

Abstract
Intracellular tau deposits are characteristic of several neurodegenerative disorders called tauopathies. The tau protein regulates the stability and assembly of microtubules by binding to microtubules through three or four microtubule-binding repeats (3R and 4R). The number of microtubule-binding repeats is determined by the inclusion or exclusion of the second microtubule-binding repeat encoded by exon 10 of the TAU gene. TAU gene mutations that alter the inclusion of exon 10, and hence the 4R:3R ratio, are causal in the tauopathy frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). A mutation located in exon 10 has been identified in several FTDP-17 families that present with increased exon 10 inclusion in both mRNA and protein, parkinsonism, movement disorders, and dementia. We have engineered a human tau minigene construct that was designed to allow alternative splicing of the tau exon 10. Here we demonstrate that transgenic mice expressing human tau protein with this mutation develop neurodegeneration as result of aberrant splicing. The mice recapitulate many of the disease hallmarks that are seen in patients with this mutation, including increased tau exon 10 inclusion in both mRNA and protein, motor and behavioral deficits, and tau protein accumulation in neurons and tufted astrocytes. Furthermore, these mice present with degeneration of the nigrostriatal dopaminergic pathway, suggesting a possible mechanism for parkinsonism in FTDP-17. Additionally, activated caspase-3 immunoreactivity in both neurons and astrocytes implicates the involvement of the apoptotic pathway in the pathology of these mice.
Introduction
A variety of progressive neurodegenerative diseases called tauopathies are characterized by intracellular aggregates of the tau protein. Tauopathies are clinically distinguished by dementia and behavioral and motor dysfunction and include frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), Alzheimer's disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Pick's disease (Goedert and Hasegawa, 1999; Sergeant et al., 1999; Buee et al., 2000; Reed et al., 2001; Togo et al., 2002; Katsuse et al., 2003; Morris et al., 2003; Forman et al., 2004; Rademakers et al., 2004). Identifying the mechanisms underlying tau pathology is an important starting point for developing therapies capable of reducing neuronal dysfunction in tauopathies.
Tau protein is a neuron-specific protein that binds and stabilizes microtubules that are involved in axonal transport (Weingarten et al., 1975; Drubin et al., 1984; Binder et al., 1985; Caceres and Kosik, 1990; Ebneth et al., 1998; Stamer et al., 2002). The alternative splicing of exons 2, 3, and 10 is developmentally, physiologically, and spatially regulated. In the fetal brain, only the shortest isoform lacking exons 2, 3, and 10 is expressed. As the brain matures, differential splicing of exons 2, 3, and 10 results in the generation of six tau isoforms (mRNA and protein) (Goedert et al., 1989; Himmler, 1989; Himmler et al., 1989). These tau isoforms are typically distinguished by the presence of either three (3R) or four (4R) tandem repeats of the microtubule-binding domain. The 4R tau isoforms contain additional amino acids encoded by exon 10 that are absent in the 3R isoforms (Goedert and Jakes, 1990; Shahani and Brandt, 2002).
The identification of TAU gene mutations in families with FTDP-17 tauopathy has provided firm evidence that abnormal tau expression is sufficient to cause neurodegeneration (Foster et al., 1997; Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998; Reed et al., 2001; Rademakers et al., 2004). Mutations in the TAU gene fall under two categories: exonic mutations that directly alter the structure and function of tau protein, and intronic or exonic mutations that indirectly alter the structure and function of tau protein, most notably with respect to alternative splicing of exon 10. Mutations that alter the splicing of exon 10 consequently affect the relative ratio of 4R to 3R tau. These mutations account for approximately one-half of the mutations observed in FTDP-17 (Foster et al., 1997; Hutton et al., 1998; Reed et al., 2001; Rademakers et al., 2004).
To model tauopathy in a transgenic mouse, we have focused on the FTDP-17 asparagine-to-lysine mutation at position 279 (N279K) located within exon 10 (Wszolek et al., 1992; Kawai et al., 1993; Wilhelmsen, 1997; Clark et al., 1998; Reed et al., 1998; Delisle et al., 1999; Yasuda et al., 1999; Cheshire et al., 2002; Tsuboi et al., 2002a,b; Soliveri et al., 2003). Unlike other tau exonic mutations, in vitro studies have shown that the N279K mutation does not change the ability of tau to bind to tubulin or promote microtubule assembly and does not increase tau aggregation (Hasegawa et al., 1998; Hong et al., 1998; Nacharaju et al., 1999). The N279K mutation has been identified in several nonrelated families who present with a variety of symptoms including parkinsonism, dementia, motor dysfunction, and personality disorders, terminating in death. Neuropathologically, patients with this disease present with widespread neuronal and glial tau accumulations.
As a first step toward confirming the function of the N279K mutation in an animal model, and to further elucidate the molecular mechanisms underlying tauopathy resulting from a putative splicing mutation, we have created a transgenic mouse line expressing the N279K TAU mutation. These animals recapitulate, by multiple criteria, the tauopathy observed in N279K patients. Importantly, the inclusion of exon 10 in tau mRNA is significantly increased, intracellular tau accumulates in neurons and astrocytes, and the mice display significant behavioral impairments.
Materials and Methods
This study was conducted in accordance with the animal welfare guidelines set forth in the Guide for the Care and Use of Laboratory Animals, United States Department of Health and Human Services Publication Number 85-23 (1985). All animal procedures were previously approved by the Duke University Institutional Animal Use and Care Committee.
Generation of tau minigene transgenic mice.
The tau minigene was constructed by ligating a combination of genomic and cDNA, PCR-amplified fragments and restriction-digested cDNA fragments (Table 1). The human TAU gene PAC24i13 clone (Genome Systems, St. Louis, MO) was used as a template to amplify genomic fragments. The longest tau cDNA (441 aa) was used as a PCR template to amplify cDNA fragments. The 3′ untranslated region (UTR) consisted of a 113 bp region past the tau gene stop codon, as well as an SV40 polyadenylation signal and enhancer.
Using the SalI site in the 5′-UTR in exon 1 of the TAU gene, a 5049 bp SacI/SalI promoter fragment was subcloned in front of the minigene. This minigene consisted of nonmutated human TAU gene sequences and was thus termed the tau promoter wild-type minigene (T-WT). The T-WT construct was mutated using the in vitro Site-Directed Mutagenesis System (Promega, Madison, WI) to contain the N279K FTDP-17 mutation. This minigene consisted of mutated human TAU gene sequences and was thus termed the tau promoter N279 minigene (T-279). We modified the T-279 construct to place the N279K mutated tau minigene under the regulation of the cytomegalovirus (CMV) construct (C-279). The promoterless N279K mutated minigene was subcloned in front of the CMV promoter in the vector pcDNA3 (Invitrogen, Carlsbad, CA).
In parallel, a human tau cDNA construct (the longest isoform, which includes exons 2, 3, and 10) was engineered by subcloning the tau cDNA downstream of the TAU gene promoter at the SalI site in the 5′-UTR and the 3′-UTR region, as described above for the minigenes (see Fig. 1). Transgenic mice expressing human tau proteins were generated by microinjection of the T-WT, T-279, and C-279 constructs into pronuclei by the Duke University transgenic facility. The mice were generated on a mixed C57BL/6//SJL background. All resultant breedings were performed with nontransgenic littermates to maintain the lines on a similar mixed-strain background.
Brain tissue harvesting.
Mice were deeply anesthetized with ketamine (200 mg/kg), xylazine (10 mg/kg), and acepromazine (2 mg/kg). Then, mice were transcardially perfused with 30 ml of PBS. Brains were removed from the skull and divided into the right and left hemispheres; one hemisphere was frozen in liquid nitrogen for biochemical assays, and the second hemisphere was fixed for 24 h in 4% formaldehyde/PBS. The harvested hemisphere that had been frozen in liquid nitrogen was pulverized by a mortar and pestle over dry ice and divided into portions for use in protein and mRNA analysis.
Reverse transcription-PCR.
