

Abstract
Filamentous actin binding protein neurabin I (NrbI) targets protein phosphatase-1 (PP1) to specific postsynaptic microdomains, exerting critical control over AMPA receptor (AMPAR)-mediated synaptic transmission. NrbI-targeted synaptic PP1, which promotes synaptic depression upon long-term depression (LTD) stimuli, serves to prevent synaptic depression under basal conditions. The present studies investigate this opposite regulation of AMPAR trafficking during basal synaptic transmission and LTD by expressing NrbI or NrbI mutant, which is defective in PP1 binding, in hippocampal slice or neuron cultures. We find that expression of the NrbI mutant to interfere with PP1 targeting dramatically reduces basal synaptic transmission, which is correlated with the reduction in surface expression of AMPA subtype glutamate receptor (GluR) 1 and GluR2 subunits. Biochemical analysis demonstrates that the NrbI mutant selectively increases the phosphorylation of GluR2 at C-terminal consensus PKC site, serine 880, which is known to favor GluR2 interaction with PDZ (postsynaptic density 95/Discs large/zona occludens 1) protein PICK1 (protein interacting with C kinase-1). Inhibition of PKC activity or GluR2–PICK1 interaction completely reverses the synaptic depression in neurons expressing the NrbI mutant, suggesting that NrbI-targeted synaptic PP1 stabilizes the basal transmission by negatively controlling PKC phosphorylation of GluR2 and the subsequent PICK1-mediated decrease in GluR2-containing AMPAR surface expression. Distinct from basal transmission, blocking GluR2–PICK1 interaction or PKC activity produces minimal effects on LTD in NrbI-expressing neurons. Instead, NrbI-targeted PP1 facilitates LTD by dephosphorylating GluR1 at both serine 845 and serine 831, with GluR2 serine 880 phosphorylation unaltered. Our studies thus elucidate that NrbI-targeted PP1, in response to distinct synaptic activities, regulates the synaptic trafficking of specific AMPAR subunits.
Introduction
Besides protein phosphatase-1 (PP1) enzymatic activity, PP1 synaptic targeting is critical for the induction and maintenance of long-term depression (LTD) in hippocampus (Mulkey et al., 1993; Morishita et al., 2001; Hu et al., 2006; Jouvenceau et al., 2006). PP1 targeting proteins direct PP1 to the immediate vicinity of PP1 substrates for specific and efficient catalysis. Recent studies identify an filamentous actin (F-actin) binding protein, neurabin I (NrbI), as the critical synaptic PP1 targeting protein that promotes lasting synaptic depression on LTD stimuli (Hu et al., 2006). Surprisingly, NrbI-targeted synaptic PP1 also plays an essential role in preventing synaptic depression under basal conditions (Terry-Lorenzo et al., 2005; Hu et al., 2006). Once PP1 binding to NrbI is disrupted, the basal synaptic transmission is markedly decreased. The signaling mechanisms for this opposite regulation of synaptic strength by NrbI-targeted synaptic PP1 during basal transmission and LTD are not clear.
AMPA subtype of glutamate receptors (AMPARs) is a principle mediator of fast excitatory synaptic transmission in mammalian CNS. AMPARs interact with a series of postsynaptic PDZ (postsynaptic density 95/Discs large/zona occludens 1) proteins via the extreme cytoplasmic C-terminal tails, a molecular process that critically controls AMPAR trafficking and synaptic strength (Bredt and Nicoll, 2003; Kim and Sheng, 2004; Chen et al., 2007). Glutamate receptor 2 (GluR2)/GluR3 heteromers of AMPARs have been proposed to constitutively recycle to and from synapses, serving to maintain the concentration of synaptic AMPARs and stabilize basal synaptic transmission (Shi et al., 2001). GluR2/GluR3 subunits share a common short C tail and interact with three group II PDZ proteins: glutamate receptor interacting protein (GRIP) (Dong et al., 1997), AMPA receptor binding protein (ABP/GRIP2) (Srivastava et al., 1998), and protein interacting with C kinase-1 (PICK1) that directs protein kinase Cα (PKCα) to GluR2 (Xia et al., 1999; Perez et al., 2001). Notably, PKC phosphorylation of GluR2 at serine 880 (Ser880) eliminates GluR2 interaction with GRIP/ABP but not with PICK1 (Chung et al., 2000; Matsuda et al., 2000; Xia et al., 2000; Kim et al., 2001; Perez et al., 2001; Seidenman et al., 2003). GRIP/ABP has been hypothesized to anchor AMPARs at postsynaptic membrane (Dong et al., 1997; Srivastava et al., 1998; Osten et al., 2000), whereas PICK1 mediates the decrease in the AMPAR surface expression through either internalizing AMPARs or retaining internalized AMPARs in subsynaptic cytoplasmic compartments (Chung et al., 2000; Xia et al., 2000; Iwakura et al., 2001; Perez et al., 2001; Sossa et al., 2006).
It has been reported that PP1 can reduce PKC activity in COS cells (Bornancin and Parker, 1997) and in hippocampus after LTD induction (Thiels et al., 2000). The present work finds that NrbI-targeted synaptic PP1 negatively controls PKC phosphorylation of GluR2 at Ser880, thus preventing PICK1-mediated decrease in the surface expression of GluR2-containing AMPARs and stabilizing basal transmission. After LTD-inducing stimuli, however, NrbI-targeted synaptic PP1 dephosphorylates GluR1 subunits, leading to lasting synaptic depression independent of GluR2–PICK1 interaction. Our data suggest that NrbI-targeted PP1 stimulates multiple signaling pathways involved in AMPAR trafficking, depending on the pattern of synaptic activities.
Materials and Methods
Construction of NrbI-expressing Sindbis virus.
The cDNAs encoding the green fluorescence protein (GFP)-tagged NrbI and mutant neurabins (Fig. 1A) were subcloned into pSinRep5 vector (Invitrogen, Carlsbad, CA) to generate recombinant Sindbis virus according to the instructions of the manufacturer. Briefly, GFP–NrbI pSinRep5 plasmids were linearized using NotI, and the DH-26S helper plasmid was linearized using XhoI. The two linearized templates were subjected to in vitro transcription using Ambion (Austin, TX) mMESSAGE mMACHINE SP6 kit. The resulting RNAs were cotransfected into BHK21 cells by electroporation and incubated for 48 h. The supernatant (which contained the virus) was collected, and the viral particles were concentrated by centrifugation at 30,000 rpm for 1.5 h.
Cell and slice culture, transfection, immunocytochemistry, and microscopy.
The experimental protocols were approved by the Institutional Animal Care and Use Committee of Louisiana State University Health Sciences Center. Primary hippocampal neurons in culture were prepared from postnatal day 0 (P0) to P1 Sprague Dawley rat pups (Deisseroth et al., 1996). Cells were plated on Matrigel-coated coverslips and grown in culture for 15–18 d before experiments. The transverse hippocampal slices (350 μm thick) were prepared from P9–P11 Sprague Dawley rat pups as described previously (Hu et al., 2006). Sindbis virus encoding recombinant NrbI or its mutants was injected immediately after slice preparation using a Nitrogen-based Picospritzer III (Parker Hannifin, Fairfield, NJ). To detect the synaptic targeting of NrbI, the hippocampal slice cultures were fixed 24 h after virus injection with 2.5% paraformaldehyde plus 4% sucrose in PBS for imaging. To examine the role of endogenous NrbI in the synaptic transmission, Lipofectamine 2000 (Invitrogen) was used to transfect primary cultured hippocampal neurons with the pNrbI–OFF plasmid (a gift from Dr. Shirish Shenolikar, Duke University, Durham, NC), which coexpressed short hairpin RNA (shRNA) against NrbI (under an H1 promoter) along with enhanced GFP (EGFP) (under a cytomegalovirus promoter). Another plasmid pZOFF–EGFP (a gift from Shirish Shenolikar), which expressed EGFP alone, was used as a control. The pNrbI–OFF plasmid has been shown to successfully knockdown the expression of endogenous NrbI in hippocampal neurons (Terry-Lorenzo et al., 2005). For GluR1 and GluR2 surface immunostaining, primary hippocampal neurons were transfected with cDNAs encoding GFP, GFP–NrbI, and GFP–NrbI mutant. The transfected neurons were incubated live with diluted rabbit primary antibody (1:50; Calbiochem, San Diego, CA) against the extracellular N terminus of GluR1 and mouse primary antibody (1:500; Chemicon, Temecula, CA) against the extracellular N terminus of GluR2 for 20 min at 37°C as described previously (Xia et al., 2002). After fixation, neurons were incubated with blocking buffer (5% BSA and 5% normal goat serum in PBS) for 1 h. Then Alexa647-conjugated secondary anti-rabbit antibody (Invitrogen) and Alexa568-conjugated secondary anti-mouse antibody (Invitrogen), both of which were diluted to 1:500 in the blocking buffer, were used for incubation with neurons for 1 h. In some experiments, NMDA receptor (NMDAR) inhibitor d-APV (100 μm; Sigma, St Louis, MO) and PKC inhibitor chelerythrine (5 μm; Sigma) or protein phosphatase-1 inhibitor tautomycin (10 μm; Calbiochem) were used to treat cells 24 and 2–4 h, respectively, before incubating the neurons live with the primary antibodies. Immunoreactivity was acquired by a Zeiss (Toronto, Ontario, Canada) 510 Meta confocal laser scanning microscope equipped with a 63× oil immersion objective.
Biochemical analysis.
Immunoprecipitation of GFP–NrbI with PP1 was performed in HEK293T cells transfected with cDNAs encoding GFP–NrbI or GFP–NrbI mutant using Lipofectamine 2000. At 24 h after transfection, cells were lysed in TEST buffer (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 20 mm EDTA, and 1 mm PMSF) for 15 min on ice. The lysate was subjected to centrifugation at 16,000 × g for 10 min at 4°C. The supernatant was incubated with anti-GFP antibody (BD Biosciences, Bedford, MA) for 1 h at 4°C, followed by incubation with protein G-Sepharose (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h. Then, the beads were washed with the TEST buffer before dissolving the immunoprecipitates in SDS sample buffer and analysis on SDS-PAGE. Immunoblotting was performed with anti-GFP and anti-PP1 antibodies (BD Transduction Laboratories, Lexington, KY).
