

Abstract
Adenosine is arguably the most potent and widespread presynaptic modulator in the CNS, yet adenosine receptor signal transduction pathways remain unresolved. Here, we demonstrate a novel mechanism in which adenosine A1 receptor stimulation leads to p38 mitogen-activated protein kinase (MAPK) activation and contributes to the inhibition of synaptic transmission. Western blot analysis indicated that selective A1 receptor activation [with N6-cyclopentyladenosine (CPA)] resulted in rapid increases in phosphorylated p38 (phospho-p38) MAPK immunoreactivity in membrane fractions, and decreases in phospho-p38 MAPK in cytosolic fractions. Immunoprecipitation with a phospho-p38 MAPK antibody revealed constitutive association of this phosphoprotein with adenosine A1 receptors. Phospho-p38 MAPK activation by A1 receptor stimulation induced translocation of PP2a (protein phosphatase 2a) to the membrane. We then examined the actions of p38 MAPK activation in A1 receptor-mediated synaptic inhibition. Excitatory postsynaptic field potentials evoked in area CA1 of the rat hippocampus markedly decreased in response to adenosine (10 μm), the A1 receptor agonist CPA (40 nm), or a 5 min exposure to hypoxia. These inhibitory responses were mediated by A1 receptor activation because the selective antagonist DPCPX (8-cyclopentyl-1,3-dipropylxanthine) (100 nm) prevented them. In agreement with the biochemical analysis, the selective p38 MAPK inhibitor SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole] (25 μm) blocked the inhibitory actions of A1 receptor activation, whereas both the inactive analog SB202474 [4-ethyl-2-(p-methoxyphenyl)-5-(4′-pyridyl)-1H-imidazole] (25 μm) and the ERK 1/2 (extracellular signal-regulated kinase 1/2) MAPK inhibitor PD98059 [2′-amino-3′-methoxyflavone] (50 μm) were ineffective. In contrast, the p38 MAPK inhibitors did not inhibit GABAB-mediated synaptic depression. These data suggest A1 receptor-mediated p38 MAPK activation is a crucial step underlying the presynaptic inhibitory effect of adenosine on CA3–CA1 synaptic transmission.
- adenosine
- A1 receptor
- hippocampus
- p38 mitogen-activated protein kinase
- hypoxia
- synaptic depression
- presynaptic inhibition
- field EPSP
Introduction
Extracellular brain adenosine concentrations increase 30- to 100-fold after pathological trauma such as head injury, epileptic seizures, and hypoxia/ischemia (During and Spencer, 1992; Bell et al., 1998; Von Lubitz, 1999). Adenosine inhibits glutamate release via A1 receptors (Fowler, 1989; Katchman and Hershkowitz, 1993; Zhu and Krnjevic, 1993) and may act as an endogenous neuroprotectant by preventing glutamate excitotoxicity (Dolphin and Prestwich, 1985; Dunwiddie and Masino, 2001).
p38 MAPK is a member of the mitogen-activated protein kinases (MAPKs) that mediate inflammation, cell proliferation, and apoptosis (Seger and Krebs, 1995; Cobb, 1999), and can be activated by the adenosine A1 receptor via a G-protein-dependent pathway (Robinson and Dickenson, 2001). MAPKs are also activated by synaptic activity and are essential for some forms of synaptic plasticity (Impey et al., 1999). In rat hippocampus, p38 MAPK activation mediates metabotropic glutamate receptor-dependent long-term depression (LTD) (Bolshakov et al., 2000; Rush et al., 2002), Rap-dependent removal of AMPA receptors during LTD (J. J. Zhu et al., 2002), and inhibition of long-term potentiation by β-amyloid (Saleshando and O'Connor, 2000; Wang et al., 2004) and interleukin-1β (Coogan et al., 1999; Vereker et al., 2000).
In heart tissue, adenosine activates p38 MAPK (Haq et al., 1998), and plays an important role in cardioprotection during ischemia (Weinbrenner et al., 1997; Baines et al., 1998, 1999). A1 receptor activation triggers early and delayed ischemic preconditioning (Thornton et al., 1992; Tsuchida et al., 1993) and mediates myocardial protection by activating p38 MAPK (Zhao et al., 2001; Schulte et al., 2004). Recent studies have confirmed that A1 receptor-mediated delayed preconditioning against myocardial infarction is dependent on p38 MAPK in vivo (Lasley et al., 2005).
Synaptic transmission in the hippocampus is inhibited by A1 receptor-mediated depression of presynaptic voltage-dependent calcium channels (Wu and Saggau, 1994; Manita et al., 2004). p38 MAPK has also been implicated in presynaptic inhibition, because p38 MAPK activation is necessary for inhibition of N-type calcium current in a G-protein-dependent pathway (Wilk-Blaszczak et al., 1998). Although both A1 receptors (Lee et al., 1983; Fastbom et al., 1987; Tetzlaff et al., 1987; Ochiishi et al., 1999) and p38 MAPK (Lee et al., 2000; Maruyama et al., 2000) are widely expressed in brain tissue, it is not known whether A1 receptors can activate p38 MAPK in the brain, or whether p38 MAPK activation plays a role in A1 receptor-mediated synaptic depression.
In this study, we tested whether p38 MAPK activation contributes to the synaptic depression induced by A1 receptor stimulation in the hippocampal CA1 region. We show that A1 receptor stimulation rapidly elevates phosphorylated p38 (phospho-p38) MAPK and that A1 receptors and phospho-p38 MAPK both exist in the same signaling complex. Protein phosphatase 2a (PP2a) rapidly translocates from the cytosol to the membrane after A1 receptor stimulation in a p38 MAPK-dependent pathway. We also show that p38 MAPK contributes to adenosine A1 but not GABAB receptor-mediated depression of CA3–CA1 synaptic transmission, suggesting that p38 MAPK-dependent synaptic depression is selective for A1 receptors.
Materials and Methods
Hippocampal slice preparation.
Sprague Dawley rats [postnatal day 21 (P21) to P28] were anesthetized with halothane and decapitated according to protocols approved by the University of British Columbia committee on animal care. Brains were rapidly extracted and placed into ice-cold oxygenated dissection medium containing the following (in mm): 87 NaCl, 2.5 KCl, 2 NaH2PO4, 7 MgCl2, 25 NaHCO3, 0.5 CaCl2, 25 d-glucose, and 75 sucrose. Hippocampal slices (400 μm thick) were cut using a vibrating tissue slicer (VT1000S; Leica, Nussloch, Germany) and maintained for 1–5 h at 24°C in artificial CSF (aCSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 26 NaHCO3, 2.5 CaCl2, and 10 d-glucose, and aerated with 95% O2/5% CO2. For electrophysiological recordings, slices were submerged in a recording chamber and allowed to equilibrate for at least 1 h. The bath solution was perfused with aerated aCSF, pH 7.3, at a rate of 1.5–2 ml/min. All electrophysiological recordings and preincubation of slices with drugs for subsequent biochemical analyses (see below) were performed at room temperature (22−24°C).
Electrophysiology.