Frozen, pulverized tissue samples were homogenized. Total RNA was extracted with Trizol reagent (Invitrogen) according to the manufacturer's protocol. One microgram of total RNA was treated with RQ1 DNase (Promega) to remove DNA contamination. The RNA sample was then reverse transcribed using AMV transcriptase (Promega) and random primers. The reverse transcription reactions were incubated at room temperature for 10 min and then at 42°C for 40 min. The transcriptase was subsequently inactivated by heating at 99°C for 5 min and cooling to 4°C for 5 min. The inclusion of exon 10 was determined by PCR of the reverse-transcribed DNA using primers specific to human tau exon 9 and exon 11 (primer 3, 5′-CTCCAAAATCAGGGGATCGC-3′; primer 4, 5′-CCTTGCTCAGGTCAACTGGT-3′). An amplified fragment of 388 bp can be expected when exon 10 is included, whereas an amplified fragment of 295 bp can be expected when exon 10 is absent. The inclusion of exons 2 and 3 was determined by reverse transcription (RT)-PCR using primers to exon 1 and exon 4 (primer 1, 5′-TTGGGGGACAGGAAAGATCAG-3′; primer 4, 5′-GCGGGGTTTTTGCTGGAATC-3′). The sizes of the expected amplified fragments were as follows: minus exons 2 and 3, 299 bp; minus exon 3, 387 bp; plus exons 2 and 3, 475 base pairs each. Relative intensities of the two PCR fragments resulting from the exon 10 amplification remained the same through cycles 28–50. Therefore, 40 amplification cycles were used. The Kodak (New Haven, CT) Digital Science Image Station and software were used to quantify the RT-PCR results. Data were obtained from a minimum of three separate experiments. Statistical significance was determined using the GraphPad (San Diego, CA) Prism software Student's t test function.
The resulting cDNA templates were also used in real-time RT-PCR. The Taqman Universal PCR Master Mix and primer/probe sets from Assay-on-Demand Gene Expression Assays from Applied Biosystems (Foster City, CA) were used for total human tau (assay Hs00213491), for a common sequence of all isoforms. The PCR amplification was performed on the ABI 7000HT Sequence Detection System. The sample was heated at 95°C for 10 min, followed by 40 cycles of denaturing at 95°C for 15 s and annealing/extending at 60°C for 1 min. The threshold cycle (CT, the cycle number at which the amount of amplified target gene reaches a fixed threshold) was subsequently determined.
Relative quantification of mRNA expression was calculated by the comparative CT method with the amount of target = 2−ΔΔCT (Livak and Schmittgen, 2001). The values of the target were first normalized to that of the endogenous control (18S) and then normalized relative to a calibrator. Data are presented as the fold change, where T-cDNA was used as the calibrator and was determined to be 1 and all other genotypes were normalized to the calibrator. Triplicate samples for each animal were run for at least three different animals per genotype. An average fold change was calculated.
Antibodies.
The following antibodies were used: human-specific highly sensitive Tau13 (residues 9–18) (Garcia-Sierra et al., 2003) (dilutions, immunocytochemistry, 1:33,000; Western analysis, 1:100,000 for T-279 and 1:5,000,000 for T-WT and C-279), TauC3 specific for cleaved tau at Asp421 (residues 412–421) (Gamblin et al., 2003a) [dilution, 1:10,000; both Tau13 and TauC3 were kind gifts from Dr. Lester Binder (Northwestern University, Evanston, IL], human-specific HT7 (residues 159–163; dilution, 1:3000; Pierce, Rockford, IL), human- and mouse-specific Tau5 (residues 210–236) (dilution, 1:50; Abcam, Cambridge, MA), tau protein AT8 (phosphorylated residues Ser202 and Thr205; dilution, 1:3000; Innogenetics, Alpharetta, GA), tau protein AT100 (phosphorylated Ser212 and 214; dilution, 1:200; Pierce), tau protein AT180 (phosphorylated Thr231 and Ser235; dilution, 1:100; Pierce), tau protein AD2 (Buee-Scherrer et al., 1996) [phosphorylated residues Ser396 and Ser404; a gift from Dr. Delacourte (INSERM Research Center, Lille, France)], tau protein MC-1 [pathological tau conformation specific; a kind gift from Dr. Peter Davies (Albert Einstein College of Medicine, New York, NY)], astrocyte-specific anti-GFAP antibody (clone 6F2; dilution, 1:300; Dako, Carpinteria, CA), tyrosine hydroxylase (TH) antibody (clone TH2; dilution, 1:2500; Sigma-Aldrich, St. Louis, MO), activated caspase-3 antibody (dilution, 1:100; Cell Signaling Technology), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (dilution, 1,000,000; Advanced Immunochemical, Long Beach, CA).
Western blotting.
Western analysis using various tau antibodies was used to confirm the presence of the transgenic proteins and to semiquantitatively determine the expression level of tau protein. As described by Dawson et al. (2001), frozen, pulverized mouse brain tissue was homogenized in mixture containing protease inhibitors (Complete Mini; Roche, Indianapolis, IN) and centrifuged twice at 4°C and 10,000 × g for 15 min each. The amount of protein in each fraction was determined by the BCA protein assay (Pierce). The supernatants were mixed with Laemmli sample buffer, heated, and separated by electrophoresis. The sample was visualized using the ECL Plus kit (GE Healthcare, Piscataway, NJ) sensitive to antigens in the picogram range.
The level of human tau protein expression in T-279 mice was determined semiquantitatively by immunoblotting brain homogenate from three T-279 mice with Tau13 and comparing it to a standard curve of purified recombinant human tau protein (EMD Biosciences, San Diego, CA) loaded on the same Western blot (see Fig. 2A). The level of human tau protein expression in T-WT and C-279 mice was analyzed the same way (see Fig. 2E,F). Similarly, the level of endogenous mouse tau protein expression in nontransgenic mice was determined semiquantitatively by immunoblotting brain homogenate from three wild-type mice with Tau5 and comparing it to a standard curve of purified recombinant human tau protein (EMD Biosciences) loaded on the same Western blot (see Fig. 2B). The intensity of staining was quantified by densitometry using the Kodak Image Station 440 CF and Kodak 1D image analysis software. Standard curves and their corresponding R2 values of recombinant tau protein were calculated by Microsoft (Seattle, WA) Excel software, and the amounts of human and mouse tau protein were calculated from the standard curves.
Because the levels of human tau protein in T-279 mice were so low, we also compared the tau expression levels to the T-cDNA mice on a mouse tau knock-out background (Table 1) (Dawson et al., 2001). Using Western blot analysis (see Fig. 2C), we determined that the levels of human tau protein in the T-cDNA mice were equal to endogenous mouse tau protein levels in wild-type nontransgenic mice. Using human tau-specific antibodies, we were then able to show the level of expression of human tau protein expressed in T-279 mice to tau levels expressed in the T-cDNA mice (see Fig. 2D).
Immunohistochemistry.
Forty micrometer sagittal tissue sections were cut using a freezing microtome. For immunostaining, brain sections were first boiled in SSC and antigen retrieval buffer (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Then, tissue was incubated in 1% hydrogen peroxide, permeabilized by 0.1% saponin, and blocked with 10% goat serum. Primary antibodies were diluted to the final working concentration (see antibodies section) in 1% goat serum and then reacted overnight at 4°C. The Vectastain Elite ABC kit/DAB (diaminobenzidine) staining kit (Vector Laboratories) was used according to the manufacturer's instructions. Brain sections were dehydrated and mounted using DPX mounting media (Fluka, Milwaukee, WI). Brain sections were examined using a Nikon (Tokyo, Japan) Eclipse TE200 microscope, and images were recorded using a Nikon digital still camera (DXM1200).
The Gallyas silver stain was used to identify fibrillar tau in brain sections (Iqbal et al., 1991). Brain slices were washed with PBS and treated with 0.25% potassium permanganate. The sections were then exposed to 5% oxalic acid, treated with alkaline silver iodide (4% NaOH, 1% KI, and 0.035% AgNO3), and rinsed in aqueous acetic acid. A blue-black color was developed by the addition of developing solution containing a mixture of 2.5% sodium carbonate, 0.1% ammonium nitrate, 0.1% silver nitrate, and 0.5% tungstosilicic acid plus 0.13% formaldehyde. Sections were rinsed in acetic acid and toned in a 0.09% gold chloride solution. Then, sections were exposed to 1% sodium thiosulfate.
Fluoro-Jade B is a polyanionic fluorescein derivative that binds to degenerating neurons (Schmued and Hopkins, 2000). To mark degenerating neurons and their processes, slides were immersed in 80% ethanol with 1% NaOH for 5 min followed by 2 min in 70% alcohol and 1 min in distilled water. The slides were then transferred to a solution of 0.06% potassium permanganate for 15 min followed by a 1 min rinse in distilled water. Sections were stained in a 0.0004% Fluoro-Jade B (Histochem, Jefferson, AR) solution prepared in 0.1% acetic acid for 30 min and rinsed three times for 1 min each in distilled water.