To detect the synaptic abundance of PP1 in synaptoneurosome fraction (Johnson et al., 1997), CA1 minislices were microdissected out (Nayak et al., 1996) from hippocampal slice cultures infected with GFP, GFP–NrbI, or GFP–NrbI mutant. The minislices were homogenized in ice-cold incubation buffer (in mm: 20 HEPES, pH 7.4, 140 NaCl, 5 KCl, 5 NaHCO3, 1.2 Na2HPO4, 10 dextrose, 1 MgCl2, 1.8 CaCl2, and 10 EDTA) plus the inhibitors of proteases and phosphatases (4 mm EGTA, 50 mm NaF, 10 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 0.2 mm PMSF, 10 U/ml aprotinin, 0.0001% chymostatin, 0.0001% leupeptin, 0.0001% antipain, and 0.0001% pepstatin) by seven strokes in a glass–glass tissue homogenizer (B. Braun, Melsungen, Germany). The homogenates were filtered through two layers of 100-μm-pore nylon net filters (Millipore, Billerica, MA), followed by two layers of 10-μm-pore Mitex membrane filters (Millipore). The filtered particulates were spun at 1000 × g for 15 min at 4°C. The pellets were resuspended with the incubation buffer and spun again to obtain the washed synaptoneurosome for immunoblotting with anti-PP1 antibody and anti-synaptophysin antibody (Upstate, Charlottesville, VA). The relative amount of PP1 in synaptoneurosome was determined by the ratio of PP1 signal to synaptophysin signal.
To examine the phosphorylation of AMPA receptors, primary hippocampal neurons transfected with GFP, GFP–NrbI, or GFP–NrbI mutant cDNAs were lysed in the TEST buffer. The lysate was subjected to SDS-PAGE and immunoblotting with phosphorylation site-specific antibodies against GluR1 serine 845, GluR1 serine 831, or GluR2 serine 880 (Upstate) and the antibodies against the C terminus of GluR1 or GluR2. In some experiments, d-APV (100 μm) and chelerythrine (5 μm) were added to neurons for 24 and 2–4 h, respectively, before lysis. The relative amount of phosphorylated GluR1 and GluR2 was determined by the ratio of phosphorylation signals to total GluR1 and GluR2 signals, respectively. To assay the phosphorylation of AMPARs after LTD induction, low-frequency stimuli (1 Hz) were first delivered to the Schaffer collateral afferents in CA1 minislices infected with GFP or recombinant GFP–NrbI with rake electrodes, which were used to increase the fraction of synapses undergoing plasticity (Nayak et al., 1996); then the homogenates were prepared by sonicating the minislices for 30 s in the incubation buffer (in mm: 10 sodium phosphate, pH 7.0, 100 NaCl, and 10 EDTA) plus the inhibitors of proteases and phosphatases. The homogenates were centrifuged at 12,000 × g for 10 min at 4°C. The crude membrane pellets were resuspended in the incubation buffer and centrifuged again at the same speed. The pellets were then resuspended in SDS sample buffer and were subjected to SDS-PAGE and immunoblotting with phosphorylation site-specific antibodies against GluR1 serine 845, GluR1 serine 831, GluR2 serine 880, and with the anti-synaptophysin antibody. To examine the effect of PP1 inhibitor tautomycin on GluR2 serine 880 phosphorylation, non-infected CA1 minislices were incubated with tautomycin (10 μm) for 2–4 h before homogenization. The relative amount of phosphorylated AMPAR subunits was determined by the ratio of phosphorylation signals to synaptophysin signals. The ratios of the control and the matching test samples were used for statistic analysis. For display purposes, these ratios were normalized to the control value.
Electrophysiological recordings.
Whole-cell or field potential recordings were performed in hippocampal slice cultures 24 h after virus injection with an Axopatch-700A amplifier (Molecular Devices, Palo Alto, CA). The recording chamber was perfused at room temperature with artificial CSF that contained the following (in mm): 119 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 d-(+)-glucose, and 0.1 picrotoxin (bubbled with 5% CO2–95% O2, pH 7.4). For miniature EPSCs (mEPSCs) recordings performed at 30–32°C, tetrodotoxin (0.5 μm; Sigma) was added to the external solution and MgCl2 was decreased to 0.5 mm. Recording pipettes (3–5 MΩ) were filled with the internal solution containing the following (in mm): 115 cesium methanesulfonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 Na2ATP, 0.4 Na3GTP, 0.6 EGTA, 10 sodium phosphocreatine, and 0.1 spermine, pH 7.25 (osmolarity, 290–295 mOsm). For field potential recordings, the pipettes were filled with the external solution. EPSCs were evoked by bipolar tungsten electrodes placed over Schaffer collateral fibers ∼75–150 μm away from the CA1 cell body layer. Series and input resistances were monitored on-line throughout each experiment. In pairwise comparison experiments, recordings were first made in an infected or uninfected cell, and, afterward, second recordings were obtained immediately from an adjacent uninfected or infected cell without changes in the location and intensity of stimulus (Hayashi et al., 2000; Shi et al., 2001; Marie et al., 2005). Infected and uninfected control neurons were visually identified using an Olympus Optical (Tokyo, Japan) BX51WIF microscope fitted with a 60× water immersion objective under fluorescence and transmitted light illumination. LTD was induced by pairing postsynaptic depolarization at −45 mV with presynaptic stimulation at 1 Hz for 5 min. LTD of field potentials was induced by low-frequency stimuli (1 Hz) in slices infected with GFP or recombinant GFP–NrbI. Current signals were filtered at 2 kHz, sampled at 5 kHz, and collected with Igor Software (WaveMetrics, Lake Oswego, OR). The mEPSC signals were analyzed with mini-analysis software (Synaptosoft, Fort Lee, NJ). In peptide application experiments, a 15 amino acid peptide KKEGYNVYGIESVKI (p-SVKI) (Invitrogen) or KKEGYNVYGIEEVKI (p-EVKI) (AnaSpec, San Jose, CA) was included (200 μm) in the internal solution along with two protease inhibitors, leupeptin and bestatin (100 μm each; Sigma). For some pharmacological experiments, d-APV or high concentration of MgCl2 (10 mm) was added to the culture medium immediately after virus injection. Protein kinase inhibitors KN-62 (1-[N,O-bis(5-isoquinolinesulphonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine) (Sigma), Rp-cAMP (Sigma), chelerythrine (Sigma), and protein phosphatases inhibitors calyculin A (Sigma) or tautomycin (Calbiochem) were added into the culture medium 2–4 h before recordings, and they were also included in the internal solution. All of the data were presented as mean ± SEM, and statistical significance was set at p < 0.05 using Wilcoxon's and paired t tests for paired measurements and Mann–Whitney and unpaired t tests for unpaired measurements, respectively.
Results
NrbI targeted PP1 at synapses
One of the most important functions of NrbI is its presumptive ability to target PP1 at specific postsynaptic compartments, a crucial process that allows PP1 to exert catalytic effects in close proximity of its substrates. NrbI contains several functional domains (Fig. 1A). However, which domains are relevant to the PP1 targeting profile of NrbI has not been fully understood. The N-terminal region of NrbI has an F-actin-binding domain (ABD) and a conserved PP1 binding motif (KIKF motif), which localizes NrbI to dendritic spines (Zito et al., 2004) and forms complex with PP1 (McAvoy et al., 1999), respectively. We speculated that the N terminus that encompassed ABD and KIKF motif was sufficient to direct NrbI to spines and, at the same time, target its associated PP1 at postsynaptic compartment. To test this hypothesis, we deleted the C terminus to generate NrbI mutant protein, GFP–NrbI (1–490). The truncated GFP–NrbI (1–490) was indeed concentrated at dendritic spines (Fig. 1C). Immunoprecipitation experiments showed that GFP–NrbI (1–490) exhibited the similar PP1 binding property (Fig. 1B) as full-length wild-type NrbI (Hu et al., 2006). More importantly, the concentration of PP1 in synaptoneurosome fraction significantly increased in GFP–NrbI (1–490)-infected hippocampal slices relative to that in GFP-infected slices (Fig. 1D). These results suggested that the N terminus of NrbI targeted endogenous PP1 at synapses. GFP–NrbI (1–490, F460A), in which phenylalanine 460 within the core PP1 binding motif was substituted with alanine (Fig. 1A), also concentrated itself at spines like GFP–NrbI (1–490) (Fig. 1C) [spine-to-shaft GFP signal ratio normalized to that in GFP–NrbI (1–490)-expressing neurons, 95 ± 3%; p > 0.05; n = 12]. However, GFP–NrbI (1–490, F460A) failed to interact with PP1 (Fig. 1B); as a result, the synaptic abundance of PP1 was dramatically reduced in GFP–NrbI (1–490, F460A)-infected slices as illustrated by immunoblotting of PP1 in synaptoneurosome preparation (Fig. 1D). These results suggested that GFP–NrbI (1–490, F460A) interfered with the PP1 targeting at synapses. Because the N terminus of NrbI was sufficient to fulfill PP1 targeting function, we used GFP–NrbI (1–490) and GFP–NrbI (1–490, F460A) in the subsequent experiments to focus on NrbI-mediated PP1 targeting in synaptic transmission.

