffer. Brain slices (300 μm) prepared with a microslicer were postfixed in 1.0% osmium tetroxide, dehydrated, infiltrated with QY2, and then embedded in Araldite. Ultrathin sections were cut with an ultramicrotome, stained with uranyl acetate and lead citrate, and observed under a LEO912AB transmission electron microscope (Zeiss, Oberkochen, Germany) at 80 kV.
Statistical analysis.
All data were shown as means ± SEM (Student's t test; p < 0.05 was considered to be significant).
Results
Cultured Purkinje cells from IP3R1 KO mice show abnormal dendritic outgrowth
We removed cerebellar cells from P1–P2 WT and IP3R1KO mice, cultured them for 14 d, and immunostained them with anti-calbindin antibody to visualize the dendritic morphology of the Purkinje cells. Interestingly, as shown in Figure 1 A, the Purkinje cells from the IP3R1KO mice showed abnormal dendritic outgrowth when compared with Purkinje cells from WT mice. To quantify the differences, we measured various parameters of the dendritic morphology of Purkinje cells. The maximal length of the primary dendrite of IP3R1KO Purkinje cells was 1.4-fold longer than that of the WT Purkinje cells [WT, 71.7 ± 1.8 μm (n = 32); IP3R1KO, 101.6 ± 2.0 μm (n = 26); n, total number of Purkinje cells examined; p < 0.001]. Dendritic area, defined as the area surrounded by straight lines connecting the ends of all terminal dendritic tips of a single Purkinje cell, was 4.8 ± 0.2 (× 103 μm2) (n = 35) for the WT Purkinje cells and 10.4 ± 0.6 (× 103 μm2) (n = 23) for IP3R1KO Purkinje cells (p < 0.001). The primary dendrites of the IP3R1KO Purkinje cells tended to be slightly thinner than those of the WT Purkinje cells [WT, 4.2 ± 0.2 μm (n = 44); IP3R1KO, 2.7 ± 0.2 μm (n = 34); p < 0.001]. There were fewer branch points on the primary dendrite of the IP3R1KO Purkinje cells than of the WT Purkinje cells [WT, 6.5 ± 0.3 (n = 32); IP3R1 KO, 4.6 ± 0.2 (n = 26); p < 0.001], resulting in the longer intervals between adjacent branch points in IP3R1KO Purkinje cells [WT, 11.3 ± 0.5 μm (n = 31); IP3R1KO, 23.9 ± 1.6 μm (n = 25); p < 0.001]. This abnormal dendritic morphology was specific to the IP3R1KO mice, because it was not seen in cultured Purkinje cells from IP3R2/3 double KO mice (Futatsugi et al., 2005). The maximal length of the primary dendrites of IP3R2/3 double KO Purkinje cells was slightly less than in WT Purkinje cells, but the difference was not statistically significant (data not shown).
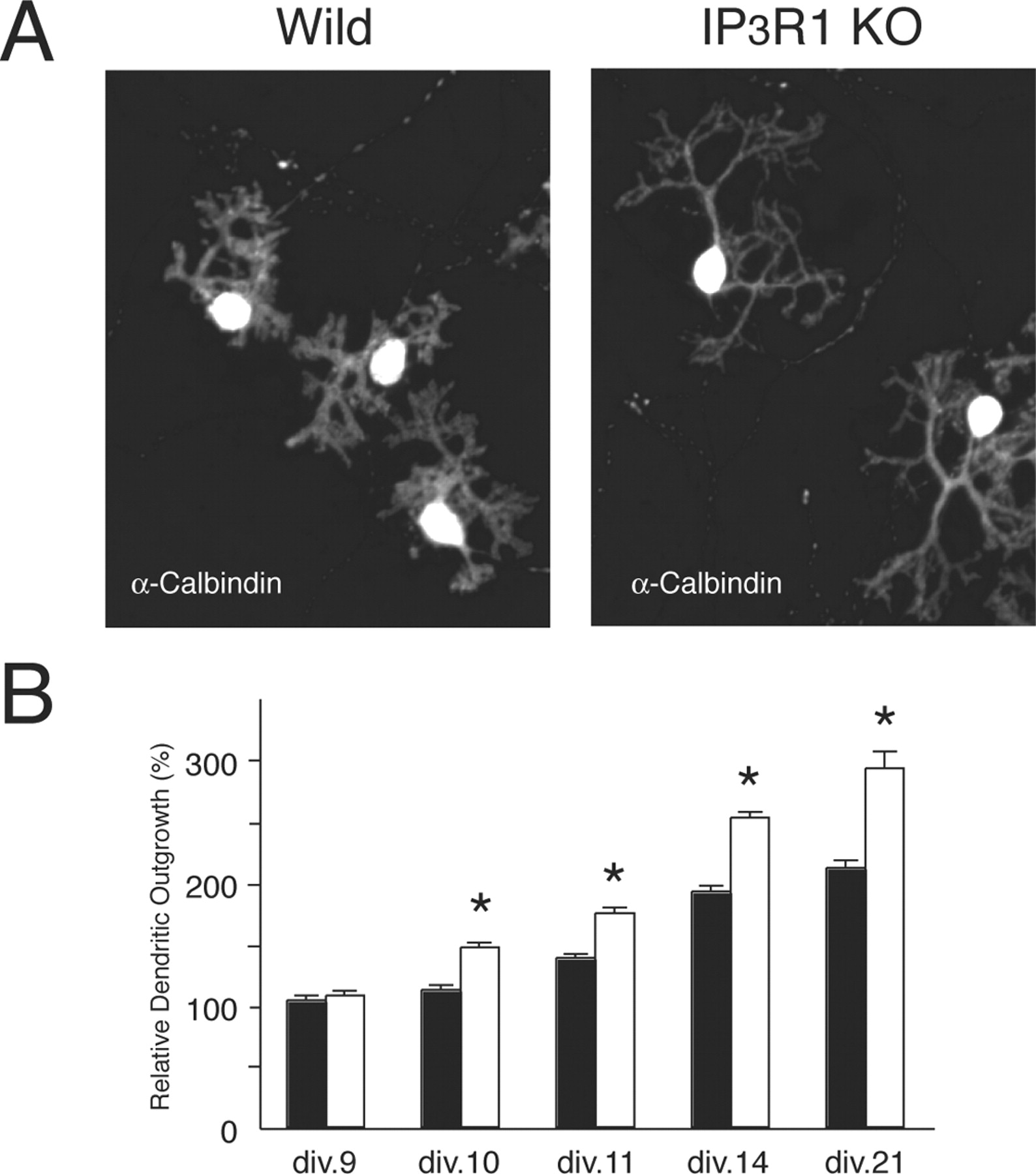
Abnormal dendritic outgrowth of cultured Purkinje cells from IP3R1 KO mice. A , Morphology of WT and IP3R1 KO Purkinje cells stained with anti-calbindin antibody. B , Time course of the dendritic outgrowth by Purkinje cells from WT and IP3R1 KO mice. Data expressed have been normalized to the primary dendrite length of wild-type Purkinje cells at 9 div. div.9: WT, 100 ± 3.5 (n = 139), IP3R1 KO, 103.4 ± 3.8 (n = 130); div.10: WT, 108.2 ± 3.4 (n = 139), IP3R1 KO, 141.6 ± 3.7 (n = 116); div.11: WT, 133.1 ± 3.5 (n = 129), IP3R1 KO, 168.5 ± 4.6 (n = 104); div.14: WT, 185.3 ± 4.9 (n = 96), IP3R1 KO, 243.0 ± 4.4 (n = 96); div.21: WT, 204.2 ± 5.9 (n = 90), IP3R1 KO, 282.1 ± 13.5 (n = 55). n, Number of Purkinje cells used for analysis. All data are presented as mean ± SEM. *p < 0.001.