Stereology.
The brains of 11 T-279 and six WT mice were sectioned sagittally (40 μm) on a freezing microtome. Because of the changing architecture of the substantia nigra compacta, three sections matched for location were selected at distances 960, 1080, and 1200 μm from the midline according to the Paxinos and Franklin (2001) mouse brain atlas. The entire substantia nigra was outlined with a 4× objective using the Stereo Investigator 7.0 software (MicroBrightField, Williston, VT). TH-immunopositive neurons with visible nuclei were identified with a 20× objective, and the total numbers were estimated with the optical fractionator method (West et al., 1991). An average of 20 counting frames was analyzed per section, and the counting frame dimensions were 80 × 80 × 13 μm. Group averages for control and experimental animals were generated and compared using the Student's t test function (GraphPad Prism software).
Behavioral testing.
The rotarod protocol adapted from Hamm et al. (1994) was used to assess motor coordination and balance (Crawley, 2000; Motoi et al., 2004). The mice were first conditioned to the rotarod at a constant speed (16 rpm) for 60 s. This was done twice with a 5 min interval between trials. For testing, mice were placed on the rotarod. The rotarod speed accelerated at a rate of 7.2 rpm2 for 5 min starting at 4 rpm. Performance was assessed by measuring the latency time to either falling from the rod or turning 360° twice (stationary and holding on to the rod).
The radial arm water maze (Gordon et al., 2001, Morgan et al., 2000) measures learning and memory performance in mice. The radial arm water maze tests the animal's ability to find a round (7.5 cm diameter) submerged escape platform in a 105 cm diameter pool within a 60 s interval. The pool is divided into six arms (swim paths), each of which had aluminum sides. There was an open center in the middle of the pool, where the arms came together. The platform was placed at the end of one arm. Spatial cues were present on walls of the testing room. The mice were given the opportunity to learn the location of the submerged platform in four consecutive acquisition trials followed by a retention trial 30 min later (trial 5). The number of errors the mouse made before locating the platform was recorded. An error constituted the entry into a nonplatform arm or entering the platform arm but leaving without climbing on to the platform. This test was conducted on 4 consecutive days. The platform was placed in a different arm on each consecutive day and was always submerged. By the fourth day, nontransgenic mice consistently located the platform on the fourth trial. Because the measure of learning acquisition and memory retention was based on the number of errors it takes the mouse to find the platform in 1 min, rather than the latency, slight motor deficits will not bias the results toward showing more memory deficits.
Results
Generation of splicing mutation mice
To study the relationship between splicing mutations and tauopathy, we created a transgenic mouse that expresses human tau protein with a FTDP-17 N279K mutation that alters splicing of exon 10 in tau mRNAs. To permit differential splicing, the expression of gene products was programmed with a human TAU minigene that was engineered to contain the intronic sequences that normally surround the alternatively spliced TAU exons 2, 3, and 10 (Fig. 1A). To further evaluate the function of the authentic TAU promoter, the minigene was placed under the regulation of the human TAU promoter (including 5 kb of upstream sequence) to permit human-promoter-driven expression of the tau protein. The N279K mutation was then incorporated into the minigene. For comparative purposes, the mutated minigene was placed under the regulation of the CMV promoter for ubiquitous, nonspecific expression of tau protein.

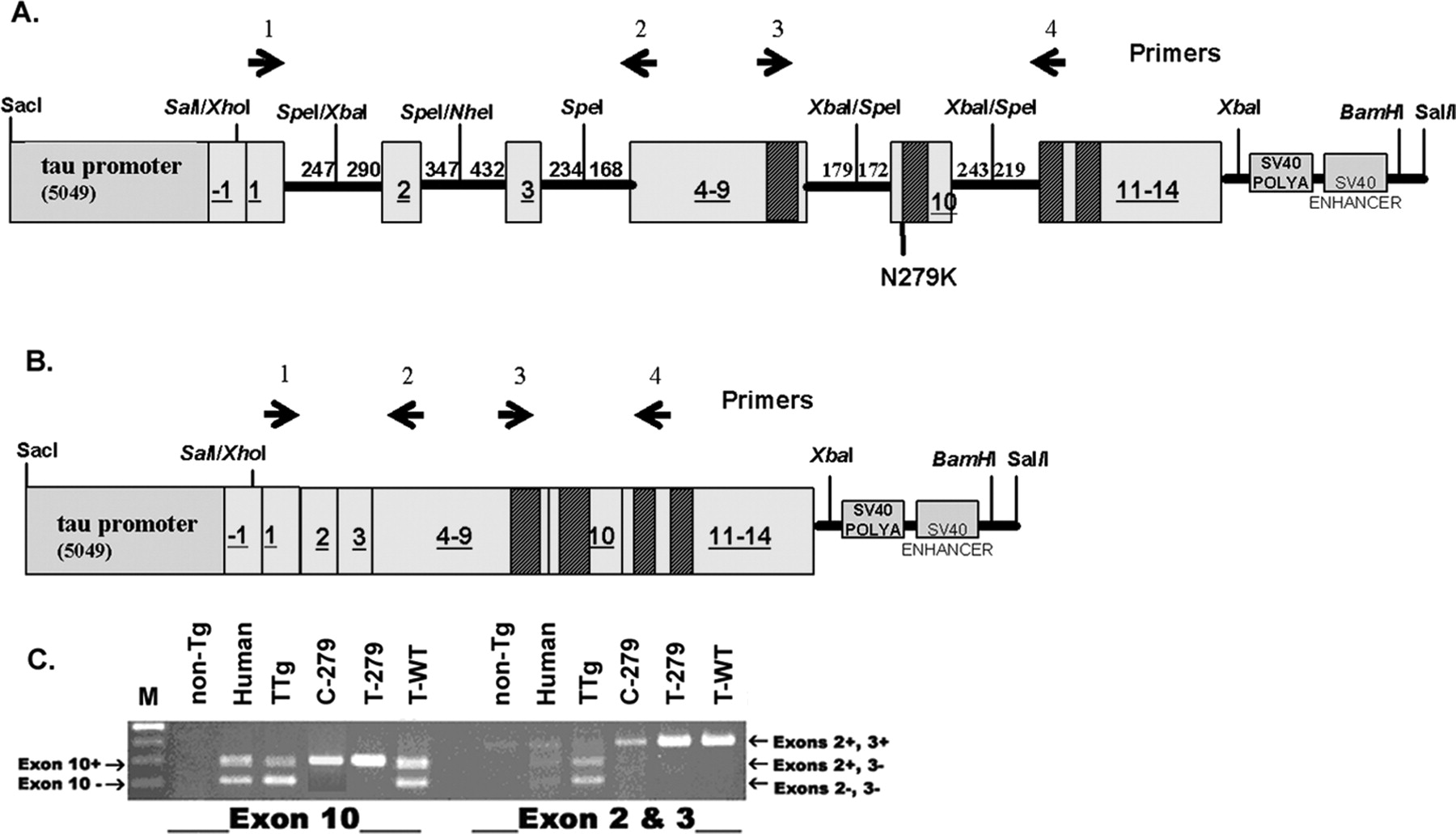
Schematic of tau constructs and splicing. A, The tau minigene construct. The first box on the left represents the putative TAU promoter region, and numbered boxes represent exons with the corresponding numbers. The hatched rectangles represent the locations of the four microtubule-binding domains. The thick black lines connecting the exons represent intronic sequences. The numbers above the lines indicate the number of intronic nucleotides included in the minigene. In the C-279 construct, the CMV promoter was inserted into the SalI/XhoI site instead of the TAU promoter. The position of the N279K mutation in the T-279 and the C-279 constructs is indicated below the construct. The last two boxes represent the SV40 polyadenylation (POLYA) and enhancer sequences, respectively. B, The tau cDNA construct is identical to the tau minigene constructs except it lacks the intronic sequences. The primers used for RT-PCR are indicated as arrows above each of the constructs in A and B, Primers 3 and 4 were used for RT-PCR of exon 10, whereas primers 1 and 2 were used for RT-PCR of exons 2 and 3. The figures are not drawn to scale. C, Representative data from RT-PCR of exons 2 and 3 and exon 10 using template mRNA from adult mouse and human brain homogenates. The lanes are marked and abbreviated as follows: markers (M), nontransgenic mouse (non-Tg), human control (human), TTg, C-279, T-279, and T-WT (see Table 1). Primers 3 and 4 are human specific and show the inclusion of exon 10 (top band) and exclusion of exon 10 (bottom band). RT-PCR of exons 2 and 3 shows the inclusion of both exons 2 and 3 (top band), inclusion of exon 2 only (middle band), and absence of both exons 2 and 3 (bottom band).