NrbI targeted PP1 to dendritic spines via its N terminus. A, Schematics of NrbI polypeptides analyzed in this study. Wild-type NrbI (1095 amino acids), fused with GFP at its N terminus, has an F-actin-binding domain (black), PP1-binding motif (KIKF), PDZ homology domain, coiled-coil (CC) domain, and sterile α motif (SAM). GFP-tagged truncated NrbI (1–490) and NrbI (1–490, F460A), in which phenylalanine 460 was substituted with alanine, were also shown. B, NrbI associated with PP1 via PP1 binding motif. HEK293 cells were transfected with cDNAs encoding GFP–NrbI (1–490) or GFP–NrbI (1–490, F460A). The protein extracts were immunoprecipitated (IP) with anti-GFP antibodies. Immunoprecipitates were subjected to SDS-PAGE and immunoblotting (IB) with anti-GFP and anti-PP1 antibodies. Note that GFP–NrbI (1–490, F460A) did not bind to PP1. C, Both GFP–NrbI (1–490) (top) and GFP–NrbI (1–490, F460A) (bottom) were concentrated at dendritic spines (marked by arrows) in CA1 pyramidal cells from organotypic hippocampal slice cultures. Scale bar, 5 μm. D, Expression of GFP–NrbI (1–490) increased, whereas GFP–NrbI (1–490, F460A) decreased, the PP1 concentration in synaptoneurosome fraction from hippocampal slice cultures. Left, Western blots of PP1 in synaptoneurosome from slices infected with GFP, GFP–NrbI (1–490), or GFP–NrbI (1–490, F460A). Equal protein loadings were indicated by immunoblotting of synaptophysin. Right, Graph showed the mean percentage changes of PP1 concentration in synaptoneurosome. The relative amount of PP1 was determined by the ratio of PP1 signals to synaptophysin signals. These ratios were normalized to the control (GFP) value. *p < 0.05 when compared with GFP-infected slices.
NrbI-targeted synaptic PP1 functioned to stabilize the basal synaptic transmission
Previous studies have examined the role of PP1 in the basal synaptic transmission, finding that inhibition of all pools of PP1 activities by chemical reagents (such as calyculin A) or disruption of PP1 interaction with all of its regulatory/targeting proteins has no effects on the basal synaptic transmission (Mulkey et al., 1993; Herron and Malenka, 1994; Morishita et al., 2001). The present study confirmed these observations by showing that inhibition of all intracellular PP1 activities with either calyculin A (1 μm) or tautomycin (10 μm) did not change the AMPAR-mediated basal synaptic transmission in hippocampal slice cultures (Fig. 2A). It appears that PP1 is “inactive” in regulating synaptic responses. Surprisingly, once GFP–NrbI (1–490, F460A) was expressed to specifically interfere with NrbI-mediated PP1 targeting and decrease the PP1 abundance at synapses, AMPAR-mediated synaptic transmission was dramatically suppressed. As shown in Figure 2A, the frequency and amplitude of mEPSCs were reduced in GFP–NrbI (1–490, F460A)-expressing neurons (Fig. 2A) [amplitude: GFP, −13.6 ± 0.9 pA, n = 10; GFP–NrbI (1–490, F460A), −8.9 ± 0.3 pA, n = 13, p < 0.05; frequency: GFP, 0.21 ± 0.02 Hz, n = 10; GFP–NrbI (1–490, F460A), 0.12 ± 0.02 Hz, n = 13, p < 0.05]. Pairwise comparison of evoked EPSCs, recorded sequentially from infected and neighboring uninfected neurons in the same slice, also illustrated the synaptic depression in infected neurons (Fig. 2C) [uninfected, −42.9 ± 4.6 pA; GFP–NrbI (1–490, F460A), −21.7 ± 2.3 pA, n = 23, p < 0.05]. The critical role of NrbI in the basal synaptic transmission was further confirmed by performing RNA interference in primary cultured hippocampal neurons to knockdown endogenous NrbI. When transfected on 13 d in vitro (DIV13) with pNrbI–OFF encoding shRNA against NrbI (Terry-Lorenzo et al., 2005), neurons on DIV18 exhibited marked reduction in mEPSC amplitude and frequency relative to those transfected with control pZOFF–EGFP plasmid encoding EGFP alone (Fig. 2B). In contrast, infected neurons with GFP–NrbI (1–490) in hippocampal slices exhibited normal basal transmission, with the frequency and amplitude of mEPSCs comparable with those in GFP-expressing neurons (Fig. 2A) (amplitude, −14.8 ± 1.0 pA, n = 11, p > 0.05 compared with GFP-expressing neurons; frequency, 0.26 ± 0.06 Hz, n = 11, p > 0.05). The evoked EPSCs also displayed the similar amplitudes to those in adjacent control neurons (Fig. 2C) [uninfected, −41.5 ± 4.5 pA; GFP–NrbI (1–490), −44.9 ± 4.3 pA, n = 19, p > 0.05]. These results suggested that the pool of NrbI-targeted synaptic PP1 played a critical role in stabilizing AMPAR-mediated basal synaptic transmission. The differential effects of tautomycin and GFP–NrbI (1–490, F460A) on the basal transmission suggested that the pool of NrbI-targeted synaptic PP1 and the remaining pool of synaptic PP1, targeted by other PP1-binding proteins, might play opposite roles in regulating synaptic transmission. Tautomycin nonselectively inhibited all pools of PP1, leaving the net synaptic transmission unaltered. If this was the case, application of tautomycin to further inactivate the remaining pool of synaptic PP1 in GFP–NrbI (1–490, F460A)-expressing neurons should reverse the synaptic depression. Indeed, pretreatment of GFP–NrbI (1–490, F460A)-expressing neurons with tautomycin (10 μm) significantly increased the mEPSC amplitude and frequency in hippocampal slice cultures (Fig. 2A) [amplitude, −13.5 ± 0.8 pA, n = 8, p < 0.05 when compared with GFP–NrbI (1–490, F460A) alone; frequency, 0.21 ± 0.03 Hz, n = 8, p < 0.05]. Consistent with this, the decreased surface expression of AMPARs in GFP–NrbI (1–490, F460A)-expressing neurons was also restored (see Fig. 5E). These results suggested that the effect of NrbI-targeted synaptic PP1 on the basal synaptic transmission was tightly specific, and distinct pools of PP1 exerted diverse modifications of AMPAR-mediated synaptic transmission.
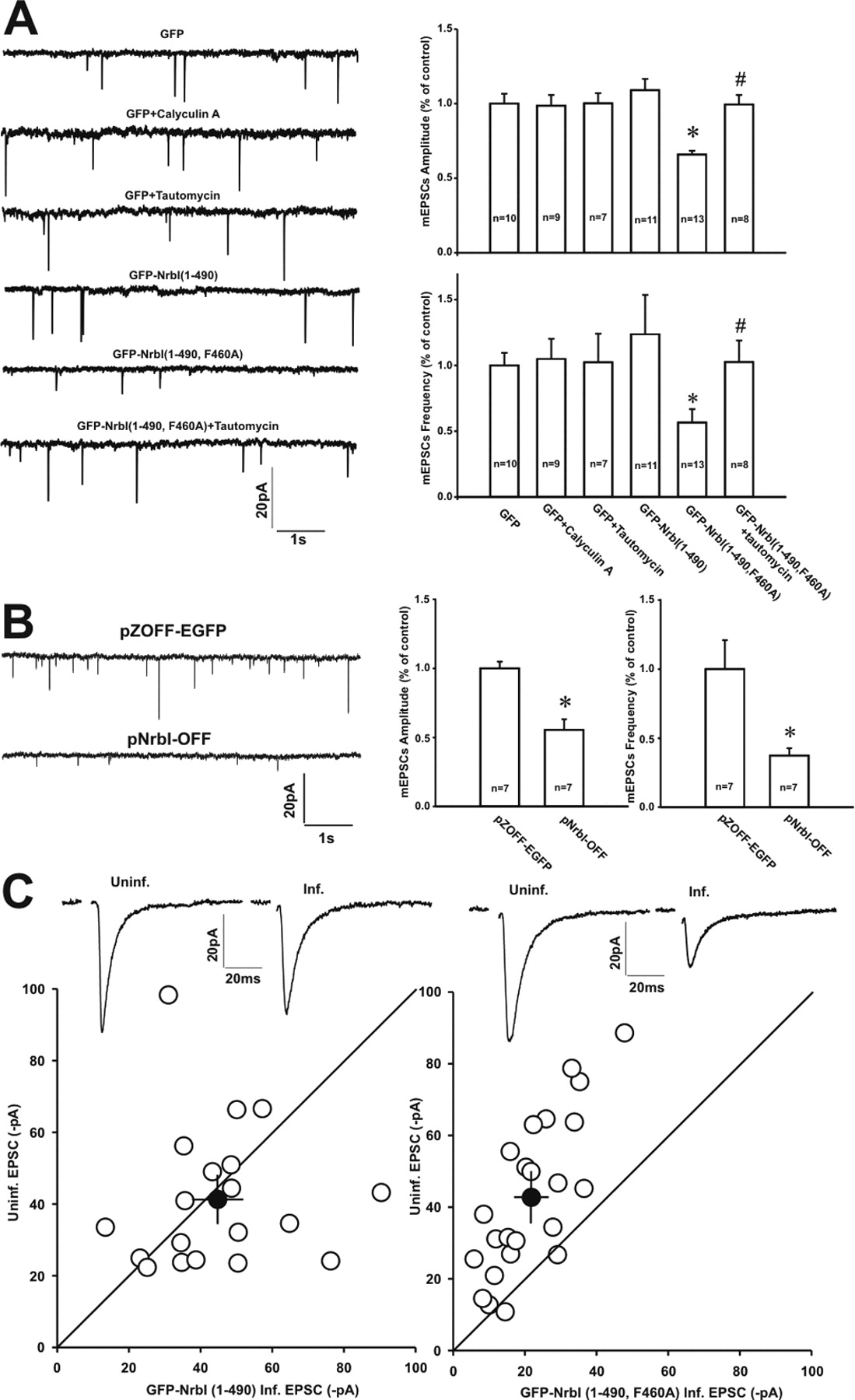
Disruption of PP1 binding to NrbI decreased the basal synaptic transmission. A, GFP–NrbI (1–490, F460A), which contained a point mutation in the core PP1 binding motif to abolish PP1 binding, decreased the frequency and amplitude of mEPSCs recorded in CA1 pyramidal neurons in organotypic hippocampal slice cultures. Left, Sample traces of mEPSCs recorded in neurons expressing GFP, GFP–NrbI (1–490), or GFP–NrbI (1–490, F460A). Note that pretreatment of GFP-expressing neurons with PP1 inhibitor tautomycin (10 μm) or calyculin A (1 μm) had no effects on mEPSCs. However, tautomycin increased the frequency and amplitude of mEPSCs in GFP–NrbI (1–490, F460A)-expressing neurons. Right, Graph showed the mean percentage changes in the mEPSCs amplitude (top) and frequency (bottom). *, #p < 0.05 when compared with neurons expressing GFP (*) or GFP–NrbI (1–490, F460A) (#). B, Primary cultured hippocampal neurons transfected with pNrbI–OFF that coexpressed shRNA against NrbI along with EGFP to knockdown endogenous NrbI displayed significant decrease in the amplitude and frequency of mEPSCs relative to those transfected with pZOFF–EGFP that expressed EGFP alone. Left, Sample traces of mEPSCs recorded in neurons expressing pZOFF–EGFP (top) and pNrbI–OFF (down). Right, Graph showed the mean percentage changes in the mEPSC amplitude and frequency. C, Disruption of NrbI-mediated PP1 targeting depressed the evoked basal synaptic transmission in CA1 pyramidal neurons in hippocampal slice cultures. Left, Pairwise comparison of synaptic response amplitudes between GFP–NrbI (1–490)-expressing neurons and neighboring uninfected neurons in the same slice. Right, Summary data of pairwise comparisons of synaptic response amplitudes between GFP–NrbI (1–490, F460A)-expressing neurons and neighboring uninfected neurons. Averaged sample traces of synaptic transmission on an uninfected cell (Uninf.) and an adjacent infected cell (Inf.) from a pairwise experiment were also shown at the top of each panel.