Field EPSPs (fEPSPs) were evoked by orthodromic stimulation of the Schaffer collateral pathway using a bipolar tungsten-stimulating electrode. Glass micropipettes filled with aCSF (resistance, 1–3MΩ) were used to measure CA1 fEPSPs in stratum radiatum. fEPSP signals were amplified 1000 times with an AC amplifier, bandpass filtered at 1000 Hz, digitized at 10 kHz using a Digidata 1320A interface board (Molecular Devices, Foster City, CA), and transferred to a computer for analysis. Data were analyzed using Clampfit 9.0 (Molecular Devices). Baseline synaptic responses were established by evoking fEPSPs every 30 s (0.03 Hz) for at least 20 min. The fEPSP slope was normalized to the mean of the 20 sweeps (10 min) immediately preceding drug perfusion. The mean normalized fEPSP slope was plotted as a function of time with error bars representing SEM. Sample traces are the average of five sweeps from a recording that was included in the plot of the mean normalized fEPSP slope. All bar graphs show the mean normalized percent inhibition from baseline ± SEM. Statistical significance was assessed using Student's t test (p < 0.05).
Immunoprecipitation, coimmunoprecipitation, and Western blot analysis.
For biochemical studies, rat hippocampal slices were first incubated with various treatments (see below), and then lysed in a solubilization buffer (30 min; 4°C) that contained 1% NP-40; 20 mm MOPS (4-morpholinepropanesulfonic acid), pH 7.0; 5 mm EDTA; 2 mm EGTA; 1 mm phenylmethylsulfonyl fluoride (PMSF); 10 μg/ml aprotinin; 10 μg/ml leupeptin; 10 μg/ml pepstatin A; 1 mm Na3VO4; 30 mm NaF; 40 mm β-glycerophosphate, pH 7.2; 20 mm sodium pyrophosphate; and 3 mm benzamidine. The tissue homogenates were then centrifuged at 13,000 × g (20 min; 4°C) to remove cellular debris, and then protein concentrations of the crude lysates were determined by performing a Bradford assay with the DC Protein Assay dye (Bio-Rad, Mississauga, Ontario, Canada). In some experiments, the membrane and cytosolic fractions from hippocampal slices were separated by centrifugation at 13,000 × g for 1 h at 4°C by omitting the detergent from the solubilization buffer. The proteins from the particulate (membrane) fraction were resolved in normal solubilization buffer (as above) after removal of the cytosolic extract. Hippocampal homogenates were diluted with 1× Laemmli sample buffer and boiled for 5 min. The proteins were resolved in 10% polyacrylamide gel and electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Cambridge, Ontario, Canada). The blots were blocked with 5% nonfat milk in TBST for 1 h, and the membranes were incubated with primary antibody (see below) overnight at 4°C. After four washes with TBST, the membranes were incubated with the appropriate secondary antibody conjugated to horseradish peroxidase (1 h; room temperature). The membranes were then washed three to four times (15 min) with TBST, and proteins were visualized using enhanced chemiluminescence (ECL) (Amersham Biosciences, Arlington Heights, IL).
To examine interactions between adenosine A1 receptors and phospho-p38 MAPK, coimmunoprecipitation was performed by first incubating 1 mg extract from hippocampal homogenates with a goat or rabbit IgG (1 h; 4°C), and then goat or rabbit IgG agarose beads (Sigma, St. Louis, MO) were added to the homogenates for an additional 1 h. In some experiments, ∼250 μg of lysates from the membrane or cytosolic fractions were used for the coimmunoprecipitation. The agarose beads were removed by pulse spinning at 6000 rpm for 5 s, and the supernatant was subsequently reacted with an immunoprecipitating antibody overnight at 4°C. A1 receptor was immunoprecipitated using either a polyclonal goat anti-A1 receptor (5 μg; Santa Cruz Biotechnology, Santa Cruz, CA) or a polyclonal rabbit anti-A1 receptor (5 μg; Sigma). After overnight preincubation of lysates with a polyclonal rabbit anti-phospho p38 MAPK antibody (5 μg; Cell Signaling, Beverly, MA), the p38 MAPK antigen was immunoprecipitated by incubating the immune complexes for >6 h at 4°C with agarose beads conjugated to secondary antibody (rabbit or goat anti-IgG). Agarose beads were then collected by pulse spins, and washed four times with wash buffer (solubilization buffer containing 0.1% NP-40). Proteins from the agarose beads were eluted with 60 μl of 1× Laemmli sample buffer (Bio-Rad), boiled for 5 min, and resolved in 10% polyacrylamide gels. Proteins were then electrotransferred to PVDF membranes (Millipore, Cambridge, Ontario, Canada).
PVDF membranes were blocked with 5% nonfat milk in TBS (50 mm Tris, pH 7.4; 150 mm NaCl) containing 0.1% Tween 20 (TBST). The membrane was incubated overnight (4°C) with the appropriate primary antibody diluted in 5% nonfat milk in TBST containing 0.025% sodium azide. The antibody dilutions are as follows: polyclonal rabbit anti-A1 receptor (1:1000; Sigma), polyclonal rabbit anti-phospho-p38 MAPK (1:500; Cell Signaling), polyclonal rabbit panspecific p38 MAPK antibody (1:500; Cell Signaling), and mouse anti-PP2a (1:500; Cell Signaling). To normalize the protein bands from the membrane and cytosolic fractions, we used a polyclonal rabbit anti-β-actin (1:8000; RBI, Natick, MA) or a polyclonal goat anti-GAPDH (1:300; Santa Cruz Biotechnology). The PVDF membranes were washed with TBST for 15 min three times, and then incubated with a mouse, goat, or rabbit horseradish peroxidase-conjugated secondary antibody against IgG (1:3000; Santa Cruz) in 5% nonfat milk. After three to four washes with TBST, labeled protein bands were visualized using ECL and Fluor-S-Max imaging software. Densitometry analysis was performed using ImageJ software.
Drugs.
Adenosine, N6-cyclopentyladenosine (CPA), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), and baclofen were obtained from Sigma. Tetrodotoxin (TTX) was purchased from Alomone Labs (Jerusalem, Israel). 4-(4-Fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole (SB203580), trans-1-(4-hydroxycyclohexyl)-4-(4-fluorophenyl)-5-(2-methoxy-pyrimidin-4-yl)imidazole (SB239063), 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole (SB202190), 4-ethyl-2-(p-methoxyphenyl)-5-(4′-pyridyl)-1H-imidazole (SB202474), and 2′-amino-3′-methoxyflavone (PD98059) were obtained from Calbiochem (La Jolla, CA) and made up as stock in DMSO before being added to aCSF. The final concentration of DMSO was always ≤0.1%.
Results
p38 MAPK activation increases after A1 receptor stimulation
The selective A1 receptor agonist CPA activates p38 MAPK in rat ventricular myocytes within minutes (Liu and Hofmann, 2003). In the present study, we tested whether CPA treatment increased the level of phosphorylated p38 MAPK in rat hippocampus. Hippocampal slices were treated with either normal aCSF or CPA (500 nm) for time periods ranging from 2 to 30 min. The activation of p38 MAPK was determined using an antibody that only recognizes p38 MAPK when it is dually phosphorylated at threonine 180 and tyrosine 182. When samples of homogenized hippocampal slices were centrifuged to separate the membrane and cytosolic fractions, we observed that p38 MAPK activation increased in the membrane fraction and decreased in the cytosolic fraction (Fig. 1A,B). In the membrane fraction, the response was maximal after 10 min of CPA exposure (500 nm) and returned partly back to baseline by 30 min (the longest time point tested). In the cytosolic fraction, phospho-p38 MAPK decreased maximally at 30 min. Specifically, in membrane fraction, CPA increased phospho-p38 MAPK immunoreactivity to 148 ± 12% (n = 5; p < 0.01) of baseline after 2 min, 135 ± 6% (n = 11; p < 0.01) after 5 min, 158 ± 15% (n = 11; p < 0.05) after 10 min, and 124 ± 35% (n = 5; p > 0.05) after 30 min. In the cytosolic fraction, CPA decreased phospho-p38 MAPK to 98 ± 6% (n = 5; p > 0.05) of baseline after 2 min, 85 ± 5% (n = 15; p > 0.05) after 5 min, 78 ± 6% (n = 9; p < 0.05) after 10 min, and 64 ± 5% (n = 5; p < 0.01) after 30 min.