The following three transgenic minigene mouse lines were generated: (1) control mice expressing human tau protein under the regulation of the human TAU promoter from the nonmutated minigene (T-WT mice); (2) experimental mice expressing human N279K mutated tau protein from the mutated minigene under the regulation of the human TAU promoter (T-279 mice); and (3) experimental mice expressing human N279K mutated tau protein from the mutated minigene under the regulation of the CMV promoter (C-279 mice) (Table 1).
Tau transgenic mice and their abbreviations
In addition to these novel minigene mice, a previously generated TAU gene transgenic (TTg) mouse created from the human TAU gene PAC24i13 clone (Genome Systems) and was included as a control for human tau mRNA expression (Table 1) (Dawson et al., 2001). An additional mouse line was generated in which WT human tau (derived from the longest 441 aa coding cDNA) was expressed from the minigene human tau promoter (T-cDNA mouse). This mouse line was placed on a murine tau knock-out background (Dawson et al., 2001) such that only human tau protein was expressed. This line served as a control to compare human tau protein expression in the minigene mice (Table 1).
The N279K mutation increased the inclusion of exon 10 in Tau mRNA
The N279K mutation has been previously shown to favor the inclusion of exon 10 in human patients with tauopathy. This mutation resides within a region of DNA containing the exon 10 splicing enhancer and is thought to favor the inclusion of exon 10 in mRNA transcripts (D'Souza et al., 1999; D'Souza and Schellenberg, 2000, 2002, 2005). Thus, the level of exon 10 expression was used as an indirect indicator of disease potential in our various transgenic mouse lines. Total RNA was isolated from the brains of adult animals. The levels of 4R tau mRNA (Yasojima et al., 1999) and 3R tau mRNA (no exon 10) were quantified using RT-PCR. As predicted, the three minigene mouse lines (T-WT, T-279, and C-279), as well as the control animals (TTg and T-cDNA), expressed human tau mRNA, although they differed in their ratio of 4R to 3R. Like the expression reported in humans, 4R and 3R tau were expressed in approximately equal amounts in human, TTg, and T-WT mRNA, whereas 4R was almost exclusively expressed in T-279 and C-279 mRNA (Fig. 1C, Table 2). RT-PCR of exons 2 and 3 in all three minigene transgenic mice showed only one band corresponding to the inclusion of both exons in tau mRNA (Fig. 1C). Control RT-PCR of RNA from humans and TTg mice showed three bands that correspond to the tau mRNA isotypes resulting from the differential splicing of exons 2 and 3 (Fig. 1C).
Expression of 4R tau mRNA in fetal and adult transgenic mice
Tau protein expression
To verify that human tau mRNA expressed in the transgenic mouse lines was translated into tau protein, Western blot analysis was performed on whole-brain homogenates using antibodies specific for human tau. As depicted in Figure 2, A, E, and F, an immunoreactive band for human tau protein was detected in brain lysates from all three minigene mouse lines. To compare the expression level of tau protein in the minigene mice to endogenous mouse tau expression in mWT mice, we generated a standard curve using human recombinant protein of the longest tau isoform (tau 441). Two different concentrations of brain homogenate from three T-279, three T-WT, three C-279, and three nontransgenic mice were analyzed by Western blot analysis using three different amounts of recombinant tau protein (Fig. 2). The T-279, T-WT, and C-279 Western blots were probed with the human-specific Tau13 antibody (Garcia-Sierra et al., 2003), whereas the mWT Western blot was probed with the Tau5 antibody that recognizes all tau protein (made to bovine tau). The intensity of the resultant bands for recombinant tau was used to generate Tau13 and Tau5 standard curves, from which the corresponding amounts of tau protein in the transgenic mice were calculated (Table 3). The expression of endogenous mouse tau protein in 52-week-old WT was much greater than human tau protein in either 23- or 52-week-old T-279 mice (Table 3, Fig. 2A1,A2,B). Similar results were obtained with the human-specific HT7 antibody using the same method described above (Fig. 2A3). From this semiquantitative Western blot analysis, we conclude that the expression of human tau protein in the T-279 mice is ∼1–2% of endogenous mouse tau.

Expression of human tau protein in transgenic mice. A–F, The numbers above the lanes indicate the amount of total protein loaded in micrograms (A–D) and nanograms (E, F) for brain homogenate and in picograms for recombinant human tau (r-TAU). A1, A comparison of brain homogenates from 52-week-old T-279 mice and r-TAU protein, probed with the human-specific Tau13 antibody. A2, A comparison of brain homogenates from 23-week-old T-279 mice and r-TAU probed with the human-specific Tau13 antibody. For A1 and A2, 4 and 6 μg of brain homogenates from three animals and 50, 100, and 200 pg of r-TAU were loaded for comparison. A3, Same three 52-week-old mice as in A1 probed with human-specific HT7 antibody. A4, Three 1-d-old and three 5-d-old T-279 mice and one 52-week-old T-279 mouse probed with HT7 depicting the developmental shift in tau protein isotopes with (top band) and without (bottom band) exon 10. B, Brain homogenates from three 52-week-old WT nontransgenic mice (0.2 and 0.3 μg for each animal) and 400, 800, and 1600 pg of r-TAU were probed with the Tau5 antibody that recognizes both mouse and human tau protein. C, Brain homogenate from nontransgenic (mWT) and T-cDNA mice were loaded using equal amounts of total protein and probed with Tau 5. The TcDNA mouse was created on a tau knock-out background and thus only expresses human tau. The levels of expression can therefore be compared with the levels of mouse tau in nontransgenic mice. The blots were reprobed with an anti-GAPDH antibody to ensure equal loading. D, Human tau protein in brain homogenate from a T-cDNA mouse was compared with human tau protein from two T-279 mice probed with Tau13. Equal amounts (2, 4, and 6 μg) of total protein were loaded for each animal. The blot was probed with Tau13. The blots were reprobed with an anti-GAPDH antibody to ensure equal loading. E, F, A comparison of brain homogenates from three 52-week-old T-WT (E) and C-279 (F) mice (each two lanes labeled with 20 and 30 ng represent one animal) and r-TAU probed with the human-specific Tau13 antibody. M, Markers.
Expression tau protein levels in WT and minigene mice
Because the calculated human tau levels were so low, we compared the expression of human tau in T-279 mice to T-cDNA mice. The T-cDNA mice were created on a mouse tau knock-out background such that they express only human tau protein. Equal amounts of brain homogenate from nontransgenic mice and T-cDNA mice were immunoblotted with Tau5, and the tau band intensities were compared with a GAPDH internal standard. It was determined by semiquantitative analysis that the T-cDNA mice expressed approximately the same amount of tau protein as WT mice (Fig. 2C).
Equal amounts of brain homogenate from 52-week-old T-279 mice and T-cDNA mice were then immunoblotted individually with Tau13 and GAPDH to control for equal loading of total protein (Fig. 2D). The immunoblot clearly shows the large difference between the expression of human tau protein in T-279 mice and tau protein in T-cDNA and hence WT mice.
Next, using the same technique as described above, we compared the amount of human tau in brain homogenates from T-WT and C-279 mice to recombinant tau using the Tau13 antibody (Fig. 2E,F, Table 3). The expression of human tau in the 52-week-old T-WT and C-279 mice was greater than the expression of endogenous mouse tau protein in 52-week-old mWT (Table 3). Surprisingly, the expression level of human tau in T-WT and C-279 was much greater than in either 23- or 52-week-old T-279 mice (Table 3, Fig. 2A1,A2,B).
Because of the large differences in the human tau protein expression levels in the transgenic mice, we assessed the mRNA levels of tau in the different transgenic mice. Using real-time RT-PCR, the levels of human tau mRNA in all transgenic mouse lines were determined and normalized to 18 s mRNA. The level of tau expression was calculated as a ratio of mRNA expressed in the T-cDNA mice (where T-cDNA human tau mRNA expression = 1). There was an eightfold decrease in the expression of tau mRNA in T-279 mice, an 11-fold increase in tau expression in T-WT, and a ninefold increase in tau mRNA expression in C-279 mice.