To test whether the synaptic depression on GFP–NrbI (1–490, F460A)-expressing neurons was activity dependent, the NMDAR inhibitor d-APV (100 μm) was included in the culture medium for 24 h before recordings. The result showed that d-APV application abolished the decrease in the synaptic transmission on neurons expressing GFP–NrbI (1–490, F460A) (Fig. 3A) [uninfected, −27.0 ± 3.9 pA; GFP–NrbI (1–490, F460A), −30.6 ± 4.0 pA, n = 13, p > 0.05]. Incubation of slices with high concentration of Mg2+ (10 mm), which blocked the spontaneous neural activity (Zhu et al., 2000), produced the similar effect as d-APV (Fig. 3B) [uninfected, −39.9 ± 7.2 pA; GFP–NrbI (1–490, F460A), −35.0 ± 5.9 pA, n = 13, p > 0.05]. These results suggested that the pool of NrbI-targeted synaptic PP1 had a tonic inhibition of activity-dependent decrease in AMPAR-mediated synaptic transmission.

The synaptic depression in neurons expressing GFP–NrbI (1–490, F460A) was prevented by the NMDA receptor antagonist d-APV and high concentration of Mg2+. Hippocampal slice cultures were incubated in the culture medium containing d-APV (100 μm) or Mg2+ (10 mm) immediately after injection of Sindbis virus encoding GFP–NrbI (1–490, F460A). A, B, Pairwise comparison of synaptic transmission on uninfected and in-slice adjacent infected cells in the presence of d-APV (A) and high Mg2+ (B). Averaged sample traces from an uninfected (Uninf.) and an infected (Inf.) neuron in the same slice were shown on the right side of each panel.
Inhibition of PKC activity prevented the synaptic depression in neurons expressing GFP–NrbI (1–490, F460A)
Because viral expression of GFP–NrbI (1–490, F460A) decreased the basal synaptic transmission, we hypothesized that this construct-induced reduction of synaptic PP1 concentration might increase the activities of certain kinases in the vicinity of the NrbI/PP1 complex, which contributed to this synaptic depression via phosphorylation events. We set out to probe this by pretreating slices with the blockers of various candidate kinases. As shown in Figure 4, the decrease of EPSC amplitudes in GFP–NrbI (1–490, F460A)-expressing neurons could not be prevented by either the calmodulin-dependent protein kinase II (CaMKII) inhibitor KN-62 (5 μm) (Fig. 4B) [uninfected, −34.8 ± 6.0 pA; GFP–NrbI (1–490, F460A), −16.6 ± 3.2 pA, n = 17, p < 0.05] or protein kinase A (PKA) inhibitor Rp-cAMP (50 μm) (Fig. 4C) [uninfected, −49.8 ± 6.5 pA; GFP–NrbI (1–490, F460A), −25.4 ± 2.8 pA, n = 19, p < 0.05]. However, in the presence of the PKC inhibitor chelerythrine (5 μm), viral expression of GFP–NrbI (1–490, F460A) failed to reduce the synaptic responses compared with those in uninfected control neurons (Fig. 4A) [uninfected, −37.4 ± 4.1 pA; GFP–NrbI (1–490, F460A), −40.7 ± 3.4 pA, n = 23, p > 0.05]. These data suggested that NrbI-targeted synaptic PP1 negatively controlled PKC-mediated depression in the basal synaptic transmission.
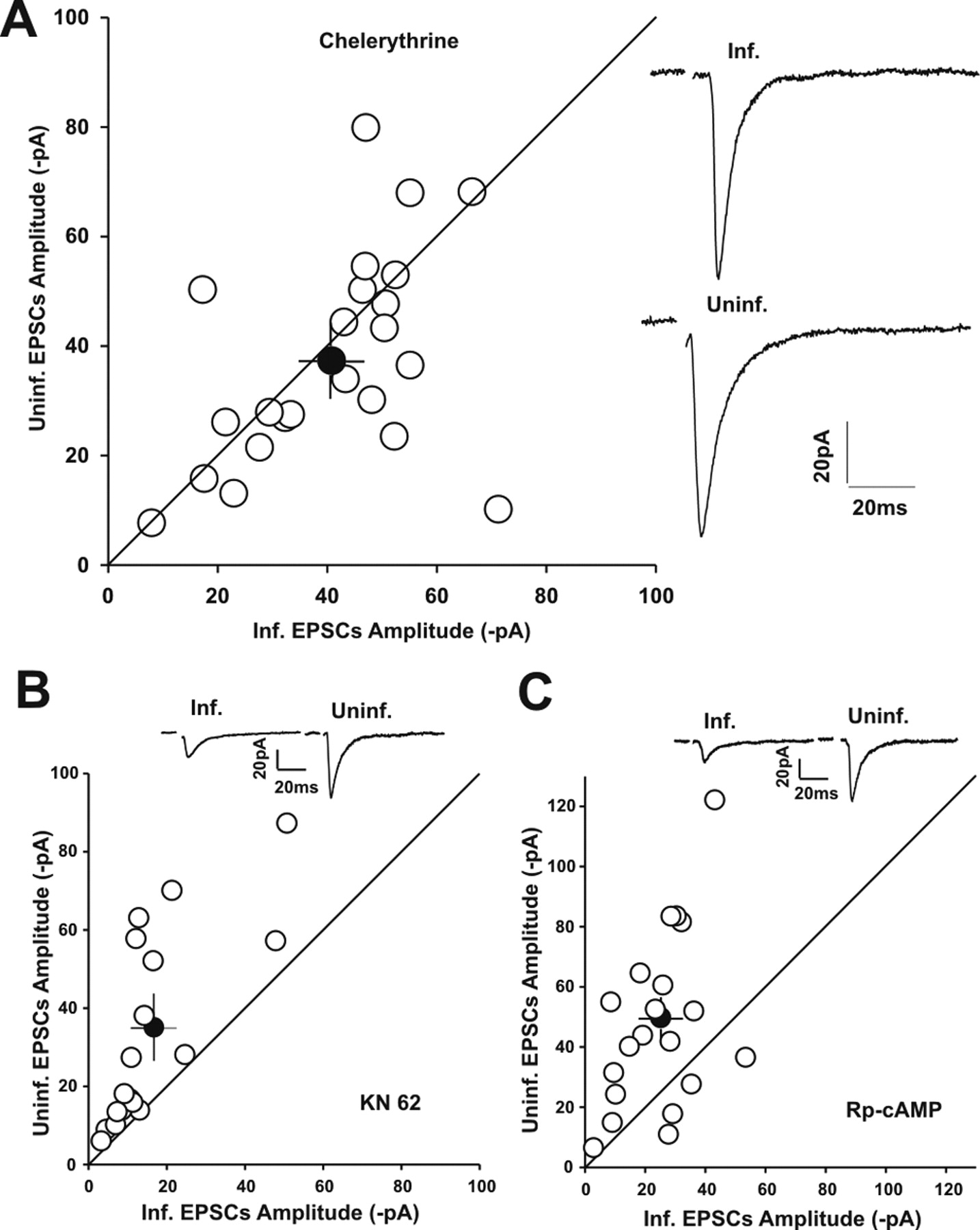
The synaptic depression in neurons expressing GFP–NrbI (1–490, F460A) that interfered with PP1 synaptic targeting was selectively prevented by the PKC inhibitor chelerythrine (5 μm) but not by the CaMKII inhibitor KN-62 (5 μm) or the PKA inhibitor Rp-cAMP (50 μm). A, Left, Summary data of pooled amplitudes of synaptic transmission on GFP–NrbI (1–490, F460A)-expressing neurons and in-slice adjacent control neurons from slices pretreated with chelerythrine for 2–4 h before recordings. Chelerythrine (5 μm) was also included in the recording pipettes. Right, Average sample traces from a GFP–NrbI (1–490, F460A)-expressing neuron (Inf.) and an adjacent uninfected control neuron (Uninf.) in the same slice. B, C, Pairwise comparison of synaptic response amplitudes between GFP–NrbI (1–490, F460A)-expressing neurons and neighboring uninfected neurons in the presence of KN-62 (B) and Rp-cAMP (C). Insets, Representative synaptic responses obtained from a pair of uninfected and infected neurons in each panel.