CPA increased p38 MAPK phosphorylation in membrane fractions and decreased p38 MAPK phosphorylation in cytosolic fractions. Hippocampal slices were exposed to either normal aCSF or CPA (500 nm) for 2, 5, 10, or 30 min. The membrane and cytosolic fractions of hippocampal homogenates were separated by centrifugation for Western blot analysis. Whole-cell hippocampal lysates were used for immunoprecipitation. A, Representative Western blots showing phosphorylated p38 MAPK (p-p38) and β-actin in the membrane and cytosolic fractions. B, Quantitative representation of multiple Western blots showing phospho-p38 MAPK immunoreactivity (mean ± SEM) in the membrane fraction [control (n = 5), 2 min (n = 5), 5 min (n = 11), 10 min (n = 11), and 30 min (n = 5)] and cytosolic fraction [control (n = 5), 2 min (n = 5), 5 min (n = 15), 10 min (n = 9), and 30 min (n = 5)]. Data were normalized to the level of protein phosphorylation at time 0 after CPA treatment, and β-actin immunoreactivity was used as a loading control. C, Top panel, Representative Western blot showing that the total amount of p38 did not change in the membrane fraction in response to CPA treatment (500 nm; 10 min). C, Bottom panel, Quantitative representation of total p38 immunoblots (mean ± SEM) showing no significant change in the total amount of p38 (n = 5). D, E, Proteins were immunoprecipitated (IP) with or without immunoprecipitating antibodies, and proteins were subsequently detected on Western blots (WB) with the indicated primary antibodies. D, Coimmunoprecipitation showing an association between p-p38 and the adenosine A1 receptor (A1R) (lane 3). E, Coimmunoprecipitation showing an association between the A1R and p-p38 (lane 2, reverse of D). No association between these proteins was detected when the immunoprecipitating antibodies were omitted (lane 1 of D and E). F, Phospho-p38 MAPK immunoprecipitates from membrane (lane 1) and cytosolic (lane 2) hippocampal lysates also showed immunoreactivity to A1 receptor antibody. Statistical significance compared with control was assessed using Student's t test (*p < 0.05; **p < 0.01).
To test whether the total amount of p38 MAPK was increasing in the membrane fraction, we performed a parallel experiment using a panspecific p38 MAPK (pan-p38 MAPK) antibody. We normalized the pan-p38 MAPK signal to β-actin immunoreactivity and found that CPA (500 nm; 10 min) did not change the total amount of p38 present in the membrane fraction (100 ± 11% of control; n = 5; p > 0.30) (Fig. 1C).
The adenosine A1 receptor and phospho-p38 MAPK are physically associated
We performed coimmunoprecipitation experiments to determine whether the adenosine A1 receptor exists in the same signaling complex as phospho-p38 MAPK. The adenosine A1 receptor corresponds to a band at ∼37 kDa in the rat hippocampus (Rebola et al., 2003). Immunoprecipitation with the adenosine A1 receptor antibody pulled down the adenosine A1 receptor in whole-cell hippocampal lysates (Fig. 1D, lane 2). Immunoprecipitation with the phospho-p38 MAPK antibody (probed with a polyclonal rabbit anti-A1 receptor antibody) revealed the A1 receptor (Fig. 1D, lane 3). No such bands were present when the immunoprecipitating antibody was omitted (Fig. 1D, lane 1). The reverse immunoprecipitation also confirmed the presence of phospho-p38 MAPK in the adenosine A1 receptor immunoprecipitates (Fig. 1E, lane 2). A phospho-p38 MAPK band is also present in the positive control lane (Fig. 1E, lane 3). Immunoprecipitation with anti-phospho-p38 MAPK pulled down the A1 receptor in lysates prepared from both membrane and cytosolic fractions (Fig. 1F). These data suggest that the adenosine A1 receptor and p38 MAPK physically interact in the hippocampus.
A1 receptor-dependent increases in p38 MAPK activation are blocked by p38 MAPK inhibition and A1 receptor antagonism
The pyridinyl imidazole compound SB203580 is a relatively selective inhibitor of p38 MAPK activity (Cuenda et al., 1995). In heart tissue, increases in p38 MAPK phosphorylation attributable to adenosine A1 receptor activation are blocked by SB203580 (Liu and Hofmann, 2003). We tested whether A1 receptor-induced phosphorylation of p38 MAPK was also blocked by SB203580 in the brain. Hippocampal slices were either incubated in normal aCSF or SB203580 (25 μm) for 1 h. Slices from each condition were either left untreated or treated with 40 or 500 nm CPA for 10 min, and then homogenized and centrifuged to separate the membrane and cytosolic fractions. Forty nanomolar CPA (10 min) and 500 nm CPA (10 min) increased phospho-p38 MAPK immunoreactivity to 118 ± 7.2% (n = 4; p < 0.05) and 151 ± 11.7% (n = 11; p < 0.01) of control, respectively. SB203580 by itself decreased phospho-p38 MAPK immunoreactivity to 65.8 ± 8.4% (n = 4) of control (p < 0.05) (Fig. 2A,B). Neither 40 nm CPA nor 500 nm CPA had an effect in slices pretreated with SB203580, because phospho-p38 MAPK immunoreactivity decreased to 63.1 ± 6.4% (n = 4) and 62.3 ± 7.8% (n = 4) of control, respectively. Neither condition was statistically different from slices treated with SB203580 alone (p > 0.30).

The increase in p38 MAPK phosphorylation induced by CPA was blocked by p38 MAPK inhibition and A1 receptor antagonism. Hippocampal slices were either preincubated with normal aCSF or SB203580 (25 μm) for 1 h, and then exposed to CPA (40 or 500 nm) for 10 min. The membrane fraction was separated by centrifugation for Western blot analysis. A, Representative Western blots showing phosphorylated p38 MAPK (p-p38) and β-actin in response to CPA in the presence and absence of SB203580 (25 μm). B, Quantified phosphorylated p38 MAPK immunoreactivity (mean ± SEM) for control (n = 4), control plus SB203580 (n = 4), 40 nm CPA (n = 4), 40 nm CPA plus SB203580 (SB) (n = 4), 500 nm CPA (n = 15), and 500 nm CPA plus SB203580 (n = 4). C, Representative Western blots showing that the CPA-induced increase in phospho-p38 MAPK was blocked in the presence of DPCPX. D, Quantified phospho-p38 MAPK immunoreactivity for control (n = 10), 500 nm CPA (n = 10), and 500 nm CPA plus 500 nm DPCPX (n = 10). E, Quantified p38 MAPK immunoreactivity showing that adenosine (Ad) (10 μm) increased p38 MAPK phosphorylation and that this increase was prevented by pretreating slices with either 500 nm DPCPX (n = 6) or 5 μm SB239063 (SB) (n = 6). In all experiments, data were normalized to β-actin immunoreactivity. Statistical significance compared with control was assessed using Student's t test (*p < 0.05; **p < 0.01).