We assessed the mRNA levels in 2-d-old, 18-week-old, and 52-week-old T-279 mice. There was no significant difference in the levels of tau mRNA expression between these ages. However, the human tau protein levels in 23-week-old mice were two times higher than in 52-week-old mice (Table 3). Several reasons could account for the difference in tau protein expression between the 52-week-old and the 23-week-old mice. First, a decrease in tau expression at 52 weeks could be a downregulation of tau expression as a result of a negative feedback in response to tau protein accumulation. Second, the decrease in tau protein in brain homogenate at 52 weeks could be a result of cell loss. In fact, our data showed a 30% loss of neurons of the substantia nigra (data described below). Last, because of the semiquantitative nature of Western blot analysis and the large differences in the total protein loaded between brain homogenate and recombinant tau, the calculated difference in tau protein levels in the two age groups may be a result of technical limitations.
In general, the human tau mRNA levels corresponded to the human tau protein levels. The T-279 mice express much less tau mRNA than all of the other transgenic mice, whereas the T-WT and C-279 mice express the most tau mRNA. However, these results show that tau protein expression may also be regulated at the posttranscription level because the mRNA levels are higher in T-WT and C-279 mice and lower in T-279 than what was expected from the protein analysis.
The human tau promoter regulates exon 10 splicing
The differential splicing of exon 10 sequences in tau mRNA, which affects the ratio of 3R to 4R tau expression and protein production, is developmentally regulated in humans and mice. Only 3R tau mRNA and protein are expressed in the fetal brains of humans and mice, whereas in the adult human brain, both 3R and 4R tau isoforms are expressed at approximately equal levels. In the mouse brain, however, the majority of tau is of the 4R isoform (Goedert et al., 1989; Dawson et al., 2001). We wanted to determine whether expression of these tau mRNA isoforms would be similarly regulated by the presence or absence of the N279K mutation, the promoter driving the expression, or a combination of the two.
To address this issue, RT-PCR was used to measure the levels of 4R tau mRNA expressed as a percentage of total tau mRNA in homogenates from fetal [embryonic day 16 (E16)] and adult (8–30 weeks of age) mouse brains (Table 2). Representative results are shown in Figure 3. Expression of 4R mRNA in TTg mice expressing tau from the whole human tau gene was significantly higher in the adult brain relative to fetal brain (35.8 and 4.0%, respectively; p = 0.0037), confirming previously published results (Takuma et al., 2003) that exon 10 is differentially spliced during development in the murine brain. The expression level of 3R and 4R tau in adult TTg mice was comparable with that reported previously (Duff et al., 2000).

Comparison of 4R tau in adult and fetal tau from transgenic mice. A representative gel (bottom) and a graph form (top) of RT-PCR depict the inclusion of tau exon 10 (4R tau) in adult (A) and fetal (F) human tau using tau mRNA from T-WT, T-279, C-279, and TTg transgenic mice as a template. Human mRNA (Hu; bottom) was used as a control to indicate the expression of 4R (top band) or 3R (bottom band) tau. The leftmost column of the gel depicts markers. The expression of 4R tau was statistically decreased in fetal tau compared with adult tau in TTg and T-279 mice. The expression of 4R did not change between fetal and adult mice in T-WT and C-279 mice. A minimum of three experiments were averaged.
Surprisingly, no significant difference in 4R tau mRNA expression was detected between fetal and adult brain in the T-WT mice (Table 2). It is possible, however, that the T-WT mice, which harbor the smaller tau minigene, lack the required regulatory sequences present in TTg animals that are necessary for the developmental regulation of tau.
Messenger RNA from E16 fetal T-279 mice showed significant less expression of 4R tau mRNA relative to mRNA from adult T-279 mice (percentage of total tau mRNA = 49.4 and 96.6%, respectively; p < 0.0001) (Table 2). However, the actual level of 4R fetal tau in T-279 animals was comparable with the level 4R fetal tau in T-WT animals (49.4 vs 44.3%, respectively), further strengthening the theory that the smaller tau minigene constructs lack the required regulatory sequences to completely suppress the expression of 4R tau in the mouse fetal brain.
These findings were corroborated by the inclusion of exon 10 in the human tau protein in T-279 mice (Fig. 2A4). Brain homogenate from three 1- and 5-d-old T-279 mice and one 52-week-old T-279 mouse was dephosphorylated and analyzed by Western blot analysis using the HT7 antibody. As predicted by the RT-PCR, the expression of human tau protein with exon 10 increases with age until tau protein containing exon 10 is almost exclusively expressed.
Interestingly, the expression of 4R tau mRNA in fetal and adult brains remained unchanged in the C-279 mice (Table 2) (p = 0.2564), which express mutated tau regulated by the CMV promoter. To rule out the possibility that suppression of fetal neuronal mRNA remained undetected in C-279 animals as a result of the ubiquitous expression of tau in non-neuronal cells, separate control experiments were performed using the hippocampi isolated from E16 animals, a developmental stage in which the hippocampi are composed primarily of pyramidal neurons (Craig and Banker, 1994). RT-PCR confirmed that fetal 4R tau suppression does not occur in C-279 animals (data not shown). Given that the T-279 and C-279 mouse lines differ only by the promoter driving tau expression, these results suggest that the tau promoter plays an important role in the differential splicing of exon 10 and subsequent expression of 4R mRNA. Furthermore, these data suggest that it is not the constant overexpression of 4R tau alone that leads to pathology but that an aberrant regulation of a changing 3R-to-4R tau ratio is also required; when tau with 4R is increased but the same ratio with 3R is maintained as in the T-cDNA mice, no pathology is observed.
Disease-like neuropathology in T-279 mice
Patients with the N279K mutation present with widespread intracellular aggregates of tau protein in neurons in the cortex, basal ganglia, brainstem, and white matter. To test whether the N279K mutation reproduces the human pathology in mice, whole-brain sections from T-279, C-279, and T-WT transgenic mice were immunostained using Tau13, a human-specific antibody to the N terminus of tau (Garcia-Sierra et al., 2003). In normal human subjects, light Tau13 immunostaining was only present in the neuropil (data not shown). An example of tangle and neuritic tau pathology in the cortex from an Alzheimer's disease patient immunostained with Tau13 is represented in Figure 4A. Tau13 immunostaining was weakly present, which correlates with the theory that the N terminus of tau protein is cleaved in late Alzheimer's disease (Garcia-Sierra et al., 2003).

Tau pathology in CNS neurons of T-279 mice but not in T-WT or C-279 mice. A, A representative image (60×) of human Alzheimer's brain immunostained with the Tau13 human-specific antibody revealed neuronal and neuritic staining. B, H, I, Representative images of the frontal cortex of T-279, T-WT, and C-279 mice, respectively, immunostained with Tau13 (20×). C, Representative image of the frontal cortex corresponding to B, immunostained with AT8, a phosphorylation-dependent antibody (20×). D–F, Representative images of brain slices of the pons (D; 40×), olfactory bulb (E; 60×), and hypothalamus (F; 20×) from T-279 mice showing Tau13-immunostained neurons and neuronal processes. G, Representative Gallyas silver-stained brain slice from the hypothalamus of a T-279 mouse (60×). All animals represented in these images were 52 weeks of age.
Immunoreactive tau accumulation was detected in neurons of the cortex, thalamus, hypothalamus, olfactory bulb, basal ganglia, cerebellum, midbrain, brainstem, and spinal cord in 52-week-old T-279 mice (Fig. 4 B–F). Neuronal accumulation of tau was present in various patterns, ranging from focal accumulations near the apical dendrite to dense staining surrounding the nucleus and extending into the dendrites (Fig. 4 B–F). Tau-immunoreactive material completely filled the soma of some neurons, whereas others displayed granular accumulations of tau (Fig. 4D). A number of neurons in the olfactory bulb exhibited intense tau staining that extended into the dendrites and dendritic spines (Fig. 4E). A subset of neurons was positive with the Gallyas silver stain that detects abnormal filaments such as those composed of aggregated tau (Fig. 4G). In contrast to the T-279 animals, no tau accumulations were detected in either the T-WT or C-279 mice (Fig. 4H,I). The lack of tau accumulation in the C-279 animals was particularly surprising given that T-279 mice express only a very small fraction of the mutated tau expressed in C-279 mice.