Expression of GFP–NrbI (1–490, F460A) increased GluR2 phosphorylation at serine 880 and decreased the surface expression of GluR2-containing AMPARs
A great number of documents have indicated that PKC can phosphorylate AMPAR GluR2 subunit at Ser880 within the cytoplasmic PDZ ligand (Matsuda et al., 1999; Xia et al., 2000; Perez et al., 2001). Ser880 phosphorylation disrupts GluR2 interaction with GRIP/ABP but not with PICK1, which induces the decrease in the surface expression of GluR2-containing AMPARs and the reduction in the synaptic transmission (Perez et al., 2001; Seidenman et al., 2003). Therefore, PKC phosphorylation of GluR2 at Ser880 represents one of the pivotal mechanisms for AMPAR internalization. Because GFP–NrbI (1–490, F460A), which dispersed PP1 away from synapses (Fig. 1D), conferred PKC-dependent synaptic depression, we speculated that NrbI-targeted synaptic PP1 regulated synaptic strength by controlling PKC phosphorylation of GluR2 and PICK1-mediated internalization of GluR2-containing AMPARs. Previous studies have indicated that protein phosphatases indeed regulate PKC phosphorylation of GluR2 because inhibition of phosphatases activities increases the Ser880 phosphorylation level in acute hippocampal slices (Kim et al., 2001). Under our conditions, treatment of cultured hippocampal slices with tautomycin (10 μm) also enhanced Ser880 phosphorylation (151.4 ± 7.9% of control; n = 5; p < 0.05). These data suggest that PP1 is functionally coupled with PKC phosphorylation of GluR2. To address whether it was NrbI-targeted synaptic PP1 that played the negative control over GluR2 Ser880 phosphorylation, we interfered with PP1 targeting by expressing GFP–NrbI (1–490, F460A) in primary cultured hippocampal neurons and tested the change in GluR2 phosphorylation at basal conditions. Immunoblotting analysis showed that Ser880 phosphorylation substantially increased when neurons were transfected with GFP–NrbI (1–490, F460A) (Fig. 5A). Pretreatment of GFP–NrbI (1–490, F460A)-expressing neurons with d-APV (100 μm) or chelerythrine (5 μm) markedly decreased Ser880 phosphorylation (Fig. 5B). As a control, we assayed the phosphorylation of the AMPA receptor GluR1 subunit at serine 831, which can also be phosphorylated by PKC (Roche et al., 1996). Interestingly, expression of GFP–NrbI (1–490, F460A) did not increase the phosphorylation level of GluR1 Ser831 when compared with GFP or GFP–NrbI (1–490) (Fig. 5C). Similarly, we did not detect any difference in the phosphorylation level of GluR1 at Ser845, a consensus PKA recognition site, between GFP-expressing, GFP–NrbI (1–490)-expressing, and GFP–NrbI (1–490, F460A)-expressing neurons (Fig. 5D). These results strongly implicated the specificity of precisely targeted PP1 by NrbI in the negative control over PKC phosphorylation of GluR2 under basal conditions. We also examined the surface expression of AMPARs by performing immunocytochemistry in cultured hippocampal neurons. Using subunit-specific antibodies against the extracellular episodes of AMPA receptor subunits under impermeable conditions, we showed that GFP–NrbI (1–490, F460A) indeed decreased the surface level of GluR2 when compared with GFP or GFP–NrbI (1–490) (Fig. 5E). Meanwhile, GluR1 surface expression was also reduced (Fig. 5E). Consistent with the electrophysiological data, both d-APV (100 μm) and chelerythrine (5 μm) eliminated the reduction in the surface expression of GluR2 and GluR1 caused by GFP–NrbI (1–490, F460A) (Fig. 5E). These results suggested that NrbI-targeted synaptic PP1 negatively controlled NMDAR-dependent phosphorylation of GluR2 by PKC, thus preventing the decrease in the surface expression of GluR2-containing AMPARs.

GFP–NrbI (1–490, F460A), which interfered with PP1 synaptic targeting, specifically increased the phosphorylation of AMPA receptor GluR2 subunits at Ser880 and decreased the surface expression of GluR2-containing AMPARs in primary cultured hippocampal neurons, which could be prevented by the NMDA receptor antagonist d-APV and the protein kinase C inhibitor chelerythrine. A, Expression of GFP–NrbI (1–490, F460A) significantly increased the phosphorylation of GluR2 at Ser880. Left, Western blots (WB) of GluR2 Ser880 phosphorylation using anti-Ser880 phospho-specific antibody. The equal protein loading was verified by immunoblotting with the antibody against GluR2 C terminus. Right, Graph showed the percentage changes in the phosphorylation level of GluR2 Ser880 in neurons expressing GFP, GFP–NrbI (1–490), and GFP–NrbI (1–490, F460A). The relative amount of phosphorylated GluR2 at Ser880, expressed by the ratio of phosphorylation signals to total GluR2 signals, was normalized to the control (GFP) value. *p < 0.05 when compared with GFP-expressing neurons. B, Pretreatment of GFP–NrbI (1–490, F460A)-expressing neurons with d-APV (100 μm) or chelerythrine (5 μm) decreased the phosphorylation level of GluR2 Ser880. *p < 0.05 when compared with GFP–NrbI (1–490, F460A)-expressing neurons. C, D, Expression of GFP–NrbI (1–490, F460A) did not increase the phosphorylation of AMPA receptor GluR1 subunits at either Ser831 (C) or Ser845 (D). E, Expression of GFP–NrbI (1–490, F460A) decreased the surface expression of GluR2-containing AMPARs. Left, Surface GluR2 and GluR1 were labeled with antibodies against the extracellular N terminus of GluR2 and GluR1 under impermeable conditions before fixation and immunostaining with second antibodies in cultured hippocampal neurons transfected with GFP, GFP–NrbI (1–490), or GFP–NrbI (1–490, F460A). GluR2 and GluR1 staining in the presence of d-APV, chelerythrine, and tautomycin (10 μm) was also shown in GFP–NrbI (1–490, F460A)-expressing neurons. Scale bar, 5 μm. Right, Quantitative analysis of GluR2 and GluR1 surface immunostaining intensity. *p < 0.05 when compared with GFP-expressing neurons; n > 14 for each condition.
Disruption of GluR2 interaction with PICK1 rescued the synaptic depression in GFP–NrbI (1–490, F460A)-expressing neurons
Phosphorylated GluR2 by PKC preferentially interacts with PICK1, leading to GluR2-containing AMPAR internalization or retaining AMPARs at subsynaptic compartments. To directly examine whether PICK1 interaction with the phosphorylated GluR2 caused by GFP–NrbI (1–490, F460A) was responsible for the synaptic depression, we loaded neurons, via recording pipettes, with a synthetic peptide (KKEGYNVYGIESVKI; p-SVKI) that corresponded to the last 15 amino acids of the GluR2 C terminus. This peptide has been shown to disturb GluR2 interaction with PDZ proteins, including PICK1 and GRIP/ABP (Daw et al., 2000; Kim et al., 2001; Terashima et al., 2004). Because GFP–NrbI (1–490, F460A) substantially increased GluR2 phosphorylation at Ser880, we assumed that this peptide predominantly disrupted GluR2–PICK1 interaction in GFP–NrbI (1–490, F460A)-expressing neurons. We monitored EPSC amplitudes between the infected and in-slice neighboring uninfected neurons by performing pairwise comparison. In GFP–NrbI (1–490, F460A)-expressing neurons, intracellular loading of p-SVKI for 30 min dramatically increased AMPAR-mediated synaptic responses to 214.9 ± 14.4% of the initial 2 min responses (Fig. 6A1) (n = 7; p < 0.05). In closely adjacent uninfected neurons, however, synaptic responses only slightly increased to 128.0 ± 7.9% (Fig. 6A1) (n = 7; p > 0.05). As a result, the absolute EPSCs amplitudes at the end of 30 min recordings had no significant difference between GFP–NrbI (1–490, F460A)-expressing neurons and in-slice control neurons (Fig. 6A2) (−61.6 ± 9.1 vs −61.9 ± 4.8 pA; n = 7; p > 0.05), although the initial responses in infected neurons were significantly smaller than those in uninfected neurons (Fig. 6A2) (−25.1 ± 5.3 vs −45.6 ± 5.2 pA; n = 7; p < 0.05). These results indicated that disruption of GluR2–PDZ interaction completely rescued the synaptic depression induced by GFP–NrbI (1–490, F460A). We next probed whether selective disruption of GluR2–PICK1 interaction produced the same effect as p-SVKI. To test this, neurons were loaded with another peptide KKEGYNVYGIEEVKI (p-EVKI), which was identical to the last 15 amino acids of the GluR2 C terminus, except that serine 880 was substituted with glutamic acid to mimic the phosphorylation (Daw et al., 2000). Perfusion of p-EVKI to specifically disturb GluR2–PICK1 interaction substantially increased the synaptic responses at 30 min to 210.6 ± 33.1% of the initial 2 min responses in GFP–NrbI (1–490, F460A)-expressing neurons (Fig. 6B1) (n = 19; p < 0.05), whereas the synaptic responses in adjacent uninfected neurons only increased to 114.1 ± 7.4% (Fig. 6B1) (n = 19; p > 0.05). Like p-SVKI, p-EVKI totally reversed the synaptic depression in GFP–NrbI (1–490, F460A)-expressing neurons at the end of recordings (Fig. 6B2) (infected vs uninfected, −53.2 ± 6.2 vs −57.6 ± 8.3 pA; n = 19; p > 0.05). Thus, our data suggested that NrbI-targeted synaptic PP1 stabilized the basal synaptic transmission by inhibiting GluR2 Ser880 phosphorylation and GluR2–PICK1 interaction.