We also tested whether the A1 receptor antagonist DPCPX prevented the CPA-induced increase in the amount of phospho-p38 MAPK in the membrane fraction. Because DPCPX increases glutamate release in brain slices (Sehmisch et al., 2001; Marcoli et al., 2003), which could potentially activate p38 MAPK through metabotropic glutamate receptors (Rush et al., 2002), we treated slices with TTX (1.2 μm) for 20 min to block action potentials and minimize glutamate release, thus ruling out the potential major contribution of glutamate receptor-mediated p38 MAPK activation with DPCPX application. We found that, in the presence of TTX, CPA (500 nm; 10 min) increased phospho-p38 MAPK to 143 ± 15% (n = 10; p < 0.01) of control levels in the membrane fraction (Fig. 2C,D). This CPA-induced increase in phospho-p38 MAPK was completely blocked (98 ± 15% of control; n = 10; p > 0.80) by pretreatment with DPCPX (500 nm; 20 min) before CPA application (Fig. 2C,D).
Finally, we tested whether the endogenous ligand adenosine also increased the levels of phospho-p38 MAPK. Phospho-p38 MAPK immunoreactivity was elevated to 118 ± 8% (n = 8; p < 0.05) of control in slices treated with 10 μm adenosine for 10 min (Fig. 2E). This increase could be blocked by the addition of either 500 nm DPCPX or 5 μm SB239063: in DPCPX, p38 MAPK immunoreactivity was decreased to 90 ± 7% (n = 6; p > 0.05); in SB239063, the level of p38 MAPK was decreased to 80 ± 9% (n = 6; p < 0.05) of control.
A1 receptor activation induced translocation of protein phosphatase 2a to the plasma membrane
Adenosine A1 receptor activation causes PP2a translocation and activation in ventricular myocytes (Liu and Hofmann, 2002) in a pathway requiring p38 MAPK (Liu and Hofmann, 2003). We tested whether CPA treatment activated PP2a in rat hippocampal slices. Slices were treated with either normal aCSF or CPA (500 nm) for time periods ranging from 2 to 30 min, and homogenized and centrifuged to isolate the membrane fraction. Using an antibody recognizing the C-subunit of PP2a, we determined that CPA treatment increased PP2a immunoreactivity within minutes in membrane fractions (Fig. 3A,B). Specifically, PP2a immunoreactivity increased to 123 ± 10% (n = 7; p < 0.05) of baseline after 2 min, 145 ± 13% (n = 7; p < 0.05) after 5 min, 139 ± 13% (n = 15; p < 0.01) after 10 min, and 121 ± 12% (n = 7; p > 0.05) after 30 min. The response was maximal after 5 min of CPA exposure and returned partly back to baseline by 30 min.

CPA increases PP2a levels in the membrane fraction. Hippocampal slices were exposed to either normal aCSF or CPA (500 nm) for 2, 5, 10, or 30 min. The membrane fraction was extracted by centrifugation for Western blot analysis. A, Representative Western blot showing the time course of CPA-induced changes in PP2a immunoreactivity. The antibody detects both PP2a-α and PP2a-β isoforms, which gives rise to the double bands. B, Quantified PP2a immunoreactivity, showing that PP2a increases within minutes in response to CPA treatment [control (n = 7), 2 min (n = 7), 5 min (n = 7), 10 min (n = 15), and 30 min (n = 7)]. C, Representative Western blot showing that the increase in PP2a levels after exposure to CPA (500 nm; 10 min) was blocked in the presence of DPCPX (500 nm). D, Quantification of C (n = 8). E, Western blot showing that the p38 MAPK inhibitor SB239063 (5 μm) prevented CPA from increasing PP2a levels. F, Quantification of E (n = 6). All data were normalized to β-actin immunoreactivity. Values are means ± SEM. Statistical significance compared with control was assessed using Student's t test (*p < 0.05; **p < 0.01).
In addition, we tested whether DPCPX prevented the increase in PP2a immunoreactivity in membrane fractions induced by treating slices with CPA. All slices were incubated in TTX (as above), and then exposed to either CPA (500 nm; 10 min) alone or in the presence of DPCPX (500 nm). CPA increased the amount of PP2a in the membrane fraction to 145 ± 21% (n = 8; p < 0.05) of control (Fig. 3C,D). However, in the presence of DPCPX, the CPA-induced increase in PP2a was blocked (86 ± 16% of control; n = 8; p > 0.30) (Fig. 3C,D). Finally, we also determined that p38 MAPK inhibition prevented the CPA-induced increase in PP2a immunoreactivity in membrane fractions (Fig. 3E,F). We incubated slices in either normal aCSF or the second-generation p38 MAPK inhibitor SB239063 (5 μm) for 1 h before CPA treatment (500 nm; 10 min). In slices treated with SB239063, CPA failed to increase levels of PP2a in the membrane fraction (91 ± 6% of control; n = 6; p > 0.15). Together, these data suggest that p38 activation is required for the A1 receptor-mediated translocation of PP2a to the membrane.
Adenosine-induced depression of synaptic transmission is mediated by the A1 receptor subtype and is sensitive to p38 MAPK inhibition
To investigate the functional significance of A1 receptor-mediated p38 MAPK activation in the brain, we used extracellular fEPSP recordings to monitor the effect of p38 MAPK inhibition on the action of adenosine in area CA1 of the rat hippocampus. We confirmed that adenosine-induced depression of synaptic transmission in area CA1 is mediated by the A1 receptor subtype in area CA1 (Dunwiddie and Fredholm, 1989; Wu and Saggau, 1994; Johansson et al., 2001). In normal aCSF, perfusion of 20 μm adenosine onto the slice for 10 min decreased the mean normalized fEPSP slope to 33 ± 4% (p < 0.01; n = 8) of baseline (Fig. 4A). Perfusion of 100 nm DPCPX, a specific A1 receptor antagonist (Bruns et al., 1987; Haleen et al., 1987), onto the same slice for 20 min increased the fEPSP slope to 125 ± 4% (p < 0.01; n = 8) of baseline, presumably because of removal of tonic A1 receptor-mediated inhibition of glutamate release. Perfusion of 20 μm adenosine in the presence of 100 nm DPCPX had no effect on fEPSPs (98 ± 3% of baseline; p > 0.15; n = 8), indicating that the A1 receptor subtype mediates the inhibition of fEPSPs caused by adenosine in rat CA1.

Adenosine-induced depression of CA1 fEPSPs is mediated by the A1 receptor subtype and sensitive to p38 MAPK inhibition. A–C, Averaged sample traces and plot of the mean fEPSP slope (±SEM) over time normalized to baseline. fEPSPs (evoked every 30 s) were recorded in CA1 stratum radiatum. A, Adenosine reversibly decreased the mean normalized fEPSP slope. After recovery to baseline, the A1 receptor antagonist DPCPX (100 nm) was bath applied, resulting in an increase in the mean fEPSP slope. A second perfusion of adenosine in the presence of 100 nm DPCPX had no effect on the mean normalized fEPSP slope (n = 8). B, Perfusion of SB20380 (25 μm) slightly increased baseline synaptic transmission and partially inhibited the ability of adenosine (10 μm) to decrease fEPSPs (n = 5). C, The effect of adenosine (10 μm) was the same whether applied in the presence of 0.1% DMSO (50 min perfusion) or normal aCSF (n = 10). D, Summary of A–C showing that SB203580 attenuates adenosine-induced depression, whereas DMSO does not. Calibration: 10 ms, 1 mV. *Denotes statistical significance using Student's t test (p < 0.01).