Neuronal pathology was examined in more detail in 52-week-old T-279 mice. As shown in Figure 5A, tau-immunopositive degenerating neuronal processes found in the CNS appeared as beaded swellings, sausage-shaped swellings, and spheroids. FluoroJade B-positive staining (Fig. 5B), which is indicative of degenerating neuronal cells (Schmued and Hopkins, 2000), showed beaded and sausage-like processes similar to those in Figure 5A. Gallyas silver staining revealed argyrophilic processes (Fig. 5D) in the axonal bundles of the striatum indicating abnormal filament accumulation in the neuronal processes. Adjacent sections immunostained with Tau13 revealed similar structures (Fig. 5C). Numerous Tau13-positive structures in the granular layer of the cerebellum were observed in all sections examined (Fig. 5E). Under high-power magnification, these structures were identified as mossy fiber cerebellar rosettes (Fig. 5F), which constitute the principal mode of termination of cerebellar afferent systems (Carpenter, 1991). The tau immunostaining detected in T-279 mice closely resembled the swelling and beading of neuronal processes typically seen in tauopathies and is indicative of neuronal degeneration.
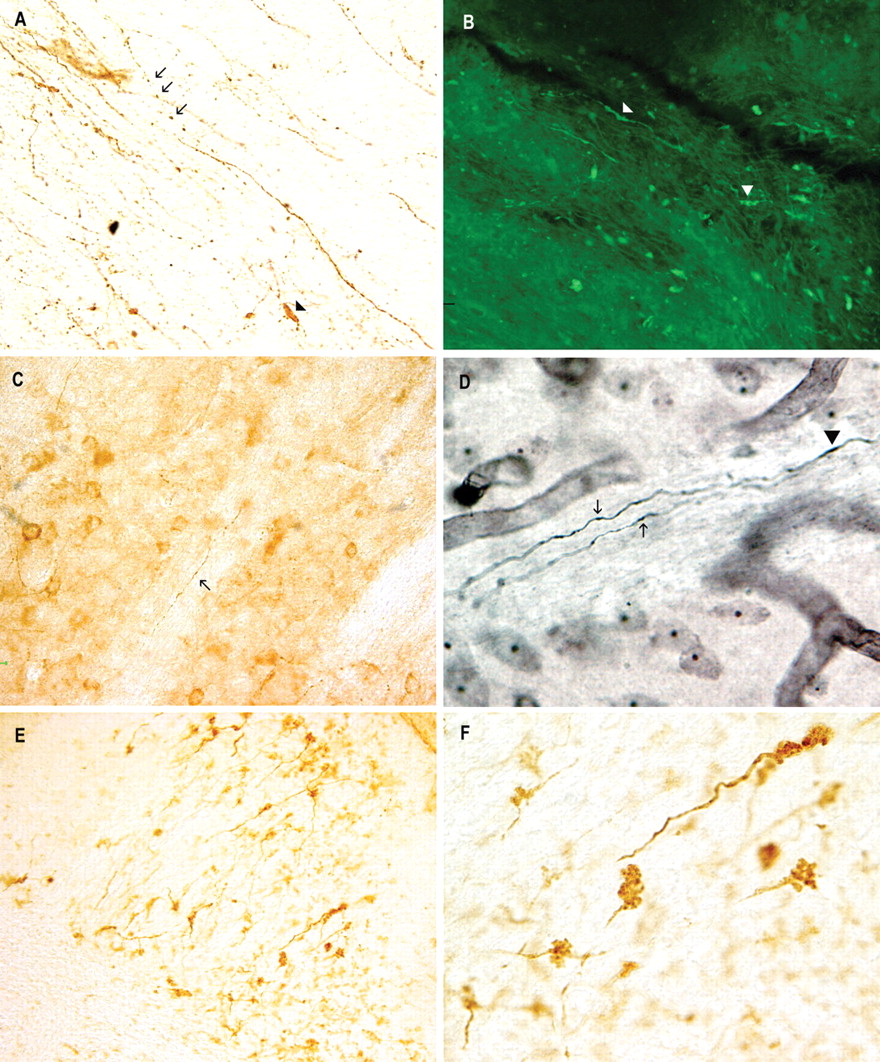
Tau accumulation and degeneration of neuronal processes in T-279 mice. A, B, Tau13 immunostain (20×; A) and FluoroJade B histochemical stain (60×; B) of beaded and swollen neuronal processes in the midbrain. C, D, Tau13-immunopositive (C; 40×) and Gallyas silver-positive (60×; D) axons in the striatum. E, F, Representative image of Tau13-immunopositive staining of structures in the cerebellum (20×; E), which when examined at high power (60×; F) were identified as cerebellar rosettes. Arrows indicate beaded neuronal processes, and arrowheads indicate swollen neuronal processes. All animals represented in these images were 52 weeks of age.
Tau accumulation in T-279 mice was dependent on the age of the animal, ranging from diffuse immunoreactivity in neurons in 18-week-old animals to much denser immunoreactivity by 52 weeks of age (a minimum of three animals per age group were examined) (Fig. 6). Degenerative neurites containing many dense bodies immunopositive for Tau13 were observed in neurons from 18-week-old animals in a characteristic “beads on a string” pattern commonly associated with neurodegeneration (Yamaguchi et al., 1991). Tau-immunopositive processes in 52-week-old mice progressed to a severely degenerated appearance with disrupted processes depicted by dense spheroids lacking connecting neurites. The numbers of visible tau-immunoreactive neurons and neuronal processes were greatly increased in the 52-week-old mice compared with 18-week-old mice.

Tau accumulation and neurodegeneration is age progressive. A, C, E, Images of Tau13-immunostained brain slices from 18-week-old T-279 mice. B, D, F, Images of Tau13-immunostained brain slices from 52-week-old T-279 mice. A, B, Lateral hypothalamic area. C, D, Lateral septal nucleus. E, F, Cerebellum. There was an age-dependent increase in tau accumulation in neurons and neuronal processes as well as an age-dependent degeneration of processes, as evidenced by beaded and disrupted neurites.
Tau-immunopositive astrocytes are a unique feature of some tauopathies (Hauw et al., 1990; Botez et al., 1999; Oyanagi et al., 2000; Takanashi et al., 2002; Togo and Dickson, 2002; Morris et al., 2003; Yang et al., 2003; Motoi et al., 2004; Wilhelmsen et al., 2004) and have been identified in patients with the N279K mutation (Delisle et al., 1999, 2000; Yasuda et al., 1999; Arima et al., 2000). Tufted astrocytes demonstrate tau immunoreactivity throughout the soma and proximal and distal processes, appearing as clumps of thin fibers (Hauw et al., 1990; Yamada et al., 1992, 1993; Togo and Dickson, 2002), whereas normal astrocytes exhibit no tau immunoreactivity.
Tau13-immunopositive cells morphologically resembling astrocytes demonstrated “tufted” processes. Tau13-immunopositive cells were observed in the cortex, hippocampus, thalamus, amygdala, and piriform cortex of the T-279 mice as early as 18 weeks (Figure 7). Colocalization of tau13 and GFAP immunoreactivity confirmed these cells to be astrocytes (Fig. 7D). In some cases, astrocytic tau aggregates were stained with Gallyas silver stain, suggesting abnormal filament formation (Fig. 7E). Tufted astrocytes were a prominent feature of the T-279 mice and were more frequently observed than tau-immunopositive neurons in some mice. Tufted astrocytes were not observed in brains from T-WT (data not shown) and C-279 mice (Fig. 7F).

Tau protein accumulates in astrocytes. A–E, Images of brain slices from 52-week-old T-279 mice. A–C, Tau13-immunopositive tufted astrocytes in the hippocampus and cortex (A), frontal cortex (B), and amygdala (C). D, Tufted hippocampal astrocyte double immunostained with Tau13 (brown) and GFAP (blue). E, Gallyas silver-stained astrocytes in the hippocampus. F, Tau13 immunostaining of the hippocampus from a C-279 age-matched control. The animals in these images were selected because of their high numbers of tufted astrocytes.
Abnormal phosphorylation and insoluble aggregation of tau protein has long been implicated in the role of formation and/or stabilization of pathological tau species. Various antibodies specific for pathological tau were tested in 52-week-old brains from T-279 mice. A subset of neurons and astrocytes were observed using the AT8 antibody (Figs. 4B, 8B) (Biernat et al., 1992; Mercken et al., 1992) and the tau AD2 antibody (Fig. 8A,C) (Buee-Scherrer et al., 1996), which are specific for phosphorylated tau (Ser199, Ser202 and Ser396, Ser404, respectively). In addition, a subset of astrocytes, but not neurons, was immunopositive with the phosphorylation-specific tau antibody AT100 (phosphorylated tau Thr212 and Ser214) (Mercken et al., 1992). No immunopositive neurons or astrocytes were observed with phosphorylation-specific tau antibody AT180 (phosphorylated Thr231 and Ser235) or MC1, a tau antibody specific for conformation changes observed in AD. Antibodies AT8 and AD2 are specific for pathological and fetal tau but not normal adult brain tau (Matsuo et al., 1994; Buee et al., 2000) and because neurons and astrocytes are immunopositive with these antibodies, we can conclude that tau is pathologically phosphorylated in the T-279 mice. The AT100 antibody detects only insoluble, hyperphosphorylated tau (Matsuo et al., 1994; Buee et al., 2000). Although AT100-immunostained astrocytes were detected in brain slices from T-279 mice, tau protein was not detected in Sarkosyl insoluble fractions extracted from the brains of these mice (data not shown). However, considering these results and the low level of tau protein expression, it is likely that hyperphosphorylated, insoluble tau is present in the T-279 mice but is below the level of detection in the Sarkosyl insoluble fractions.

