

Abstract
Prion-related protein (PrP) is a neural cell adhesion molecule involved in neurite outgrowth, neuronal survival, and synaptic function. In search of novel binding partners for PrP, we identified the α2/β2-Na+/K+-ATPase and showed that this astroglial ATPase interacts directly with the immunoglobulin superfamily adhesion molecule basigin. In cultured astrocytes, PrP is involved in regulating lactate transport via the astroglial monocarboxylate transporter 1 (MCT1) and in conjunction with α2/β2-ATPase and basigin. Lactate transport via MCT1 is glutamate dependent and regulated by glutamate receptor 2 (GluR2)-containing AMPA receptors with which PrP interacts. The functional interplay between PrP, GluR2, α2/β2-ATPase, basigin, and MCT1 in regulating lactate transport of astrocytes may be functional in the metabolic cross talk between astrocytes and neurons, most likely under stress.
Introduction
The cellular, nonpathogenic form of prion-related protein (PrP) is a glycosylphosphatidylinositol-linked cell surface glycoprotein that is ubiquitously expressed by most, if not all, cell types in mammals, but most prominently in the brain. PrP functions as a recognition molecule and associates with the 67 and 37 kDa laminin receptors, the extracellular matrix glycoprotein laminin, and NCAM (neural cell adhesion molecule) (for references, see Santuccione et al., 2005). PrP mediates neurite outgrowth and synaptic function (Collinge, 1997; Kanaani et al., 2005; Re et al., 2006). PrP has also been implicated in neuronal survival and neural protection, most likely because of its metal ion binding ability (for review, see Milhavet and Lehmann, 2002).
In prion diseases, PrP is converted to a conformationally altered form that accumulates in the brain (for review, see Prusiner, 1998; Weissmann and Flechsig, 2003) and mutations in the PrP gene have been linked to the Gerstmann–Straussler–Scheinker syndrome, familial fatal insomnia, familial Creutzfeldt–Jakob disease, and certain forms of dementia with cerebellar disorder (Collinge, 1997). Because of the importance of PrP in normal and pathological brain functions, it appeared important to gain more insights into the cellular functions of PrP in the nervous system by identifying molecules interacting with PrP. Here, we identified the astroglial α2/β2-Na+/K+-ATPase (Peng et al., 1997) as a direct PrP interaction partner. In addition, we showed that this astroglial Na+/K+-ATPase interacts with the immunoglobulin superfamily cell adhesion molecule basigin, which was identified as an oligomannose binding protein (Heller et al., 2003).
Basigin interacts tightly with monocarboxylate transporter 1 (MCT1) and MCT4 (Wilson et al., 2005), which are predominantly expressed by astrocytes (Kirk et al., 2000; Pellerin et al., 2005). Monocarboxylate transporters allow the bidirectional transport of lactate that is from the extracellular space to the cytoplasm and vice versa depending on the lactate concentrations inside and outside of the cell. Basigin plays a role as a chaperone in translocation of MCT1 and MCT4 to the plasma membrane and is required as ancillary protein to maintain the catalytic activity of the lactate transporters (Wilson et al., 2005). These proton-coupled transporters of lactate mediate the release of lactate from astrocytes and thus are important in supplying neurons with lactate, particularly during periods of high neuronal activity, and thus protect neurons from glucose deprivation or hypoxic episodes (Schurr et al., 1997). The release of lactate from astrocytes is enhanced by the neurotransmitter glutamate, which is released from excitatory neurons during nervous system activity (Pellerin and Magistretti, 1994; Pellerin et al., 2005). The present study was aimed at the characterization of the role of PrP in regulating Na+/K+-ATPase activity and basigin-associated lactate transport using cultured astrocytes from wild-type and PrP-deficient mice. We show that PrP regulates Na+/K+-ATPase activity and glutamate-triggered lactate transport, which depends on the AMPA receptor subunit glutamate receptor 2 (GluR2) and the lactate transporter-associated immunoglobulin superfamily molecule basigin, which binds to oligomannosidic glycans carried by the astroglial β2 subunit of the glial Na+/K+-ATPase.
Materials and Methods
Animals.
Wild-type and homozygous PrP mutant mice used for this study were littermates from heterozygous breeding pairs of PrP-deficient mice (Bueler et al., 1992) backcrossed onto the C57BL/6J background for at least eight generations.