Perfusion of the p38 inhibitor, SB203580 (25 μm; 40 min), slightly increased the mean fEPSP slope to 111 ± 4.6% of baseline (p < 0.05; n = 5) (Fig. 4B). The magnitude of fEPSP depression induced by adenosine (10 μm) was attenuated by SB203580 (adenosine decreased the mean fEPSP slope by 55.0 ± 3.8% in normal aCSF but by only 27.3 ± 2.6% after SB203580 treatment; n = 5; p < 0.01) (Fig. 4B,D). Because SB203580 was dissolved in DMSO, we ensured that 0.1% DMSO (Fig. 4C) had no effect on baseline synaptic transmission nor the magnitude of adenosine induced depression (adenosine decreased the mean fEPSP slope by 51.0 ± 2.4% in normal aCSF and 51.3 ± 3.5% after 50 min of DMSO treatment in the same slice; n = 10; p > 0.30) (Fig. 4C,D).
Neither the inactive analog SB202474 nor the ERK 1/2 MAPK inhibitor PD98059 decreased adenosine-induced synaptic depression
The health of hippocampal slices, as determined by the slope of evoked fEPSP slopes, was not adversely affected by prolonged exposure to SB203580 (over 50 min in the bath perfusate). Therefore, in the next set of experiments, we preincubated slices in SB203580 instead of prolonged bath perfusions. In all subsequent experiments, matched slices from the same animal were either preincubated in SB203580 (25 μm; 1–2 h) or normal aCSF. Perfusion of adenosine (10 μm; 10 min) depressed the mean normalized fEPSP slope by 61.7 ± 2.7% (n = 5) in control, and only 30.6 ± 3.0% (n = 6) in slices preincubated in SB203580 (p < 0.01) (Fig. 5A,D). Thus, the effects of SB203580 on adenosine-mediated synaptic depression were similar in both preincubation and perfusion experiments.

Adenosine-induced synaptic depression was attenuated by SB203580, but not SB202474 (an inactive analog) nor PD98059 (ERK 1/2 MAPK inhibitor). A–C, Averaged sample traces and plot of the mean normalized fEPSP slope (±SEM) over time. A, Matched slices were either pretreated with SB203580 for 1–2 h (n = 7) or normal aCSF (n = 13). Adenosine-induced depression of fEPSPs was attenuated in the presence of SB203580. B, Matched slices were either pretreated with SB202474 for 1–2 h (n = 5) or normal aCSF (n = 7). SB202474 did not affect adenosine-induced depression of fEPSPs. C, Matched slices were either pretreated with PD98059 (n = 5) or normal aCSF (n = 4). Adenosine-induced depression of fEPSPs was not attenuated by PD98059. D, Summary of A–C showing the mean maximal effect of adenosine expressed as percentage inhibition from baseline. Calibration: 10 ms, 1 mV. *Denotes statistical significance compared with control using Student's t test (p < 0.01).
SB202474 is an inactive pyridinyl imidazole compound that has been used as a negative control in p38 MAPK studies (Lee et al., 1994; Yu et al., 2000). Slices were either preincubated with 25 μm SB202474 or with normal aCSF (control). There was no difference in the action of adenosine in slices treated with SB202474 compared with control slices (Fig. 5B,D). In control, perfusion of adenosine (10 μm; 10 min) depressed the mean fEPSP slope by 52.2 ± 6.4% (n = 7). In slices that had been pretreated with 25 μm SB202474, adenosine depressed the mean fEPSP slope by 67.0 ± 3.4% (n = 5), which was not statistically different from control (p > 0.10).
The extracellular signal-regulated kinase (ERK) 1/2 MAPK pathway is activated by synaptic activity and is required for the induction of long-term potentiation of synaptic transmission in area CA1 of the hippocampus (Impey et al., 1999; Bolshakov et al., 2000). The ERK 1/2 MAPK pathway is also required for metabotropic glutamate receptor-dependent long-term depression (Gallagher et al., 2004). We tested whether ERK 1/2 MAPK also played a role in adenosine-mediated synaptic depression. Slices were either pretreated with the selective ERK 1/2 MAPK inhibitor PD98059 (Pang et al., 1995) (50 μm) or with normal aCSF (control). In control, adenosine (10 μm; 10 min) depressed the mean fEPSP slope by 52.5 ± 5.7% (n = 4) of baseline (Fig. 5C,D). In slices that were pretreated with 50 μm PD98059, adenosine depressed the mean fEPSP slope by 49.0 ± 3.0% (n = 5) of baseline, which was not statistically different from control (p > 0.30). These data indicate that inhibiting ERK 1/2 MAPK does not modify adenosine-mediated synaptic depression in area CA1.
A1 receptor-mediated synaptic depression was decreased by p38 MAPK inhibition
The physiological ligand adenosine acts at multiple receptor subtypes and is rapidly degraded by ecto-nucleotidases and/or removed from the extracellular space via nucleoside transporters (Dunwiddie and Masino, 2001). To avoid these issues, we next used the A1 receptor agonist CPA. We tested whether synaptic depression induced by CPA was also attenuated by the p38 MAPK inhibitor SB203580. Recordings were obtained from slices that were either incubated in 25 μm SB203580 (1–2 h) or normal aCSF (control). In control, a 10 min perfusion of 40 nm CPA decreased the mean fEPSP slope by 72.8 ± 2.1% (n = 4) (Fig. 6A,B,E), confirming that selective A1 receptor activation decreases synaptic transmission. However, in SB203580, CPA perfusion only decreased the mean fEPSP slope by 10.4 ± 2.0% (n = 5; p < 0.01) (Fig. 6A,B,E). These results provide additional evidence that p38 MAPK activity is necessary for A1 receptor-mediated synaptic depression.

CPA-mediated depression of CA1 fEPSPs was attenuated by p38 MAPK inhibition. A, Representative fEPSP traces before (1) and after (2) CPA (40 nm) treatment in slices either in normal aCSF (top) or preincubated in 25 μm SB203580 (bottom). Calibration: 10 ms, 1 mV. B–D, Plots of the mean normalized initial fEPSP slope (±SEM) over time. B, Slices were either preincubated with SB203580 (n = 5) or normal aCSF (n = 4) before treatment with CPA for 10 min. C, Slices were either preincubated with the p38 MAPK inhibitor SB202190 (50 μm) (n = 5) or normal aCSF (n = 5), and then treated with CPA (10 min). D, Slices were either preincubated with the inactive analog SB202474 (25 μm) (n = 8) or normal aCSF (n = 11), and then treated with CPA (10 min). E, Summary of B–D showing the magnitude of CPA-induced depression of fEPSP slope (±SEM) as a percentage decrease from baseline in normal aCSF, SB203580, SB202190, or SB202474. CPA-mediated depression of fEPSPs was attenuated by both p38 MAPK inhibitors (SB203580 and SB202190) but not by the inactive analog (SB202474). **Denotes statistical significance using Student's t test (p < 0.01).
To further confirm that p38 MAPK activity was required for A1 receptor-mediated depression of fEPSPs, we performed an experiment using another pyridinyl imidazole compound, SB202190 (50 μm), which also blocks the activity of p38 MAPK (Davies et al., 2000). Recordings were obtained from slices that were either incubated in 50 μm SB202190 (1–2 h) or normal aCSF (control). In control, a 15 min perfusion of CPA (40 nm) decreased the mean fEPSP slope by 74.3 ± 2.3% (n = 5) (Fig. 6C,E). However, in SB202190, CPA perfusion decreased the mean fEPSP slope by only 25.0 ± 6.0% (n = 5) of baseline (p < 0.01) (Fig. 6C,E).