Because the density of granule cells surrounding Purkinje cells in culture is well known to profoundly affect the dendritic morphology of the Purkinje cells through glutamate and/or neurotrophic factors (Schilling et al., 1991; Hirai and Launey, 2000), a difference in density between the surviving granule cells in the WT and IP3R1KO cerebellar cultures might have explained the abnormal dendritic morphology of IP3R1KO Purkinje cells, but immunostaining with anti-NSE (neuron-specific enolase) antibody revealed no significant difference in granule cell density between the two groups (WT, 3.22 ± 0.21 × 107 cells/μm2, n = 14; IP3R1KO, 2.99 ± 0.16 × 107 cells/μm2, n = 15).
Next, to examine the time course of the morphological change in the dendrites of WT and IP3R1KO Purkinje cells, we measured the maximal length of the primary dendrite of Purkinje cells cultured for various periods. The length of the primary dendrite of the Purkinje cells in the two groups was not statistically different until 9 div (Fig. 1 B). At 10 div, the IP3R1KO Purkinje cells had longer primary dendrites than the WT Purkinje cells, and the difference became more evident at 11, 12, and 14 div. A difference in morphology between the WT and IP3R1KO Purkinje cells was also observed at 21 div, when the Purkinje cell dendrites were almost mature.
IP3R1 in Purkinje cells is not responsible for the abnormal dendritic outgrowth of Purkinje cells
The notion that IP3R1 is dominantly expressed in Purkinje cells (Furuichi et al., 1989, 1993) easily led us to speculate that the IP3R1 expressed in Purkinje cells contributes to the regulation of the dendritic outgrowth of Purkinje cells. However, there was another possibility that the IP3R1 expressed in other cells (e.g., granule cells and glial cells) rather than in the Purkinje cells contributed to it. To identify the source of the IP3R1 that regulated the outgrowth of Purkinje cell dendrites, we prepared mixed culture of WT and IP3R1KO cerebellar cells (chimera culture) and examined the dendritic morphology of the Purkinje cells. If the intrinsic IP3R1 in the Purkinje cells controlled the dendritic morphology of the Purkinje cells, the dendritic morphology of the IP3R1KO Purkinje cells should have remained abnormal in the chimera culture. Immunostaining of Purkinje cells with both anti-IP3R1 antibody and anti-calbindin antibody enabled us to easily distinguish between WT and IP3R1KO Purkinje cells, but as shown in Figure 2, the dendritic morphology of the IP3R1KO Purkinje cells in the chimera culture became indistinguishable from that of the WT Purkinje cells [relative dendrite length of WT and IP3R1 KO Purkinje cells: WT, 100 ± 3.0% (n = 48); IP3R1KO, 101.1 ± 2.4% (n = 48); n.s.]. This finding, showing that the abnormal dendritic shape of IP3R1KO Purkinje cells was rescued by culturing with WT cerebellar cells, suggests that the dendritic outgrowth of Purkinje cells is regulated by a factor(s) provided by other cerebellar cells, the expression of which is IP3R1 dependent.