Intracellular infusion of synthetic peptides KKEGYNVYGIESVKI (p-SVKI) and KKEGYNVYGIEEVKI (p-EVKI) rescued the synaptic depression in neurons expressing GFP–NrbI (1–490, F460A). A, p-SVKI corresponded to the extreme cytoplasmic C terminus of GluR2, which disrupted GluR2 interaction with PDZ proteins, including PICK1. The extent to which p-SVKI (200 μm) increased the synaptic transmission in cells expressing GFP–NrbI (1–490, F460A) (filled circles) was more than that in adjacent uninfected (Uninf.) control cells (open circles). The normalized amplitudes of synaptic responses were plotted versus time (A1). Insets, Sample traces recorded in an uninfected (left) neuron and a neighboring infected (right) neuron were obtained at the time points indicated by 1 and 2. Histogram showed the mean amplitudes of synaptic transmission on neurons expressing GFP–NrbI (1–490, F460A) and adjacent control neurons at 1 min and 30 min after p-SVKI infusion (A2). B, p-EVKI (200 μm), which corresponded to the extreme C terminus of GluR2 except for a point mutation that mimicked the phosphorylation of GluR2 at serine 880 to selectively disrupt GluR2–PICK1 interaction, also increased the synaptic transmission to a greater extent in cells expressing GFP–NrbI (1–490, F460A) (filled circles) than in adjacent uninfected (Uninf.) control cells (open circles). The normalized amplitudes of synaptic responses were plotted versus time (B1). Insets, Sample traces recorded in an uninfected (left) neuron and a neighboring infected (right) neuron were obtained at the time points indicated by 1 and 2. Histogram showed the mean amplitudes of synaptic transmission on neurons expressing GFP–NrbI (1–490, F460A) and adjacent control neurons at 1 min and 30 min after p-EVKI infusion (B2). Asterisks indicate the groups between which the mean amplitudes were significantly different (p < 0.05).
LTD induction in GFP–NrbI (1–490)-expressing neurons was independent of GluR2–PICK1 interaction
PP1 has been established to play a central role in NMDAR-dependent LTD in hippocampus. Given that there are several pools of synaptic PP1 that are associated with distinct targeting proteins and are involved in synaptic transmission, we decided to examine the role of NrbI-targeted PP1 in LTD. In the present study, we could elicit normal LTD in GFP-expressing neurons using low-frequency stimuli (300 pulses at 1 Hz) (Fig. 7A,F) (50.0 ± 6.7%; n = 9). Robust LTD was also elicited in neurons expressing GFP–NrbI (1–490) (Fig. 7B,F) (54.5 ± 7.4%; n = 15), similar to that in neurons expressing full-length wild-type NrbI (Fig. 7C,F) (57.2 ± 6.2%; n = 7). Even if suboptimal stimuli (150 pulses instead of 300 pulses at 1 Hz) were applied, LTD was still observed in GFP–NrbI (1–490)-expressing neurons but not in GFP-expressing neurons (Fig. 7D,F) (GFP, 97.9 ± 17.5%, n = 10; GFP–NrbI (1–490), 57.8 ± 2.3%, n = 8), suggesting that NrbI-targeted synaptic PP1 facilitated LTD induction. Once PP1 targeting was disturbed by expressing GFP–NrbI (1–490, F460A), LTD was completely abolished after 300 pulse stimuli (Fig. 7E,F) (109.5 ± 19.0%; n = 8), implicating the critical role of NrbI-targeted synaptic PP1 in LTD. One would predict that PICK1-mediated reduction of the basal synaptic transmission before low-frequency stimuli occluded the subsequent LTD in GFP–NrbI (1–490, F460A)-expressing neurons. To test this, we introduced p-EVKI into neurons through recording pipettes to specifically disrupt GluR2–PICK1 interaction. LTD-inducing stimuli were delivered 30 min after break in, a time point at which the peptide had fully diffused into the dendrites and the increase in the basal transmission had reached plateau (Fig. 6B). The results showed that LTD was still completely blocked in GFP–NrbI (1–490, F460A)-expressing neurons even if p-EVKI had rescued the synaptic depression (Fig. 8E) (113.1 ± 12.9%; n = 5), indicating that the defective PP1 signaling contributed to LTD blockade in GFP–NrbI (1–490, F460A)-expressing neurons.

NrbI-targeted synaptic PP1 facilitated LTD induction in hippocampal slice cultures. A, Pairing presynaptic stimulation at 1 Hz for 5 min with postsynaptic neurons held at −45 mV stably induced LTD in GFP-expressing neurons. Normalized mean synaptic responses before and after LTD stimuli were plotted versus time. The horizontal bar indicated the period of pairing stimuli. Insets, Sample traces were taken at the time points indicated by the 1 and 2. B, C, The pairing stimuli protocol as described above induced LTD in neurons expressing GFP–NrbI (1–490) that lacked the C terminus (B) or full-length GFP–NrbI (1–1095) (C). D, Suboptimal stimuli protocol (1 Hz for 2.5 min) induced robust LTD in GFP–NrbI (1–490)-expressing neurons (open circles), which, however, failed to do so in GFP-expressing neurons (filled circles). E, Interference with PP1 synaptic targeting by expression of GFP–NrbI (1–490, F460A) completely blocked the LTD induction after 5 min pairing stimuli. F, The graph showed the percentage changes in EPSC amplitudes within the last 10 min of recordings in each transfection in response to pairing stimuli for 5 min (open bars) or 2.5 min (suboptimal LTD-inducing stimuli; filled bars).

Intracellular perfusion of GluR2 C-terminal peptide (KKEGYNVYGIEEVKI; p-EVKI) that selectively disrupted GluR2–PICK1 interaction attenuated LTD in GFP-expressing neurons but not in GFP–NrbI (1–490)-expressing neurons. The pairing LTD-inducing stimuli (1 Hz for 5 min with postsynaptic neurons held at −45 mV) were delivered to the test pathway at 30 min after break in, a time point at which the peptide in the recording electrodes had fully diffused into the dendrites and the increase in the synaptic transmission had reached a plateau. A, Intracellular infusion of p-EVKI partially blocked LTD in GFP-expressing neurons in the test pathway (filled circles). In the control pathway to which no pairing stimuli were delivered, the EPSCs amplitudes remained stable in the presence of the peptide during the course of recordings (open circles). For comparison purposes, the percentage changes of EPSC amplitudes after LTD-inducing stimuli in GFP-expressing neurons not loaded with the peptide were also shown (dashed × symbols). B, Perfusion of p-EVKI in GFP–NrbI (1–490)-expressing neurons produced minimal effects on LTD magnitudes in the test pathway (filled circles). The EPSC amplitudes in the control pathway (open circles) changed little in the presence of p-EVKI during the course of recordings. For comparison purposes, the percentage changes of EPSC amplitudes after LTD-inducing stimuli in GFP–NrbI (1–490)-expressing neurons not loaded with the peptide were also shown (dashed × symbols). C, Suboptimal LTD-inducing stimuli (2.5 min instead of 5 min) also elicited robust LTD in GFP–NrbI (1–490)-expressing neurons loaded with p-EVKI (filled circles). For comparison purposes, the percentage changes of EPSC amplitudes in response to suboptimal stimuli were also shown in GFP–NrbI (1–490)-expressing neurons not loaded with the peptide (dashed × symbols). D, The protein kinase C inhibitor chelerythrine (5 μm) did not block LTD in GFP–NrbI (1–490)-expressing neurons. For comparison purposes, the percentage changes of EPSC amplitudes after LTD-inducing stimuli in GFP–NrbI (1–490)-expressing neurons not treated with chelerythrine were also shown (dashed × symbols). E, Expression of GFP–NrbI (1–490, F460A) blocked LTD even if p-EVKI had rescued the depressed synaptic transmission in the test pathway (filled circles). The control pathway (open circles) displayed stable EPSC amplitudes in the presence of p-EVKI during the course of recordings. For comparison purposes, the percentage changes of EPSC amplitudes after LTD stimuli were also shown in GFP–NrbI (1–490, F460A)-expressing neurons not loaded with the peptide (dashed × symbols).
Numerous documents have established that PICK1 interaction with phosphorylated GluR2 at Ser880 is critical for cerebellar LTD. In hippocampus, however, the role of the GluR2–PICK1 interaction in NMDAR-dependent LTD is far less clear because disruption of GluR2–PICK1 interaction either reduces LTD magnitudes or has no effect on LTD (Daw et al., 2000; Kim et al., 2001). The present study found that GluR2 Ser880 phosphorylation, a critical event for PICK1 to internalize AMPARs, was negatively controlled by NrbI-targeted synaptic PP1. We proposed that the contribution of the GluR2–PICK1 interaction to hippocampal LTD was governed by synaptic PP1. Indeed, intracellular perfusion of p-EVKI for 30 min before LTD-inducing stimuli partially blocked LTD in GFP-expressing neurons (Fig. 8A) (71.5 ± 4.9%; n = 9; p < 0.05 compared with that in GFP-expressing neurons not loaded with p-EVKI) in the test pathway, whereas the control pathway, to which no LTD-inducing stimuli were delivered, displayed stable EPSC amplitudes. However, disruption of the GluR2–PICK1 interaction with p-EVKI in GFP–NrbI (1–490)-expressing neurons produced minimal effects on the magnitudes of LTD induced by either 300 pulse stimuli or suboptimal (150 pulses) stimuli [Fig. 8B, 300 pulse stimuli, 54.1 ± 4.3%, n = 9, p > 0.05 when compared with that in GFP–NrbI (1–490)-expressing neurons not loaded with p-EVKI; Fig. 8C, suboptimal stimuli, 53.4 ± 2.6%, n = 5, p > 0.05]. Inhibition of PKC with chelerythrine (5 μm) also had no effect on LTD in GFP–NrbI (1–490)-expressing neurons (Fig. 8D) (56.5 ± 9.5%; n = 9; p > 0.05 when compared with that in GFP–NrbI (1–490)-expressing neurons not treated with chelerythrine). These results suggested that NrbI-targeted synaptic PP1 evoked LTD independent of GluR2–PICK1 interaction.