To control for possible nonspecific effects of the pyridinyl imidazole p38 MAPK inhibitors SB203580 and SB202190, we tested the effects of the inactive pyridinyl imidazole analog SB202474 on the depression of CA1 fEPSPs induced by CPA. Recordings were obtained from slices that were either incubated in 25 μm SB202474 (1–2 h) or normal aCSF (control). In control, a 15 min perfusion of 40 nm CPA decreased the mean fEPSP slope by 74.8 ± 3.4% (n = 11) (Fig. 6C,E). In SB202474-incubated slices, a 15 min perfusion of 40 nm CPA decreased the mean normalized fEPSP amplitude by 70.6 ± 4.4% (n = 8), which was not statistically different from control (p > 0.30) (Fig. 6D,E).
The phosphorylation of p38 in the continued presence of CPA returned to baseline levels after 30 min as shown in Figure 1, A and B. Therefore we tested whether CPA-induced synaptic depression declined when CPA was applied for 30 min. Synaptic responses were maintained at the same depressed level [17.0 ± 6.1% (n = 7) of control] at 30 min. We could see no evidence for desensitization under these conditions. It is possible that other factors in addition of p38 phosphorylation contribute to synaptic depression.
Hypoxia-induced synaptic depression was mediated by the A1 receptor and was attenuated by p38 MAPK inhibition
Hypoxia releases adenosine into the extracellular space (Fowler, 1993; Dale et al., 2000), where it activates A1 receptors. During extended periods of hypoxia, adenosine release contributes to ∼50% of the hypoxia-induced synaptic depression (Fowler, 1989). In contrast to long hypoxic insults, the synaptic depression induced by short hypoxic insults (e.g., 2–3 min) is almost completely mediated by the A1 receptor (Latini et al., 1999; Sebastiao et al., 2000). In the present study, we found that the synaptic depression associated with brief (5 min) hypoxic insult, induced by perfusion of aCSF bubbled with 95% N2/5% CO2, was almost completely blocked in the presence of the A1 receptor antagonist DPCPX (100 nm) (Fig. 7A–C). In normal aCSF, 5 min perfusion of hypoxic solution decreased the mean fEPSP slope by 46.9 ± 4.3% (n = 5) (Fig. 7A–C). In the presence of DPCPX, 5 min hypoxia did not have a significant effect, only decreasing the fEPSP slope by 5.6 ± 1.7% (n = 5; p > 0.30). This was significantly less depression than occurred in normal aCSF (n = 5; p < 0.01) (Fig. 7A–C). In Figure 7D, Hypoxia (5 min) also increased the phosphorylation of p38 similar to the effects of exogenously applied adenosine (Fig. 2E) or the A1 agonist CPA (Fig. 2C,D). The increase in hypoxia was blocked by the A1 antagonist DPCPX (100 nm) indicating that the adenosine release during hypoxia induced the increase in p38 phosphorylation.
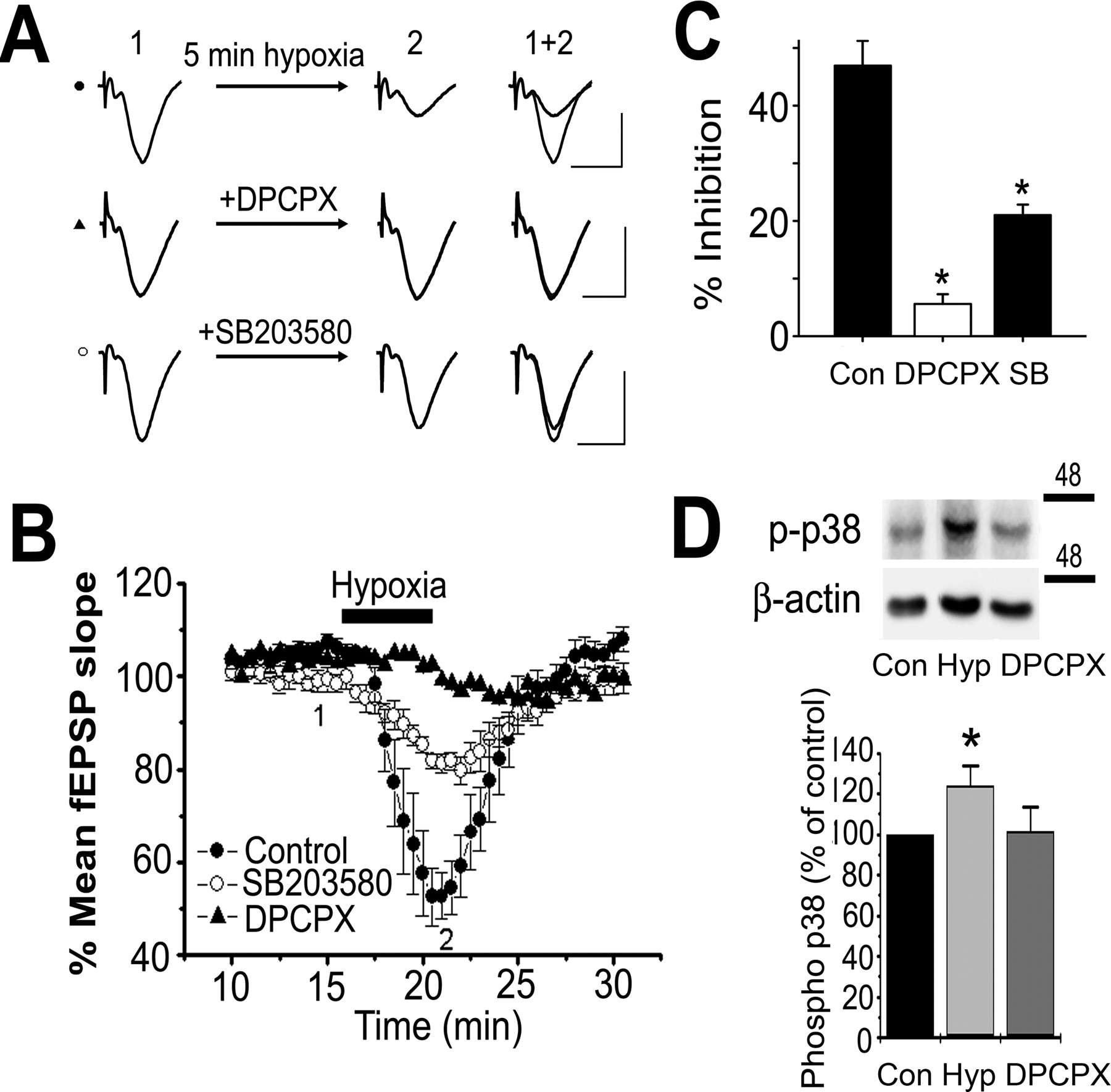
p38 MAPK inhibition attenuated hypoxia-mediated depression of CA1 fEPSPs. A, Representative fEPSP traces before (1) and after (2) treatment with hypoxia (5 min). Hypoxia was induced by bath application of aCSF bubbled with 95% N2/5% CO2. Slices were either treated with 100 nm DPCPX, 25 μm SB203580, or normal aCSF before exposure to hypoxia. Calibration: 10 ms, 1 mV. B, Plot of the mean normalized initial fEPSP slope (±SEM) over time. A1 receptor inhibition with DPCPX (n = 5) and p38 MAPK inhibition with SB203580 (n = 5) attenuated hypoxia-induced synaptic depression versus matched controls (n = 5). C, Summary of B showing the mean fEPSP slope (±SEM) as a percentage of baseline after 5 min hypoxia in normal aCSF, DPCPX, or SB203580. *Denotes statistical significance using Student's t test (p < 0.01). D, Hypoxia (5 min) increased phosphorylation of p38 as shown in the Western blots and summarized in the bar chart below (n = 5). DPCPX (100 nm) blocked the increase, indicating that the phosphorylation of p38 was attributable to the endogenous release of adenosine during hypoxia.