Morphology of IP3R1 KO Purkinje cells in mixed culture. A , B , Cerebellar cells from IP3R1 KO mice and WT mice were mixed and cultured for 14 d and then fixed and stained for anti-IP3R1 antibody (green) and anti-calbindin antibody (red), respectively. Note that when IP3R1 KO cerebellar cells were mix-cultured with WT cerebellar cells, the dendritic outgrowth of the IP3R1 KO Purkinje cells became similar to that of the WT Purkinje cells.
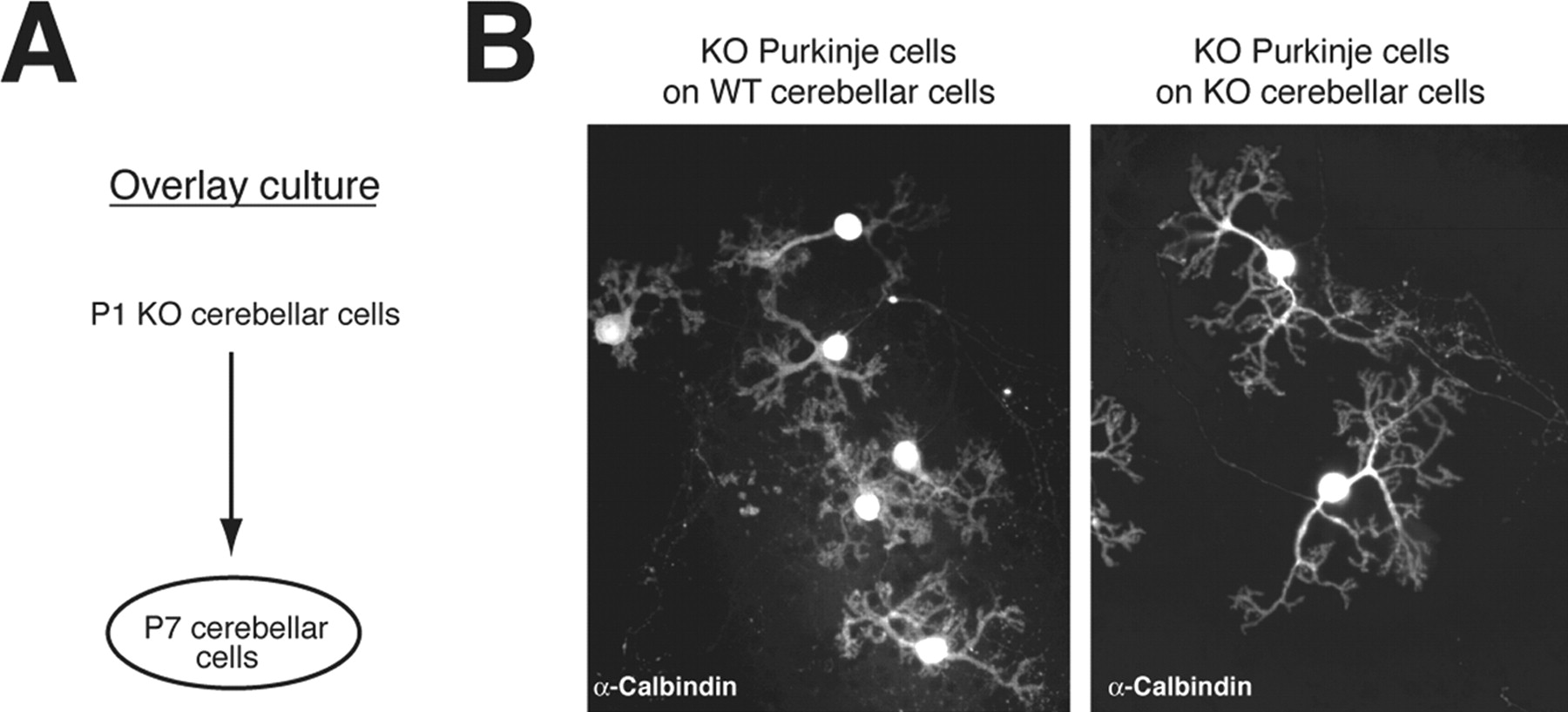
To confirm the contribution of IP3R1 expressed by cells other than Purkinje cells to the dendritic outgrowth of Purkinje cells, we performed an overlay culture by first culturing WT or IP3R1KO cerebellar cells taken from P7 mice and overlaying them with IP3R1KO cerebellar cells taken from P1 IP3R1KO mice 1 d later (Fig. 3 A). Because Purkinje cells obtained from P7 mice cerebellum do not survive in culture, P7 cerebellar cultures do not contain any Purkinje cells. We confirmed the absence of Purkinje cells in cerebellar cultures from P7 mice by immunostaining with anti-calbindin antibody (data not shown). As shown in Figure 3 B, when we overlaid P1 IP3R1KO cerebellar cells on P7 WT cerebellar cells, the dendritic outgrowth of the IP3R1KO Purkinje cells was clearly suppressed, and their dendritic morphology was similar to that of WT Purkinje cells, whereas IP3R1 KO Purkinje cells grown on P7 IP3R1 KO cerebellar cells still showed abnormal dendritic outgrowth [relative dendritic length of Purkinje cells were: IP3R1 KO Purkinje cells cultured on P7 WT cerebellar cells, 100 ± 3.9% (n = 28); IP3R1 KO Purkinje cells cultured on P7 IP3R1 KO cerebellar cells, 115.1 ± 3.8% (n = 21); p < 0.01]. These results strongly support one of the possibilities described above (i.e., that IP3R1 expressed in other cell types, not in Purkinje cells, regulates the dendritic outgrowth of Purkinje cells).