NrbI-targeted synaptic PP1 induced LTD by dephosphorylating GluR1 at serine 845 and serine 831
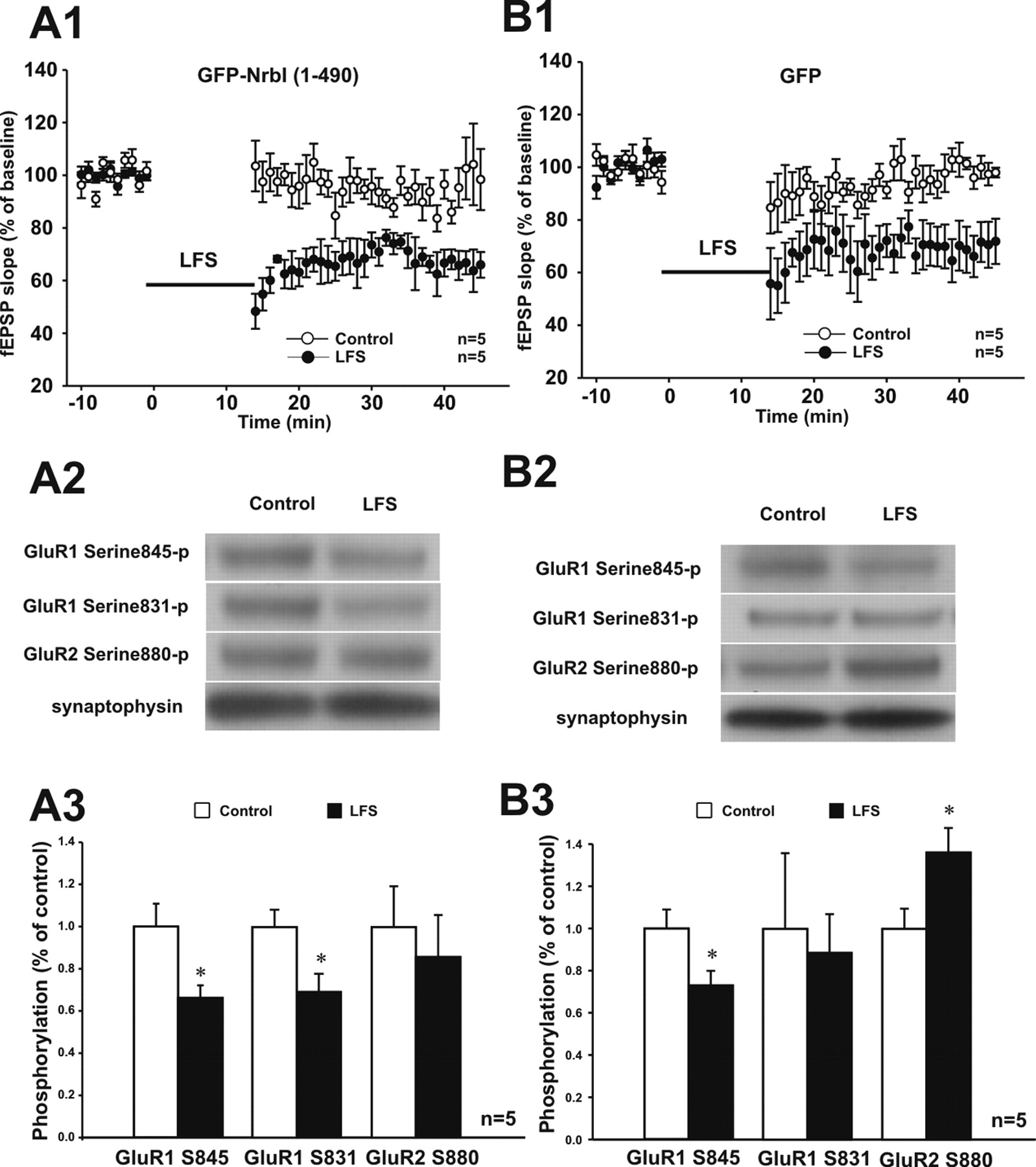
It has been implicated that the dephosphorylation of GluR1 subunits is involved in NMDAR-dependent hippocampal LTD (Kameyama et al., 1998; Lee et al., 1998, 2000). We want to address whether NrbI-targeted PP1, in response to the prolonged, LTD-inducing stimuli, could dephosphorylate GluR1, leading to persistent decrease in synaptic strength. To test this, we put in the same recording chamber two hippocampal slice cultures: one receiving low-frequency stimuli to experience LTD (LTD slice) and the other not, serving as a control (control slice). At the end of the extracellular field potential recordings in the CA1 dendritic region, the two slices were immediately frozen on dry ice for subsequent biochemical analysis. Standard LTD-inducing stimuli (900 pulses at 1 Hz) elicited LTD in both GFP-infected and GFP–NrbI (1–490)-infected slices (Fig. 9A1,B1). Concurrent with LTD in GFP-infected slices was the dephosphorylation of GluR1 at Ser845, whereas there was no significant change in the phosphorylation level of GluR1 at Ser831 (Fig. 9B2,B3). Notably, GluR2 phosphorylation at Ser880 markedly increased in GFP-infected slices after LTD induction (Fig. 9B2,B3), which was consistent with previous studies (Kim et al., 2001). Distinct from GFP-infected slices, GFP–NrbI (1–490)-infected slices exhibited significant GluR1 dephosphorylation not only at Ser845 but also at Ser831 after LTD induction (Fig. 9A2,A3). GluR2 phosphorylation at Ser880, however, showed no significant change (Fig. 9A2,A3), suggesting that NrbI-targeted PP1 prevented GluR2 from being phosphorylated during LTD. These results supported the electrophysiological data that interference with GluR2–PICK1 interaction generated little effect on LTD in GFP–NrbI (1–490)-expressing neurons. Next, we confirmed the effects of NrbI-targeted PP1 by using suboptimal LTD-inducing stimuli (450 pulses at 1 Hz), which successfully evoked LTD in slices infected with GFP–NrbI (1–490) (Fig. 10A1) but failed to do so in slices with GFP alone (Fig. 10B1). The LTD present only in GFP–NrbI (1–490)-infected slices was, therefore, attributable to the effects of additional PP1 targeted by GFP–NrbI (1–490). As shown in Figure 10A2, GFP–NrbI (1–490)-infected slices exhibited much less phosphorylation of GluR1 at Ser845 and Ser831 after LTD induction. GluR2 phosphorylation at Ser880 had no detectable change (Fig. 10A2,A3). These data suggested that the enhanced PP1 targeting by GFP–NrbI (1–490) strongly dephosphorylated GluR1 to trigger lasting synaptic depression after LTD-inducing stimuli. As expected, the suboptimal stimuli, which did not induce LTD in GFP-infected slices (Fig. 10B1), produced minimal changes in phosphorylation states of GluR1 Ser845, GluR1 Ser831, or GluR2 Ser880 (Fig. 10B2,B3). Combined with the observation under basal conditions, our results suggested that NrbI-targeted synaptic PP1 exerted dual roles in regulating AMPAR subunit trafficking in response to distinct synaptic activities.

Standard LTD-inducing stimuli in GFP-NrbI (1–490)-infected hippocampal slices reduced the phosphorylation of AMPA receptor GluR1 subunits at serine 845 and serine 831. Two hippocampal slices were put in the same recording chamber: one receiving low-frequency stimuli (LFS) (1 Hz for 15 min; LFS slice) and the other not (control slice). After recordings of the extracellular field EPSP (fEPSP), the phosphorylation of AMPA receptor subunits was detected by immunoblotting with phosphorylation site-specific antibodies against GluR1 serine 845, GluR1 serine 831, and GluR2 serine 880. A, GFP–NrbI (1–490)-infected hippocampal slices displayed robust LTD after low-frequency stimuli (A1; filled circles). GFP–NrbI (1–490)-infected control slice exhibited stable field EPSP slopes during the whole course of recordings (A1; open circles). The phosphorylation levels of GluR1 at serine 845 and serine 831 significantly decreased in low-frequency stimuli slices compared with those in control slices (A2, A3; *p < 0.05). GluR2 phosphorylation at serine 880 had no difference between low-frequency stimuli slices and control slices (A2, A3). The equal protein loadings were verified by immunostaining of synaptophysin. B, Low-frequency stimuli induced LTD in GFP-infected slices (B1; filled circles) compared with GFP-infected control slices (open circles). LTD in GFP-infected slices was coincident with the dephosphorylation of GluR1 at serine 845 but not at serine 831 (B2, B3). The phosphorylation level of GluR2 at serine 880 significantly increased after low-frequency stimulation (B2, B3).

Suboptimal LTD-inducing stimuli in GFP–NrbI (1–490)-infected hippocampal slices reduced the phosphorylation of AMPA receptor GluR1 subunits at serine 845 and serine 831. Two hippocampal slices were put in the same recording chamber: one receiving suboptimal LTD-inducing stimuli [1 Hz for 7.5 min; low-frequency stimuli (LFS) slice] and the other not (control slice). After recordings of the extracellular field EPSP (fEPSP), the phosphorylation of AMPA receptor subunits was detected by immunoblotting with phosphorylation site-specific antibodies against GluR1 serine 845, GluR1 serine 831, and GluR2 serine 880. A, GFP–NrbI (1–490)-infected hippocampal slices displayed robust LTD after suboptimal low-frequency stimulation (A1; filled circles), whereas control slices exhibited stable field EPSP slopes during the whole course of recordings (A1; open circles). The phosphorylation levels of GluR1 at serine 845 and serine 831 significantly decreased in low-frequency stimuli slices compared with those in control slices (A2, A3; *p < 0.05). GluR2 phosphorylation at serine 880 had no difference between LFS slices and control slices (A2, A3). The equal protein loadings were indicated by immunostaining of synaptophysin. B, GFP-infected slices failed to express LTD after suboptimal low-frequency stimulation (B1; filled circles). The control slices exhibited stable field EPSP slopes during the whole course of recordings (B1; open circles). B2, B3, The phosphorylation of GluR1 at serine 845 and serine 831 as well as the phosphorylation of GluR2 at serine 880 had no significant difference between low-frequency stimuli slices and control slices.