To test the contribution of p38 MAPK activity to hypoxia-induced synaptic depression, we pretreated slices with 25 μm SB203580 (1–2 h) and exposed them to hypoxia for 5 min. In SB203580-treated slices, 5 min hypoxia decreased the mean fEPSP slope by 21.0 ± 1.8% (n = 5), which was significantly less depression than occurred in matched control slices (p < 0.01) (Fig. 7A–C). Given that the synaptic depression induced by 5 min hypoxia was almost entirely blocked by the A1 antagonist DPCPX, this finding indicates that p38 MAPK contributes to the synaptic depression induced by A1 receptor activation attributable to hypoxia. Thus, the neuroprotective action of adenosine released during oxygen deprivation may result from A1 receptor-mediated activation of p38 MAPK.
The synaptic depression mediated by GABAB receptors was not affected by p38 MAPK inhibition
Finally, we set out to determine whether p38 MAPK activation is specifically coupled to A1 receptor stimulation or is a general phenomenon of pertussis toxin-sensitive Gi-protein-coupled receptor activation. To answer this question, we examined the effect of p38 MAPK inhibition on the synaptic depression mediated by baclofen, which is a GABA analog that selectively activates Gi-protein-coupled GABAB receptors (Vacher and Bettler, 2003). As has been previously reported (Olpe et al., 1982; Nathan et al., 1990), baclofen (10 μm) potently depressed fEPSPs in area CA1 (Fig. 8A–C,E,F). To determine the effect of p38 MAPK inhibition on baclofen-induced depression under conditions that attenuate CPA-induced depression, we bath applied both CPA and baclofen sequentially onto slices that had either been pretreated with SB203580 (25 μm) or normal aCSF. As expected, under normal conditions, both CPA and baclofen markedly depressed fEPSP slopes (to 42 and 21% of baseline, respectively; n = 6) (Fig. 8A). In contrast, in slices pretreated with SB203580, CPA-induced depression was attenuated (to only 75% of baseline; n = 8; p < 0.01), whereas baclofen-induced depression was unaffected (to 19% of baseline; n = 8) (Fig. 8B). In another set of experiments, we pretreated slices with another p38 MAPK inhibitor (SB239063; 5 μm) and perfused either CPA or baclofen (Fig. 8D–F). This protocol allowed us to avoid the possibility (albeit remote) of receptor interactions resulting from progressive drug applications. We found that, in the presence of SB239063, CPA-induced depression was attenuated (to 62% of baseline in SB239063; n = 7; vs to 30% of baseline in normal aCSF; n = 5; p < 0.01) (Fig. 8D), whereas baclofen-induced depression was unchanged [to 24% of baseline in SB239063 (n = 9) and to 23% of baseline in normal aCSF (n = 8)] (Fig. 8E). Figure 8F is a summary of the magnitude of CPA- and baclofen-induced depression in the absence and presence of SB239063 showing that, unlike CPA-induced depression, baclofen-induced depression is not sensitive to p38 MAPK inhibition. Thus, it appears that A1 receptor-dependent activation of p38 MAPK is specific to adenosine A1 receptors and not a general property of Gi-protein-coupled receptors.

p38 MAPK inhibition had no effect on baclofen-induced depression of CA1 fEPSPs. A, Representative fEPSP traces and plot of the mean normalized fEPSP slope (±SEM) over time. The traces are as follows: (1) before CPA (40 nm) treatment, (2) after CPA treatment, (3) after recovery from CPA depression, and (4) after baclofen (10 μm) treatment. B, Parallel experiment to A except slices were preincubated for 1–2 h in SB203580 (25 μm). C, Summary of A and B showing that CPA-induced depression of fEPSPs was attenuated by treatment with SB203580, whereas baclofen-induced depression was not. D, Representative fEPSP traces and plot of the mean normalized fEPSP slope (±SEM) over time. In slices pretreated with 5 μm SB239063 (open circles) (n = 7), CPA-induced depression was attenuated compared with matched control slices (filled circles). E, Parallel experiment to D except that baclofen was perfused onto slices instead of CPA. F, Summary of E and D showing that CPA-induced depression was attenuated in slices preincubated in SB239063, whereas baclofen-induced depression was not. **Denotes statistical significance using Student's t test (p < 0.01).
Discussion
In this study, we report a novel interaction between the adenosine A1 receptor and p38 MAPK in the CNS. We present biochemical and electrophysiological evidence showing that p38 MAPK is activated by A1 receptor stimulation and that p38 MAPK inhibition significantly attenuates A1 receptor-mediated synaptic depression. In contrast, GABAB receptor-mediated synaptic depression was insensitive to p38 MAPK inhibition, which suggests specific coupling of p38 MAPK activation to adenosine A1 receptor stimulation. Consistent with this prediction, coimmunoprecipitation studies revealed that phospho-p38 MAPK and the adenosine A1 receptor are physically associated in the hippocampus. Our results suggest that A1 receptors and p38 MAPK form a signaling complex, and that adenosine A1 receptor-mediated synaptic depression is at least partly dependent on the activation of p38 MAPK in the rat hippocampus.
Although adenosine A1 receptors and p38 MAPK are expressed both presynaptically (Maruyama et al., 2000) and postsynaptically (Bolshakov et al., 2000; J. J. Zhu et al., 2002) in the hippocampus, the primary mechanism underlying adenosine-induced synaptic depression is inhibition of calcium influx into presynaptic nerve terminals (Dunwiddie and Masino, 2001). In area CA1 of the hippocampus, as well as other areas, the adenosine A1 receptor subtype decreases neurotransmitter release from presynaptic nerve terminals primarily by inhibiting N-type calcium channels (Mogul et al., 1993; Yawo and Chuhma, 1993; Mynlieff and Beam, 1994; Wu and Saggau, 1994; Ambrosio et al., 1997; Zhang and Schmidt, 1999; Brown and Dale, 2000; Park et al., 2001; Sun et al., 2002; Wang et al., 2002; Manita et al., 2004). Therefore, our study suggests that p38 MAPK activation is a necessary step for A1 receptor activation to decrease synaptic transmission by decreasing presynaptic voltage-dependent calcium channel function. Although synaptic depression via A1 receptor activation is thought to be principally presynaptic, we cannot rule out additional contributions to p38 activation attributable to postsynaptic receptor activation. p38 MAPK activation is necessary for the depression of N-type calcium currents in neuroblastoma X glioma cells in response to bradykinin application (Wilk-Blaszczak et al., 1998).
A possible link between p38 MAPK and calcium channel modulation is our finding that A1 receptor stimulation induced PP2a translocation to the membrane requires p38 MAPK, which is in agreement with previous studies in cardiomyocytes (Liu and Hofmann, 2002, 2003). PP2a is also activated by p38 MAPK in the brain, where it can modulate serotonin transporters (Zhu et al., 2005). PP2a binds to the C terminus of α-1C calcium channels, and reverses PKA (protein kinase A) -mediated phosphorylation of serine 1928 located inside this C-terminal region (Davare et al., 2000). A recent report has also demonstrated direct interaction of the protein phosphatase 2Cα (PP2Cα) to the C terminus of both N- and P/Q-type calcium channels, and that PP2Cα reverses phosphorylation of these neuronal calcium channels by PKC (protein kinase C)(Li et al., 2005). However, it is unclear whether PP2a also physically associates with and alters the function of presynaptic α 1B (N-type) calcium channels. In the present study, we demonstrated that CPA-mediated synaptic depression accompanied increased activation of p38 MAPK and PP2a. Future biochemical and functional studies are needed to address whether this decreased synaptic transmission may be due, in part, to neuronal calcium channel inhibition by channel interaction with PP2a. Alternatively, other voltage-gated ion channels, including sodium channels (Wittmack et al., 2005) and HCN (hyperpolarization-activated cyclic nucleotide)-gated channels (Poolos et al., 2002, 2006), are newly discovered potential targets for the A1 receptor-mediated p38 MAPK activation that could play additional roles in decreasing membrane excitability and synaptic depression.