Phosphorylated tau pathology in neurons and astrocytes of T-279 mice. A, A representative image of neurons and neuronal processes in the hypothalamus immunostained with AD2. B–D, Representative images of astrocytes in the hippocampus, immunostained with AD2, AT8, and AT100, respectively. The scale bar in D is representative of all four images. All animals represented in these images were 52 weeks of age.
Dopaminergic neuron degeneration in T-279 mice
Parkinsonism is a clinical phenotype that is a prominent identifying feature of all FTDP-17 patients (Foster et al., 1997). Because Parkinson's disease is characterized by the loss of dopaminergic neurons of the substantia nigra pars compacta, we used an antibody for TH to detect dopaminergic cells in the brain of T-279 mice. TH is an enzyme commonly used as a dopaminergic neuron marker. Degeneration of neuronal processes characterized by strongly TH-immunopositive beads was observed in the substantia nigra of T-279 mice but not in WT controls (Fig. 9A,B). Furthermore, intensely TH-immunopositive varicosities and spheroids were readily identified in the nigrostriatal track and striatum of the T-279 mice (Fig. 9C,D). These neuronal changes were annotated with a significant decrease (p = 0.0344) in TH-immunopositive neurons in the substantia nigra pars compacta from 100 ± 8.7% in WT mice to 71.6 ± 10.5% in T-279 mice.

TH-positive accumulations in varicosities and spheroids of neuronal processes present in T-279 mice. A, Darkly stained varicosities immunopositive with a tyrosine hydroxylase antibody were prominent in the substantia nigra compacta of T-279 mice (arrows). B, WT control mice showed only light staining of straight processes. C, Dark, TH-positive immunostaining of the nigrostriatal tract and neuronal processes of the striatum (inset) are present in the brains of T-279 mice. Small axonal swellings immunopositive with the TH antibody are interspersed throughout the nigrostriatal tracts and the striatum (arrows). D, WT mice show very light TH-positive immunostaining in the nigrostriatal tract. C, D, Images include a partial view of the anterior commissure (top of images; asterisks) for regional comparison between animals. The scale bar in A is valid for A–D.
It is important to note that although TH-positive cell loss occurred in the T-279 mice, the cells and processes that remained stained much more intensely than their WT counterparts. Thus, the expression of the N279K mutation in the T-279 mouse elicits pathological changes in dopaminergic neurons.
Caspase-3 activation in T-279 mice
Caspase-3, a known executioner caspase involved in apoptotic cell death, has been implicated in the neuronal loss observed in tauopathies, including Alzheimer's disease (AD), and has been shown to cleave tau protein (Fasulo et al., 2000; Berry et al., 2003; Gamblin et al., 2003a; Rissman et al., 2004; Cotman et al., 2005; Newman et al., 2005). To determine whether caspase-3 is a factor in the cell damage that we observed in T-279 mice, we examined brain sections for activated caspase-3 and cleaved tau protein using immunohistochemistry. Activated caspase-3-immunopositive neurons and astrocytes were detected throughout the brain and were particularly prominent in the olfactory bulb (Fig. 10). In some cases, cortical astrocytes with disintegrating, beaded processes were also immunoreactive for activated caspase-3 (Fig. 10C,D). Increased immunostaining of cleaved tau protein was not observed in T-279 mice. Activated caspase-3 or cleaved tau protein was not observed in nontransgenic littermate control brain sections. These data suggest that caspase-3 activation may be a factor in neurodegeneration in T-279 mice, although the overall importance remains unknown.

Neurons and glia were immunopositive for activated caspase-3 in T-279 mice. A, Pyramidal neurons in layer 5 of the frontal cortex were immunopositive for activated caspase-3. B, Activated caspase-3 immunostaining was especially prominent in neurons and neuronal processes in the olfactory bulb. C, Tufted astrocytes immunopositive with the activated caspase-3 antibody were visible as a line (arrow) in layer 3 of the cortex. D, Higher magnification of C depicts the degenerated appearance of the astrocytes. Original images were taken at 20× magnification in A, B, and D and at 4× magnification in C.
Behavioral deficits in T-279 mice
Patients with the N279K tau mutation present with parkinsonism that progresses to acute motor deficits and ends in death. Approximately 25% of T-279 transgenic mice developed acute progressive motor disturbances before the age of 52 weeks. These motor deficits were observed as early as 23 weeks of age, beginning with the onset of hindlimb paresis and an abnormal wide gait waddle, progressing to front limb disturbances, total hindlimb paralysis, and eventually, death (Fig. 11). The average age of death of mice that develop acute motor deficits is 45 ± 4 weeks.

T-279 mice show motor and cognitive deficits. A, T-279 mice with acute motor deficits lost the extension reflex (left) and eventually became completely paralyzed in their hind-limbs (right). B, Mice that did not appear affected by motor deficits at 23 and 52 weeks nevertheless showed a decreased latency on the rotarod task. C, D, Twenty-three-week-old (C) and 52-week-old (D) T-279 mice did not perform as well as their nontransgenic littermates on the radial arm water maze (RAWM) task.
Animals that did not demonstrate an acute motor deficit were tested for motor dysfunction at 23 and 52 weeks using the rotarod. Twenty-three-week-old T-279 mice exhibited a statistically significant (p = 0.0025) shorter latency to falling (165 ± 21 s) than nontransgenic littermate age-matched controls (273 ± 21 s) (Fig. 11). Fifty-two-week-old T-279 mice also exhibited a statistically significant (p = 0.0002) shorter latency to falling (145 ± 11 s) than nontransgenic littermate age-matched controls (204 ± 12 s) (Fig. 11). Although overtly asymptomatic, T-279 mice demonstrate measurable motor deficits.
In addition to motor deficits, patients with the N279K mutation also present with cognitive deficits, including mental slowness and memory and attention disturbances. The radial arm water maze was used as a measurement for detecting cognitive deficits in T-279 mice (Morgan et al., 2000; Gordon et al., 2001). Learning and memory in this task is measured by the number of errors an animal makes locating a submerged platform in 1 min. The length of time it takes to find the platform is not a factor used to measure learning and memory in this task. Therefore, any motor deficits affecting the rate of swimming will not bias the results.
After rotarod testing, the same group of 23- and 52-week-old T-279 animals with no overt motor deficits along with age-matched controls were pretrained for 3 d on the radial arm water maze and tested on the fourth day for their ability to locate a submerged platform. T-279 mice with acute motor deficits were excluded from the experiment. The number of errors made by both the 23- and 52-week-old T-279 mice was statistically greater than their nontransgenic littermates (p < 0.0001 and p = 0.001, respectively, by ANOVA) (Fig. 11C,D).
Discussion
The molecular basis of the destructive action(s) of mutated tau protein in FTDP-17 remains poorly understood. The T-279 mouse expressing the N279K mutation under control of the human TAU promoter shows that selected mutations regulate tau RNA splicing patterns, which contribute to neurodegeneration. These altered patterns of RNA isotype levels then lead to altered patterns of tau protein isoform expression that appear to affect the delicate balance between stabilization of microtubule structures by tau isoforms and the contribution of microtubule structures to the movement of macromolecules within neurons and astrocytes. Consequently, overstabilization or understabilization of microtubules outside an acceptable window of dynamic activity may lead to neurodegeneration in tauopathy.