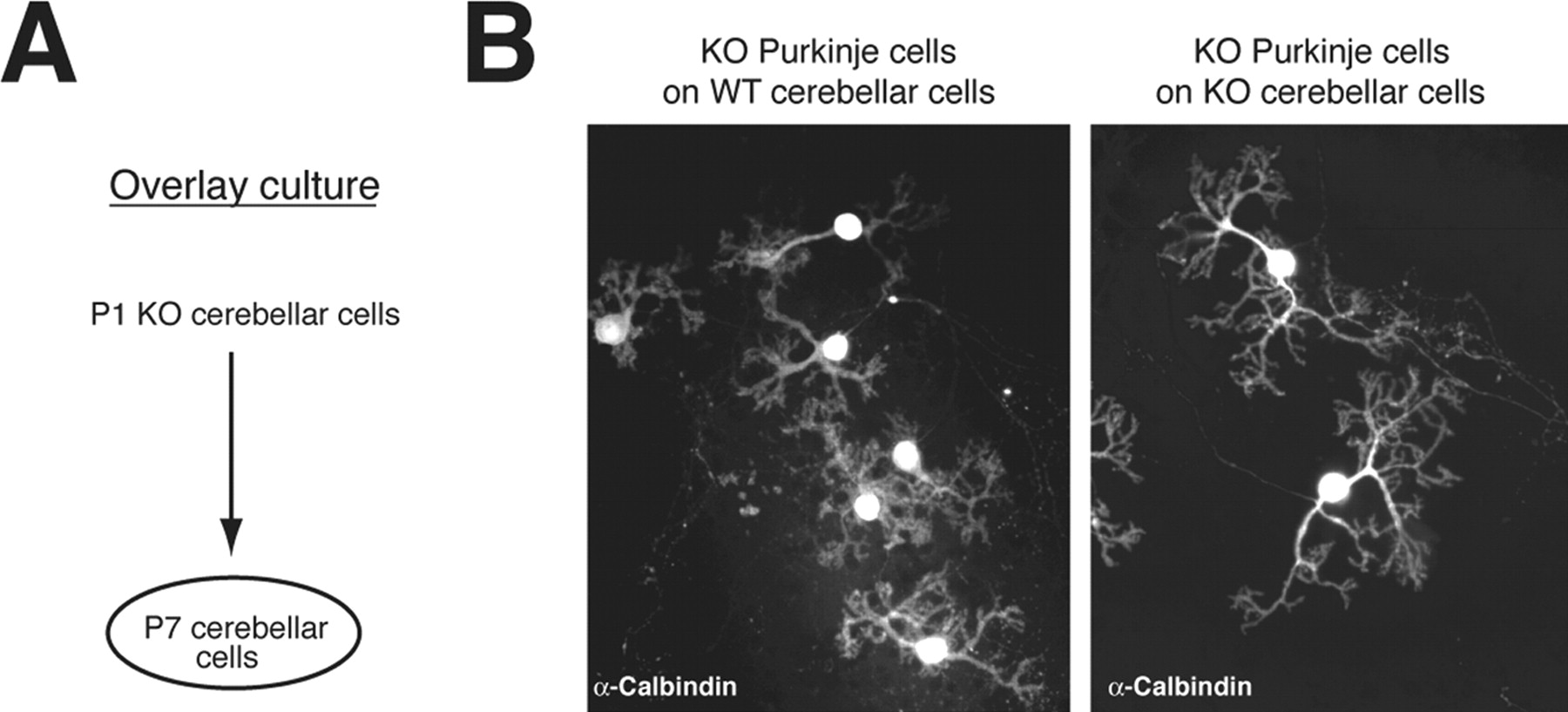
Morphology of IP3R1KO Purkinje cells in overlay culture. A , Schematic illustration of overlay culture. Cerebellar cells from WT or IP3R1 KO P7 mice were first plated on a plastic disk. The next day, cerebellar neurons from P1 IP3R1 KO mice were plated on the previously cultured cerebellar cells from the P7 mice. The cells were cultured for an additional 14 d, fixed, and stained with anti-calbindin antibody. B , The dendritic length of IP3R1 KO Purkinje cells was shorter when cultured on P7 WT cerebellar cells than when grown on P7 IP3R1 KO cerebellar cells. Relative dendritic length of Purkinje cells is shown. IP3R1 KO Purkinje cells cultured on P7 WT cerebellar cells, 100 ± 3.9% (n = 28); IP3R1 KO Purkinje cells cultured on P7 IP3R1 KO cerebellar cells, 115.1 ± 3.8% (n = 21). The difference between the primary dendrite length of IP3R1 KO Purkinje cells cultured on WT and IP3R1 KO cerebellar cells was significant: p < 0.01.