Excessive synaptic release of glutamate during trauma and hypoxia/ischemia is a major mechanism underlying neuronal cell death (Kass and Lipton, 1982; Rothman, 1983, 1984). Adenosine is an endogenous neuroprotectant during hypoxia/ischemia that decreases glutamate release in the brain via A1 receptors (Dunwiddie and Masino, 2001). The importance of adenosine as an endogenous neuroprotective agent is exemplified by the observation that brief insults of hypoxia/ischemia are more deleterious when repeated (Tomida et al., 1987; Kato et al., 1989), likely because of adenosine depletion (Pearson et al., 2001).
Adenosine A1 receptor-dependent p38 MAPK activation has been implicated in delayed ischemic preconditioning. In heart tissue, the p38 inhibitor SB203580 blocks A1 receptor agonist-induced late phase preconditioning (Carroll and Yellon, 2000; Zhao et al., 2001; Loubani and Galinanes, 2002). In addition, A1 receptor stimulation-induced p38 MAPK activity occurs in rabbit myocardium (Dana et al., 2000) and is necessary for delayed preconditioning of rat myocardium in vivo (Lasley et al., 2005).
Brain p38 MAPK is activated within minutes of transient forebrain ischemia in vivo (Sugino et al., 2000) and induces tolerance to serious ischemic insult after a brief sublethal ischemic insult in the gerbil hippocampus (Nishimura et al., 2003). Another form of ischemic preconditioning in the brain can be elicited by volatile anesthetics (Kapinya et al., 2002; Zhao and Zuo, 2004; Bickler et al., 2005; Gray et al., 2005), which requires the activation of A1 receptors (Liu et al., 2006) and p38 MAPK (Zheng and Zuo, 2004). In the present study, we showed that the inhibitory actions of adenosine A1 receptors, but not other inhibitory receptors such as GABAB receptors, were attenuated by p38 MAPK inhibitors. These results suggest that p38 MAPK activation is specific to adenosine A1 receptors and not a general property of Gi-protein-coupled receptors.
Previous studies have found that the p38 MAPK pathway is activated after hypoxia/ischemia in brain tissue (Walton et al., 1998; Y. Zhu et al., 2002). However, there is evidence that p38 MAPK activity is deleterious, mediating the cell death induced by hypoxia (Y. Zhu et al., 2002), oxygen glucose deprivation, magnesium withdrawal, and glutamate receptor agonist exposure (Legos et al., 2002). In contrast, in heart tissue, p38 MAPK plays an important role in cardioprotection during ischemia (Weinbrenner et al., 1997; Baines et al., 1998, 1999; Zhao et al., 2001; Lasley et al., 2005). The results of the present study indicate that p38 MAPK could act as a neuroprotectant by mediating the actions of adenosine in the brain.
The apparent contradiction between our results showing that p38 MAPK activation may be neuroprotective, and previous findings showing that p38 MAPK inhibition may be neuroprotective (Barone et al., 2001; Legos et al., 2002) is probably attributable to differences in the timescales analyzed. We found that A1 receptor stimulation phosphorylated p38 MAPK within minutes (Fig. 1A,B). However, most reports linking p38 MAPK activity and excitotoxic injury have analyzed cell survival in culture hours and/or days after trauma (Walton et al., 1998; Barone et al., 2001; Legos et al., 2002; Y. Zhu et al., 2002). Our results suggest that, in contrast to its longer-term deleterious actions, p38 MAPK activation may have a short-term neuroprotective role during transient oxygen deprivation. Therefore, a viable therapeutic intervention to prevent ischemic brain damage might involve the use p38 MAPK inhibitors only after reperfusion.
We have shown previously that p38 MAPK is involved in presynaptic inhibition, because it mediates the potent and long-lasting synaptic depression induced by Bz-ATP (2′,3′-O-(4-benzoylbenzoyl)-ATP) at mossy fiber–CA3 synapses (Armstrong et al., 2002). However, in mossy fiber–CA3 synapses, there is no evidence that adenosine-induced synaptic depression is attenuated by SB203580 (Armstrong et al., 2002). Thus, although p38 MAPK is involved in presynaptic inhibition at both mossy fiber–CA3 and CA3–CA1 synapses, the pathway that activates p38 MAPK is different in these two distinct areas of the hippocampus. Future studies will test whether SB203580 may differentially alter adenosine-induced synaptic depression in these hippocampal regions by modifying the interaction of p38 MAPK with a regulatory molecule or by inhibiting the constitutive association of phosphorylated p38 MAPK and A1 receptors.
It is unlikely that SB203580 exerts its effects on mossy fiber–CA3 synaptic depression by blocking adenosine transporters as previously suggested (Kukley et al., 2004). The inactive analog SB202474 (Sweeney et al., 1999) has a similar effect on nucleoside transporters as SB203580 (Huang et al., 2002). Therefore, to rule out the possibility that SB203580 and SB202190 were exerting their effects by blocking nucleoside transporters, we tested whether SB202474 affected adenosine- or CPA-induced synaptic depression. We found that SB202474 failed to affect adenosine A1 receptor-mediated synaptic depression in area CA1 (Figs. 5B,D; 6D,E). In addition, inhibitors of nucleoside transporters in brain slices actually enhance the concentration of extracellular adenosine during hypoxia or electrical stimulation (Fredholm et al., 1994). This indicates that the net effect of nucleoside transporters is the uptake of adenosine from the extracellular space (Latini and Pedata, 2001). Thus, blockade of nucleoside transporters would be expected to decrease synaptic transmission attributable to a build-up of excess adenosine in the extracellular space as has been reported (Dunwiddie and Diao, 2000). In contrast, we observed that SB203580 increased synaptic transmission (Fig. 4B).
In conclusion, we have shown in the present study that p38 MAPK physically associates with and is activated by A1 receptors within minutes and contributes to adenosine-induced synaptic depression in area CA1 of the hippocampus. Moreover, our results indicate that p38 MAPK activity contributes to the synaptic depression induced by the release of endogenous adenosine during hypoxia. Activation of p38 MAPK by adenosine A1 receptors may represent a novel mechanism underlying presynaptic inhibition of neurotransmission and ischemic preconditioning in the mammalian brain.
Footnotes
-
This work was supported by the Canadian Institutes of Health Research and the Canadian Stroke Network. B.A.M. is a Canada Research Chair in Neuroscience and Michael Smith Distinguished Scholar. T.B.B. was supported by studentships from the Natural Sciences and Engineering Research Council of Canada, Libin Neuroscience Canada Foundation (Alberta), and Alberta Heritage Foundation for Medical Research. F.S.C. is a Heart and Stroke Foundation of Canada “Focus on Stroke” and a Michael Smith Foundation for Health Research Postdoctoral Fellow.
- Correspondence should be addressed to Dr. Brian A. MacVicar, Brain Research Centre, Department of Psychiatry, University of British Columbia, 2211 Wesbrook Mall, Vancouver, BC, Canada V6T 2B5. bmacvica{at}interchange.ubc.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}