The T-279 model is unique compared with the tauopathy models previously described because (1) neurodegeneration is a result of a tau splicing mutation, (2) the tau mRNA and protein expression is regulated by the human TAU promoter, and (3) the expression of human tau is very low, making it unlikely that tauopathy is a result of high levels of tau protein. The low levels of tau protein expression were very surprising in light of the amount of tau pathology observed. It is possible that select neurons and astrocytes that accumulate tau immunoreactivity may express more tau protein than other non-immunopositive cells. However, this is unlikely, because the tau promoter regulates the expression and thus should be expressed close to the endogenous pattern. Moreover, if tau was highly expressed in a select group of cells, we would expect regional groupings of pathology, which is not the case. Instead, we favor the seeding model, in which a small amount of mutated tau protein that has adopted a unique shape can seed the growth of additional aggregates that also possess this conformation. This nucleation seeding hypothesis is well known to function in tauopathy, prion disease, and amyloid β aggregates (Vitek et al., 1994; King et al., 1999; Gamblin et al., 2000, 2003b). Makrides et al. (2003) showed that microtubules initiate oligomerization of tau protein, which occurs even at very low molar tau-to-tubulin ratios (1:200). Because 4R tau binds to microtubules threefold more tightly than 3R tau (Butner and Kirschner, 1991; Gustke et al., 1994; Goode et al., 2000; Panda et al., 2003; Levy et al., 2005), we theorize that this stronger microtubule–4R interaction disrupts the equilibrium of microtubule-bound tau and results in increased auto-oligomerization, which can then seed further accumulation of tau in the T-279 mice. Because the two control lines, T-WT, in which the human TAU gene promoter drives expression of wild-type tau, and C-279, in which the CMV promoter drives expression of the mutant tau, do not display any of the pathological features seen in the T-279 mice, it is likely that both the N279K mutation and the tau promoter are required to elicit tauopathy in these mice.
A large body of literature has shown that regulation of MT dynamics is essential for many neuronal functions, including axonal transport. The presence of tau-immunopositive degenerating neuronal processes throughout the brain strongly suggests that axonal transport is altered in the T-279 mice. Furthermore, the accumulation of TH immunoreactivity throughout axons of the substantia nigra suggests that axonal transport in dopaminergic neurons is particularly affected by this mutation. The disruption of dopamine synthesis, as suggested by the loss of TH-immunopositive neurons and the accumulation of TH-immunopositive material in the nigrostriatal pathway, may help to explain the parkinsonism that is present in all FTDP-17 patients. Similar results were previous reported in a tauopathy model based on direct mutated tau injection into the substantia nigra (Klein et al., 2005, 2006). The hypothesis that parkinsonism is a result of the disruption of the transport of the components of the dopamine synthesis pathway is especially attractive, because unlike patients with Parkinson's disease, who respond to l-DOPA treatment, the parkinsonism observed in FTDP-17 patients is mostly l-DOPA resistant. l-DOPA is a precursor of dopamine, and to restore function, the components of the dopaminergic pathway must be transported to the synapses.
Several in vitro reports have implicated important differences in how 4R and 3R tau interacts with microtubules. The N279K mutation increases the inclusion of exon 10 and hence increases the 4R-to-3R ratio in humans and mice. Thus, changes in the ratio of 4R to 3R tau caused by exon 10 splicing are likely to impact tau protein/microtubule interactions (Panda et al., 2003; Levy et al., 2005). For example, it has been proposed that the absence of exon 10 in fetal tau during development increases the plasticity of the microtubular network and facilitates structural reorganization. Consequently, elements that regulate splicing exon 10 are crucial for the proper maintenance of the 4R and 3R tau ratio, and thus mutations that alter putative exon 10 splicing elements may contribute over time to the development of tau pathology.
Our data show that C-279 mice do not demonstrate the fetal suppression of exon 10 observed in T-279 mice. Because the two tau constructs in the T-279 and C-279 mice differ only by their promoters, it is likely that the presence of the human tau promoter and/or additional five prime sequences in T-279 mice at least partly regulate the level of expression of exon 10 during development. It is possible that the insertion point of the transgenes could influence the activity of the various promoters; however, the regulation of alternative splicing by promoters has been previously reported for the ED1 exon of fibronectin (Cramer et al., 1997, 2001), exon 9 of the cystic fibrosis transmembrane regulator (Pagani et al., 2003), and the alternatively spliced exons of the CD44 gene (Auboeuf et al., 2002). Interestingly, as in FTDP-17, the incorrect expression of exon 9 of the cystic fibrosis transmembrane regulator is causal in the pathology of cystic fibrosis.
Distal intronic or gene flanking sequences in the TAU gene may also play a role in regulating exon 10 splicing during development. TTg mice that express tau from the entire human tau gene mimic the almost complete suppression of exon 10 in fetal brains, as seen in humans. However, exon 10 expression in mRNA from T-279 fetal mice is suppressed to only 50% of total tau mRNA expression. Similarly, the levels of exon 10 expression in mRNA from T-WT fetal mice also remain at 50% of total tau. Together, these data suggest that distal intronic and/or flanking regions that are in the TAU gene, but are missing from the tau minigene, also play a role in regulating exon 10 splicing.
Genetic studies found that a variant of the TAU gene called the H1 haplotype is associated with several polymorphisms and occurs with increased frequency in patients with PSP and CBD (Oliva et al., 1998; Baker et al., 1999; Ezquerra et al., 1999; Di et al., 2000; de Silva et al., 2001; Houlden et al., 2001; Liu et al., 2001; Pastor et al., 2002; Sobrido et al., 2003; Kwok et al., 2004; Pittman et al., 2004). Several of these polymorphisms are located in the human tau promoter and in intronic sequences, implicating misregulation of tau protein expression. The overabundance of 4R tau in these tauopathies suggests aberrant exon 10 splicing. Although the true mutations found in FTDP-17 are autosomal dominant, the polymorphisms associated with PSP and CBD may be weaker and therefore only predisposed to tauopathy.
Astrocytes perform several functions that are essential for normal neuronal activity, and accordingly, astrocyte function can critically influence neuronal survival. As with neurons, to be effective, astrocytic components need to be transported to the fine distal processes by the microtubular network. One pathological feature of N279K FTDP-17 patients and T-279 mice is the accumulation of tau protein in astrocytes. The accumulation of tau in astrocytic processes may disrupt transport in astrocytes and lead to astrocytic dysfunction. Previous experiments have shown that the overexpression of tau protein in primary rat astrocytes caused a clustering of kinesin, a microtubule transport molecule (Yoshiyama et al., 2003). Furthermore, a recent report by Forman et al. (2005) showed that transgenic mice that overexpress tau protein in astrocytes alone develop astrocytic pathology in the form of tufted astrocytes and are associated with focal neuronal injury. Neurons already compromised by N279K mutated tau expression may be especially vulnerable to the breakdown in the astrocytic network.
Caspases are cysteine aspartate proteases that are critically involved in the apoptosis signaling pathways. Activation of caspase-3 in both neurons and astrocytes of T-279 mice implies that neurodegeneration occurs through the apoptotic pathway. A possible link between activated caspase-3 and the pathogenesis of tauopathies was first reported in neurons from AD brains (Fasulo et al., 2000; Berry et al., 2003; Gamblin et al., 2003a; Rissman et al., 2004; Cotman et al., 2005; Newman et al., 2005) and is now further supported by the presence of activated caspase-3 in the T-279 mice.
An especially attractive aspect of this model, supported by our data implicating a regulatory role for the tau promoter and distal noncoding sequences in exon 10 expression, is that N279K FTDP-17 is a member of a larger group of tauopathies that result from the misregulation of exon 10 splicing. Specifically, this group of diseases may include PSP, CBD, argyrophilic grain disease, and other FTDP-17 diseases resulting from exon 10 splicing mutations (Mailliot et al., 1998; Sergeant et al., 1999; Arai et al., 2001; Liu et al., 2001; Togo et al., 2002; Katsuse et al., 2003; Morris et al., 2003). These tauopathies all have similar characteristics of astrocytic and neuronal tau accumulation, predominantly 4R insoluble tau and motor and cognitive deficits. However, most importantly, the T-279 mouse provides a new and unique tool to further our understanding of the role of aberrant exon 10 splicing in neurodegeneration.
Footnotes
-
This work was supported in part by Alzheimer's Association Grants 199-PRG-1709 and IIRG-02-4160 and by National Institutes of Health Grant 5R01-AG19780-05 to M.P.V. We thank the Duke University Mouse Transgenic Facility for the pronuclear microinjection of DNA constructs. We also thank Drs. Ellen Bennett and Carol Colton for their reading of this manuscript and their suggestions.
- Correspondence should be addressed to Hana N. Dawson, Division of Neurology, Duke University, P.O. Box 2900, Durham, NC 27710. dawso009{at}mc.duke.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}