To investigate whether deletion of the IP3R1 gene in glial cells contributed to the abnormal dendritic outgrowth of Purkinje cells in IP3R1KO mice, we prepared glial cell culture from P1 WT and IP3R1 KO cerebella. The morphology of the WT and IP3R1 KO glial cells was indistinguishable, and the proliferation rates of the two groups were not significantly different (data not shown). We mixed WT or IP3R1 KO glial cells with P1 IP3R1 KO cerebellar cells (in the ratio of 3:1) and cultured them for 14 d, but there was no significant morphological difference between IP3R1 KO Purkinje cells cultured with WT and IP3R1 KO glial cells (Fig. 4), suggesting that the IP3R1 expressed in glial cells, if any, does not contribute to the dendritic morphology of Purkinje cells [relative dendrite length of Purkinje cells: IP3R1 KO Purkinje cells plus WT glial cells, 100 ± 4.1% (n = 43); IP3R1 KO Purkinje cells plus IP3R1 KO glial cells, 95 ± 3.4% (n = 78); n.s.].

IP3R1 in glial cells was not involved in regulation of the dendritic outgrowth of Purkinje cells. A , Schematic illustration of the culture method. P1 cerebellar cells from IP3R1 KO mice were mixed with WT or IP3R1 KO glial cells and cultured for 14 d. Purkinje cells were stained with anti-calbindin antibodies. B , Morphology of the dendritic outgrowth of IP3R1 KO Purkinje cells that were mix-cultured with WT or IP3R1 KO cerebellar glial cells.
Abnormal Ca2+ signaling in IP3R1 KO cerebellar granule cells in response to mGluR stimulation
We next investigated the possibility that the IP3R1 expressed in cerebellar granule cells contributes to the dendritic outgrowth of Purkinje cells. First, we investigated the expression of IP3R1 in cultured cerebellar granule cells by using cerebellar cell cultures taken from P7 mice and maintained for 5–6 d in vitro, under which conditions no Purkinje cells or glial cells survive. We detected IP3R1 expression in WT cerebellar granule cells with anti-IP3R1 antibody (Fig. 5 A), but no signal was detected in IP3R1KO cerebellar granule cells. Importantly, IP3R1 was the dominant IP3R subtype in the cerebellar granule cells, because no signal was detected in the IP3R1 KO cerebellar granule cells with anti-Pan IP3R antibody, which recognizes all three types of IP3Rs equally (Hattori et al., 2004). To determine whether IP3R1 actually functions in granule cells, we stimulated granule cells with trans-(±)-1-amino-1,3-cyclopentanedicarboxylic acid (ACPD), an agonist for group 1 metabotropic glutamate receptors that are linked to the phospholipase C (PLC)-IP3 production pathway, and measured Ca2+ changes. To maximize the IP3-induced Ca2+ release, we exposed the cells to high K+ before mGluR stimulation (del Rio et al., 1994). As shown in Figure 5 B, a Ca2+ transient was observed in WT cerebellar granule cells in response to trans-ACPD (top panel), but no Ca2+ transient, only a slight increase in basal Ca2+ level, was observed in the IP3R1 KO cerebellar granule cells. Approximately 50% of the WT cerebellar granule cells responded to mGluR stimulation, whereas only 7.0% of IP3R1 KO cells did [WT, 45.1 ± 6.3% (n = 601); 1 KO, 7.3 ± 2.2% (n = 567), n, number of cells examined; p < 0.001]. Together, these results demonstrated that IP3R1 is expressed in cerebellar granule cells and plays a major role in Ca2+ signaling after mGluR stimulation.

Abnormal Ca2+ signaling of IP3R1 KO cerebellar granule cells in response to mGluR stimulation. A , Expression level of various proteins in WT and IP3R1 KO (1KO) cerebellar granule cells. Lysates of cerebellar neurons were probed with the antibodies indicated. B , Ca2+ signaling in WT and IP3R1 KO cerebellar granule cells. After depolarization with 30 mm KCl to fill the Ca2+ stores, the cells were stimulated with 100 μm trans-ACPD, an agonist for group 1 mGluRs. a , Fura-2 images of cerebellar granule cells. b–d , The ratio image of Ca2+ concentration at the times shown in the right panel.
Reduced BDNF expression in IP3R1 KO granule cells in response to AMPAR and mGluR stimulation
Because BDNF is a neurotropic factor and one of the major factors regulating dendritic morphology (Jan and Jan, 2003), we tested its effect on the abnormal dendritic morphology of IP3R1 KO Purkinje cells by adding BDNF to the culture. Interestingly, BDNF rescued the abnormal morphology (Fig. 6 A,B), suggesting possible involvement of BDNF in the dendritic shaping of Purkinje cells.