Discussion
The present study demonstrates the molecular mechanisms underlying how NrbI targets PP1 at synapses to stabilize the basal synaptic transmission and promote the LTD induction in hippocampal slices. Our data showed that NrbI targeted PP1 at synapses via its N terminus. The C terminus of NrbI was not required for PP1 to localize at dendritic spines, regulate synaptic transmission, and LTD induction, although it has been reported to interact with a number of postsynaptic proteins (Penzes et al., 2001; Ryan et al., 2005). NrbI-targeted synaptic PP1 served to prevent PKC phosphorylation of GluR2 at Ser880 and GluR2–PICK1 interaction under basal conditions, thereby stabilizing the surface expression of GluR2-containing AMPARs. During LTD process, NrbI-targeted synaptic PP1 also prevented GluR2 phosphorylation at Ser880 and minimized the contribution of the GluR2–PICK1 interaction to LTD. We showed that NrbI-targeted PP1 induced LTD by strongly dephosphorylating GluR1 subunits. Our data therefore outline two distinct pathways for NrbI-targeted PP1 to regulate AMPAR trafficking.
Role of AMPA receptor GluR2 subunits in the basal synaptic transmission
Endogenous AMPARs are tetrameric hetero-oligomers assembled from variable combinations of four subunits, GluR1–GluR4. A large number of evidence has shown that individual AMPA receptor subunit plays distinct roles in the basal synaptic transmission and plasticity because of specific trafficking features (Bredt and Nicoll, 2003). GluR2/GluR3 subunits are crucial for maintaining the basal synaptic transmission. Constitutive GluR2/GluR3 trafficking is proposed to replace the preexisting synaptic AMPARs (Shi et al., 2001); alternatively, GluR2 subunits exchange one by one with GluR1, GluR2L, and GluR4 to rescue the capability of synaptic plasticity, leaving the synaptic strength unchanged (McCormack et al., 2006). Consistent with this model, the basal synaptic transmission is impaired in GluR2/GluR3 double knock-out mice (Meng et al., 2003), although it is normal in GluR1 knock-out mice (Zamanillo et al., 1999). Our results were in agreement with this view and further demonstrated that GluR2 subunit trafficking governed by GluR2–PDZ interaction was subject to the regulation by the NrbI/PP1 complex. Expression of the NrbI mutant, which was unable to target PP1, substantially increased GluR2 Ser880 phosphorylation and decreased the basal synaptic transmission, a result similar to direct expression of GluR2 mutant mimicking Ser880 phosphorylation (Seidenman et al., 2003). Selective disruption of GluR2–PICK1 interaction increased the basal synaptic transmission to a much greater extent in neurons expressing the NrbI mutant than in control neurons. These data unraveled, for the first time, the tonic inhibition by NrbI-targeted PP1 of GluR2 phosphorylation and PICK1-mediated decrease in the surface expression of GluR2-containing AMPARs.
Role of AMPA receptor GluR2 subunits in hippocampal LTD
The role of GluR2 subunits in LTD has also been extensively studied in cerebellum and hippocampus. PKC phosphorylation of GluR2 at Ser880 is critical for LTD expression in cerebellar parallel fiber/Purkinje cell synapses (Xia et al., 2000; Chung et al., 2003) because LTD is impaired by either inhibition of PKC activity or by genetic knock-out of GluR2 in mice (Chung et al., 2003; Gao et al., 2003; Steinberg et al., 2006). The dominant role of GluR2 in cerebellar LTD is primarily attributable to the fact that the predominant form of AMPARs is GluR2/GluR3 heteromers (Zhao et al., 1997). In adult hippocampus, however, the subunit compositions are more complicated, including both GluR1/GluR2 and GluR2/GluR3 heteromers. Studies in GluR2 knock-out mice indicate that normal LTD can be induced (Jia et al., 1996; Meng et al., 2003), suggesting that GluR2 is not required for hippocampal LTD. However, disruption of GluR2–PICK1 interaction gives seemingly conflicting results, which either partially blocks LTD or produces minimal effect on LTD (Daw et al., 2000; Kim et al., 2001). These “contradictory” results make the role of GluR2–PICK1 interaction in hippocampal LTD still controversial. Our data provide a new insight into GluR2–PICK1 interaction in hippocampal LTD. We showed that NrbI-targeted synaptic PP1 determined the contribution of GluR2–PICK1 interaction to LTD by negatively controlling GluR2 Ser880 phosphorylation. Expression of GFP–NrbI (1–490) to enhance the synaptic targeting of PP1 prevented PICK1-mediated removal of AMPARs from synapses. We propose that endogenous PP1, if substantially activated and recruited into synapses by intensive stimulation, could abolish the PICK1 mechanism in LTD as expression of GFP–NrbI (1–490) did. Our data showing that NrbI-targeted PP1 facilitated LTD by strongly dephosphorylating GluR1 favor the view that GluR2 is not absolutely necessary for hippocampal LTD.
Role of AMPA receptor GluR1 subunits in LTD
GluR1 subunits appear to play a dominant role in synaptic plasticity in hippocampus. For example, LTD is abolished in phosphorylation-deficient GluR1 knock-in mice, and long-term potentiation (LTP) is blocked in GluR1 mutant mice (Zamanillo et al., 1999; Mack et al., 2001; Lee et al., 2003); in contrast, GluR2/GluR3 double knock-out adult mice, which mainly express GluR1, display robust LTD and LTP (Meng et al., 2003). It has been established that GluR1 trafficking is subjected to PP1 regulation. PP1 induces GluR1 internalization via dephosphorylation events (Ehlers, 2000). Inhibition of PP1 activity simultaneously blocks GluR1 dephosphorylation and LTD induction, suggesting that GluR1 subunit is an important target for PP1 to evoke LTD (Lee et al., 2000). Our results extend the understanding of GluR1 dephosphorylation and trafficking regulated by PP1. Under basal conditions, NrbI-targeted synaptic PP1 specifically controlled the phosphorylation of GluR2 rather than GluR1. After LTD-inducing stimuli, however, NrbI could direct PP1 to the immediate vicinity of synaptic GluR1, which was strongly dephosphorylated not only at Ser845 but also at Ser831. Dephosphorylation of GluR1 at Ser845 and Ser831 is correlated with GluR1 internalization (Kameyama et al., 1998; Lee et al., 1998, 2003; Ehlers, 2000) as well as the reduction in channel conductance (Derkach et al., 1999). In contrast, interference with NrbI-mediated PP1 targeting at synapses completely blocked the LTD induction. These results implicated that NrbI-targeted synaptic PP1 played a critical role in hippocampal LTD by regulating GluR1 phosphorylation and trafficking.
PP1-targeting proteins determine the specificities of PP1 functions
Our previous studies have indicated that loading cells with peptides (for example, GM peptide) to abolish PP1 interaction with all of its binding proteins has no effect on the basal synaptic transmission (Morishita et al., 2001), a result different from the present work showing that specific interference with NrbI/PP1 interaction reduces the basal transmission. We predict that the individual pool of synaptic PP1, targeted by different PP1 binding proteins, could play distinct or even opposite roles in regulating AMPAR trafficking, resulting in the GM peptide being “ineffective” in regulating the basal transmission. Consistent with this hypothesis, the rundown phenomena of AMPA receptor currents is primarily prevented in striatal neurons with genetic knock-out of spinophilin/neurabin II, another PP1 targeting protein, or by specific disruption of spinophilin/PP1 interaction in neostriatal neurons (Yan et al., 1999; Allen et al., 2006). In contrast, the rundown of AMPA receptor currents is intact in striatal neurons from NrbI knock-out mice (Allen et al., 2006). These results suggest that the individual targeting protein can precisely position its associated PP1 in specific subcellular compartments, determining the substrate specificity. Hence, PP1 can exert diverse modifications of many intracellular signaling cascades involved in the synaptic transmission. The roles of the different pools of synaptic PP1 in regulating synaptic strength need to be further examined in the future.
In the case of NrbI, our data suggested that it could target a subpool of PP1 close to synaptic GluR2 and its binding partner, the PICK1–PKCα complex, under basal conditions. The micro-targeted PP1 negatively controlled GluR2 phosphorylation by PICK1-bound PKCα, preventing GluR2 internalization and stabilizing the basal synaptic transmission. Therefore, the increased activity of PICK1-bound PKCα, unmasked by expression of GFP–NrbI (1–490, F460A), specifically led to the synaptic depression. By comparison, application of phorbol ester, which presumably activates all pools of PKC in neurons, has been reported to enhance AMPAR-mediated basal transmission in hippocampal slices (Carroll et al., 1998). Possibly, nonspecific activation of PKC by phorbol ester can simultaneously drive the synaptic insertion of GluR1 (Boehm et al., 2006) and the synaptic removal of GluR2 in hippocampus, leaving net synaptic responses enhanced. In cerebellar synapses that lacks GluR1 subunit, PKC activation indeed reduces the synaptic transmission (Leitges et al., 2004). These observations strongly corroborate our conclusion in hippocampus that micro-targeted PP1 by NrbI countered GluR2 phosphorylation by a specific subpool of PKC. However, it is still unclear whether NrbI-targeted PP1 could directly dephosphorylate GluR2 at Ser880. Previous documents favor the model that PP1 dephosphorylates PKCα at serine 657, reducing the kinase activity (Bornancin and Parker, 1997; Thiels et al., 2000). Experiments designed to determine the phosphorylation of PKCα in neurons expressing NrbI and its mutant constructs are underway.
Together, our studies delineate the differential signalings triggered by the NrbI/PP1 complex in stabilizing the basal synaptic transmission and facilitating the lasting synaptic depression in response to LTD stimuli. This work provides new evidence for the notion that precise anchorage of PP1 at specific subcellular compartments is crucial for the diversity and specificity of the PP1 functions.
Footnotes
-
This work was supported by National Alliance for Research on Schizophrenia and Depression, American Heart Association, and National Institutes of Health Grant P29 RR16816 (H.X.). We thank Dr. Nicolas Bazan and the Pennington Biomedical Research Institute for providing us space in conducting part of this research during the aftermath of Hurricane Katrina.
- Correspondence should be addressed to Dr. Houhui Xia, Neuroscience Center of Excellence, Louisiana State University Health Sciences Center, New Orleans, LA 70112. hxia{at}lsuhsc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}