BDNF application rescued abnormal dendritic outgrowth of IP3R1 KO Purkinje cells. A , Effect of BDNF on the dendritic morphology of WT and IP3R1 KO Purkinje cells. Cerebellar cells were exposed to BDNF (10 ng/ml) from 9 to 14 div for 5 d, fixed, and stained with anti-calbindin antibody. B , Relative dendritic outgrowth of WT and IP3R1 KO (1KO) Purkinje cells exposed to BDNF normalized by maximal dendritic length of WT Purkinje cells in the absence of BDNF. WT, 100 ± 1.5 (n = 366); WT plus BDNF, 85.8 ± 2.0 (n = 148); 1KO, 131.6 ± 1.6 (n = 248); 1KO plus BDNF, 98.0 ± 2.0 (n = 166). n, Number of cells examined. *p < 0.001. n.s., Not significant; p = 0.43. C , D , Reduced BDNF expression by IP3R1 KO cerebellar granule cells in response to AMPAR and mGluR stimulation. Cerebellar granule cells taken from P7 WT and IP3R1 KO mice were cultured for 7 d and then stimulated with 10 μm AMPA and 50 μm DHPG. After 4 h, RNAs were extracted from the cells and used for reverse transcription-PCR of BDNF and GAPDH. C , BDNF and GAPDH mRNA expression levels detected by reverse transcription-PCR. D , Relative increase in BDNF mRNA after AMPAR and mGluR stimulation. Wild, 1.7 ± 0.1 (n = 14); 1KO, 1.2 ± 0.1 (n = 13). *p < 0.02.
It is known that BDNF production by AMPAR stimulation is further augmented by costimulation with mGluRs (Baird et al., 1991; Yuzaki et al., 1994). In addition, stimulation of AMPAR increases the cytosolic Ca2+ concentration via depolarization-induced Ca2+ entry, which facilitates the activation of Ca2+-dependent proteins, PLC and IP3R, that function under mGluR signal pathway (del Rio et al., 1994; Nash et al., 2004; Young et al., 2004). Therefore, we hypothesized that IP3R1 activity regulates the expression level of BDNF in cultured cerebellar cells after stimulation of AMPAR and mGluRs. As expected, we found that stimulation with AMPA and DHPG, agonists for the AMPAR and the group 1 mGluR, respectively, increased BDNF expression by 1.7 times in WT granule cells but much less in IP3R1 KO granule cells (1.2-fold) (Fig. 6 C,D). Together, these results suggest the existence of a mechanism of IP3R1-mediated activity-dependent BDNF expression in cerebellar granule cells that regulates the dendritic shaping of Purkinje cells.
Abnormal dendritic morphology of Purkinje cells and ultrastructure of PF-PC synapses in IP3R1 KO mice in vivo
Because BDNF deficiency led to abnormality of ultrastructure of PF-PC synapses and dendritic morphology of Purkinje cells in vivo (Carter et al., 2002), we finally examined the dendritic morphology of Purkinje cells and ultrastructure of PF-PC synapses in IP3R1 KO mice in vivo. As shown in Figure 7 A, although overall PF-PC synaptogenesis appeared normal in the IP3R1KO cerebella, significant extension of the length of postsynaptic density (PSD) was observed in the IP3R1 KO PF-PC synapses [WT, 0.24 ± 0.01 μm (n = 211); IP3R1KO, 0.33 ± 0.01 μm (n = 256); p < 0.001; n, number of synapses examined]. In addition, presynaptic enlargement and increase in vesicle number in axon terminals of cerebellar granule cells were apparent in IP3R1 KO cerebella, suggesting an alteration in the synaptic transmission efficacy of the PF-PC synapses in IP3R1KO cerebella (Fig. 7 B). We also examined the shape of the dendrites of individual Purkinje cells in acute cerebellar slices by injecting a fluorescent dye by the patch-clamp technique (see Materials and Methods). As shown in Figure 7 C, whereas WT Purkinje cells extended their dendrites evenly throughout the dendritic area, the dendritic morphology of the IP3R1KO Purkinje cells was clearly abnormal, with vacant areas lacking any dendritic structures in the dendritic arbor. To quantify the abnormality, we measured the density of branch points by counting them and measuring the dendritic area of the Purkinje cells. IP3R1 KO Purkinje cells had reduced branching point density [branch numbers per unit dendritic area: WT, 3.0 ± 0.1 (n = 32); IP3R1 KO, 2.3 ± 0.1 (n = 34) (× 103 n/mm2); p < 0.001], consistent with the observation in the culture study described above. In contrast, there was no significant difference in spine density and maximal length of the primary dendrite between WT and IP3R1 KO Purkinje cells in vivo (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (data not shown). The spatial limitations on dendritic outgrowth in the cerebellar cortex may explain the reason why maximal length of the primary dendrite of IP3R1 KO Purkinje cells was longer than that of WT cells in vitro (Fig. 1) but not in vivo. We also examined the number of inhibitory interneurons that innervate and affect the excitability of Purkinje cells, but there was no apparent difference in the immunoreactivity of GAD67, one of the marker of inhibitory interneurons, between WT and IP3R1 KO cerebella (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these results suggested that IP3R1 plays an important role in shaping the dendritic morphology of Purkinje cells in vivo, possibly by changing PF-PC synaptic efficacy.

Abnormal dendritic morphology of IP3R1 KO Purkinje cells in vivo. A , Ultrastructure of the molecular layer of the cerebellum of WT and IP3R1 KO mice. Scale bar, 1.0 μm. B , Enlargement of presynaptic terminals and vesicle accumulation in PF-PC synapses in IP3R1 KO mice. Scale bar, 0.5 μm. C , Purkinje cells in acute cerebellar slices from WT and IP3R1 KO mice (∼P17–P20) were loaded with intracellular solution containing fluorescein (500 μm) by means of patch pipette in the whole-cell configuration. After 15 min, the dendritic morphology of Purkinje cells was visualized with a two-photon microscope. Typical images of the dendritic morphology of WT and IP3R1 KO Purkinje cells reconstructed from image stack of serial focal planes are shown (top panels). The bottom panels show the magnification view images of the area indicated by squares in the top panels. Scale bar, 20 μm. D , A schematic model of the regulation of dendritic morphology of Purkinje cells by IP3R1 expressed in granule cells. The BDNF production in granule cells by AMPAR stimulation is augmented by costimulation of mGluR5 through IP3R1-mediated signaling. Then, the produced BDNF increases the probability of transmitter release from presynaptic terminals of granule cells in an autocrine manner, leading to the increase of excitability of Purkinje cells and the subsequent branching of Purkinje cell dendrites.
Discussion
In this study, we demonstrated abnormal dendritic outgrowth by cultured Purkinje cells from IP3R1 KO mice. Despite the very high level of expression of IP3R1 in Purkinje cells, the following lines of evidence led us to conclude that the IP3R1 in cerebellar granule cells, not in Purkinje cells, contributes to regulation of the dendritic outgrowth of Purkinje cells. First, the dendritic morphology of WT and IP3R1 KO Purkinje cells was indistinguishable in the chimera culture when the surrounding cellular environment was same. Second, the dendritic morphology of IP3R1 KO Purkinje cells cultured on WT cerebellar cells tend to be similar to that of WT Purkinje cells. Third, of the three types of IP3Rs, cultured cerebellar granule cells dominantly express IP3R1, and cerebellar granule cells lacking IP3R1 released less Ca2+ after mGluR stimulation. Fourth, the increase in BDNF production in response to mGluR stimulation and AMPAR stimulation was significantly reduced in IP3R1 KO cerebellar granule cells, and exogenous BDNF application rescued the abnormal morphology of IP3R1 KO Purkinje cells. Thus, our findings strongly suggest a novel role of IP3R1-mediated Ca2+ signaling in activity-dependent BDNF production in cerebellar granule cells and its critical functional role on shaping the dendrite arborization of Purkinje cells.
We have shown that of the three subtypes of IP3Rs, cerebellar granule cells dominantly express IP3R1 at least for the first 2 weeks postnatally, which is consistent with our observation of much reduced mGluR-induced Ca2+ release by cerebellar granule cells lacking IP3R1 than by WT cells (Fig. 5). A small population of IP3R1 KO cerebellar granule cells (∼7.0%) still showed a Ca2+ response to ACPD, and that may be explained by the expression of other types of IP3Rs, especially IP3R2 that is expressed in granule cells at later developmental stages (A. Futatsugi, unpublished data).
Our observation of an abnormal dendritic shape in IP3R1KO Purkinje cells conflicts with the finding in a study by Matsumoto and Kato (2001), and the discrepancy may be attributable to the difference in culture conditions. For example, because development of Purkinje cell dendrites depends on the density of granule cells and the concentration of neurotrophic factors and transmitters secreted from surrounding granule cells, a difference in the density of granule cells surrounding Purkinje cells would affect dendritic outgrowth, and, in fact, we observed a tendency toward more branch points and a shorter maximal dendritic length in WT Purkinje cells as the density of the surrounding granule cells increased (data not shown). Cell density in our culture (∼1.5–2.5 × 106 cells/cm2) was much higher than in the other study (∼2.0–2.5 × 105 cells/cm2). They also reported no significant alteration in Ca2+ release by IP3R1 KO cerebellar granule cells in response to mGluR stimulation (Matsumoto and Kato, 2001), which is totally different from our own observation (Fig. 5). Although we have no clear explanation for this discrepancy, it may be explained by the difference in culture conditions or in the developmental stage of the neurons, which may affect the level of expression of IP3R2.
Several studies have reported that inhibition of mGluRs leads to changes in the dendritic morphology of Purkinje cells in cerebellar culture. Incubation of Purkinje cells with (±)-amino-4-carboxy-methyl-phenylacetic acid (MCPG), a specific inhibitor of group 1 mGluRs, resulted in abnormal dendritic morphology (Hirai and Launey, 2000), and the abnormal phenotypes of MCPG-treated Purkinje cells were very similar to those of IP3R1 KO Purkinje cells described in this paper, consistent with the notion that IP3R is a downstream molecule of group 1 mGluRs. Catania et al. (2001) investigated the role of each subtype of mGluRs in cerebellar culture by using subtype-specific inhibitors of mGluR1 and mGluR5 and found that inhibition of mGluR1 led to a reduction in the viability of Purkinje cells, whereas inhibition of mGluR5 led to changes in the dendritic morphology of Purkinje cells to fewer branch points and longer dendrites. This observation is in good agreement with our conclusion that IP3R1 expressed in cerebellar granule cells regulates the dendritic morphology of Purkinje cells, because mGluR5 is expressed in cerebellar granule cells and not in Purkinje cells. The temporal aspects of changes in the dendritic morphology in the study by Catania et al. (2001) and in our own are also consistent. Abnormal dendritic morphology appeared after 9 div in their study and at ∼10 div in our study, around which time granule cells form synapses on Purkinje cells (Schilling et al., 1991).
We have shown that exogenous BDNF rescued the abnormal morphology of IP3R1 KO Purkinje cells, and that the increase in BDNF production in response to mGluR and AMPAR stimulation was significantly reduced in IP3R1 KO cerebellar granule cells. Because the increase in BDNF production by AMPAR stimulation was augmented by costimulation of mGluR (Yuzaki et al., 1994) and IP3 production by G-protein-coupled receptors was greatly increased by costimulation of AMPAR (Baird et al., 1991; del Rio et al., 1994; Nash et al., 2004), it is plausible that AMPAR and mGluR cooperatively activate the signaling pathway for IP3R-mediated BDNF production. In contrast, it remains unknown how the BDNF produced in cerebellar granule cells regulates the dendritic morphology of Purkinje cells intercellularly. One possibility is that BDNF released by cerebellar granule cells directly regulates the dendritic morphology of Purkinje cells through its receptor (TrkB) expressed on the surface of Purkinje cells. Alternatively, because BDNF is known to increase the probability of transmitter release and to change synaptic shapes (Li et al., 1998; Tyler and Pozzo-Miller, 2001), the glutamate release mechanism in the presynaptic terminals of cerebellar granule cells may be facilitated by BDNF in an autocrine manner, which then affects the dendritic outgrowth of postsynaptic Purkinje cells. A significant enlargement of PSD and presynaptic vesicle accumulation in PF-PC synapse were observed in IP3R1 KO mice (Fig. 7 B), phenotypes very similar to those observed in BDNF knock-out mice (Carter et al., 2002), supporting the latter alternative. Moreover, the IP3R1 KO mice exhibited a reduction in paired-pulse facilitation (PPF) at PF-PC synapses, which is an index of presynaptic function [statistically significant when paired-pulse interval was 10 ms (p < 0.05; t test), and the t test p values were 0.07 and 0.08 at paired-pulse intervals of 20 and 30 ms, respectively (Matsumoto et al., 1996; our unpublished data)]. A reduction in PPF at PF-PC synapses was also seen in BDNF KO mice (Carter et al., 2002). Because electrical activity in Purkinje cell dendrites makes dendrites stop growing and start to branch (Schilling et al., 1991), the decreased activity-dependent BDNF production in IP3R1 KO cerebellar granule cells (Fig. 6 C,D) may cause reduced glutamate release from presynapses, leading to the abnormal morphology of IP3R1 KO Purkinje cells. This idea is further supported by the finding that chronic AMPAR inhibition of cerebellar cultures resulted in increased dendritic outgrowth and fewer branch points (Hirai and Launey, 2000), phenotypes very similar to those of IP3R1 KO Purkinje cells, and that the occlusive effect of chronic AMPAR blockade was observed in the dendritic morphology of IP3R1KO Purkinje cells (our unpublished data). Therefore, the evidence is strong enough to conclude that the BDNF works as an autocrine regulator in cerebellar granule cell for morphogenesis of Purkinje cell dendrites (Fig. 7 D).
During the preparation of this manuscript, Furutani et al. (2006) reported that IP3 signaling in Purkinje cells plays a role in strengthening PF-PC synapses presynaptically via BDNF in a retrograde manner (Furutani et al., 2006). Therefore, in addition to the regulation of the axon terminals of granule cells by BDNF in an autocrine manner described here, BDNF from Purkinje cells may also affect presynaptic terminals of granule cells in a retrograde manner. However, judging from the evidence that granule cells, not Purkinje cells, predominantly express BDNF (Wetmore et al., 1990; Castren et al., 1995; Neveu and Arenas, 1996), and our finding in this study that the abnormal morphology of IP3R1 KO Purkinje cells was rescued in chimera and overlay cultures with WT cerebellar granule cells, the BDNF produced in granule cells is a major player that modulates the axonal terminals of granule cells.
In summary, we demonstrated that IP3R1 in cerebellar granule cells intercellularly regulates the dendritic outgrowth of Purkinje cells through production of BDNF. Our results imply that the huge amount of IP3R1 expressed in Purkinje cells does not play a significant role in controlling the dendritic morphology of Purkinje cells. Whether the phenomenon in which the IP3R1 expressed in dendrites does not regulate dendritic morphogenesis on-site is specific to Purkinje cells is unclear. Additional studies are expected to shed light on the relationships between the IP3R1 functions and dendritic morphogenesis in various types of neurons.
Footnotes
-
This work was supported by grants from the Ministry of Education, Science, and Culture of Japan (K.M., T.I.), a Grant-in-Aid for Young Scientists (C.H.), and the Japan Science and Technology Agency. We thank all members of our laboratories, especially M. Iwai and A. Z. Suzuki for experimental materials, N. Ogawa for technical help, and Dr. A. Mizutani for fruitful discussions.
- Correspondence should be addressed to Dr. Chihiro Hisatsune, Division of Molecular Neurobiology, Institute of Medical Science, The University of Tokyo, 4-6-1, Shirokane-dai, Minato-ku, Tokyo 108-8639, Japan. chihiro{at}ims.u-tokyo.ac.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}