Antibodies and reagents.
Rat monoclonal antibody A2D2 against mouse basigin (Heller et al., 2003), rat oligomannose-reactive monoclonal antibodies L3 and L4 (Schmitz et al., 1993), and rat monoclonal antibody 426 against adhesion molecule on glia (AMOG) (Antonicek et al., 1987) (mouse β2-ATPase subunit) were described previously. Polyclonal antibodies against basigin (also called EMMPRIN) (T-18), PrP (M-20), α2 Na+/K+-ATPase (C-16), and GluR2 (N-19) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal PrP antibodies SAF34 and SAF61 directed against amino acids 79–92 and 142–160, respectively, have been described previously (Demart et al., 1999). Polyclonal antibodies against L1 have also been described (Chen et al., 1999). Antibodies against synaptophysin and glial fibrillary acidic protein (GFAP), cell culture media, and poly-l-lysine were obtained from Sigma (Taufkirchen, Germany), and 14C-lactate was supplied by Amersham Biosciences (Freiburg, Germany). Secondary and control antibodies (nonimmune IgG from goat, rat, or mouse) were from Jackson ImmunoResearch (Suffolk, UK). p-Chloromercuribenzene/sulfonic acid (pCMBS) was purchased from Toronto Research Chemicals (North York, Ontario, Canada). The production and purification of PrP-Fc, CHL1-Fc, L1-Fc, and basigin-Fc were performed as described previously (Chen et al., 1999, 2003). 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzoquinoxaline-7-sulfonamide (NBQX), d-(−)-2-amino-5-phosphonopentanoic acid (AP5), 1-trimethylammonio-5-(1-adamantane-methylammoniopentane)dibromide (IEM-1460), and l-trans-pyrrolidine-2,4-dicarboxylic acid (PDC) were from Tocris (Bristol, UK); pentobarbital was from Merial (Hallbergmoos, Germany); and ouabain was from Sigma. Oligomannosidic glycans were isolated by treatment of RNase B (Sigma) with endoglycosidase H (Sigma) and by collecting the glycans by Centricon centrifugal filter devices (Millipore, Schwalbach, Germany). The synthetic peptide NH2-CAAAGTIQTSVQEVNSKTQLTC-OH comprising amino acids 5–22 of mouse basigin was synthesized by Schafer-N (Copenhagen, Denmark). Na+/K+-ATPase purified from bovine cerebral cortex was purchased from Sigma.
Preparation of brain homogenates and subfractions.
Brains from 2- to 3-month-old C57BL/6J mice were homogenized in homogenization buffer (0.32 m sucrose, 50 mm Tris/HCl, pH 7.4, 1 mm CaCl2, 1 mm MgCl2). All procedures were performed at 4°C. The homogenate was centrifuged for 10 min at 1000 × g, and the resulting supernatant was further centrifuged at 17,000 × g. The 1000 and 17,000 × g pellets were resuspended in homogenization buffer and applied for a 1.2, 1.0, and 0.8 m sucrose step gradient and centrifuged for 2 h at 100,000 × g. The material from the 1.0/1.2 m sucrose interface was collected, diluted, and pelleted by centrifugation for 20 min at 17,000 × g. The material deriving from the 17,000 × g pellet contained no GFAP, whereas that from the 1000 × g pellet contained high levels of GFAP.
Cross-linking using sulfo-N-hydroxysuccinimidyl-2-(6-[biotinamido]-2-(p-azido benzamido)-hexanoamido)ethyl-1,3′-dithioproprionate.
A total of 0.2 mg of sulfo-N-hydroxysuccinimidyl-2-(6-[biotinamido]-2-(p-azido benzamido)-hexanoamido)ethyl-1,3′-dithioproprionate (sulfo-SBED) (Perbio, Rockford, IL) dissolved in DMSO was added to 1 mg of PrP-Fc resuspended in 500 μl of PBS and incubated for 30 min at room temperature. The sample was dialyzed using a Slide-a-lyzer mini dialysis unit (Perbio), added to 1 ml of protein A-coupled magnetic beads (Dynal Biotech, Oslo, Norway), and incubated for 1 h at room temperature. After addition of 2 mg of BS3 (Perbio) dissolved in PBS and an additional incubation for 1 h at room temperature, the beads were washed six times with TBS, pH 7.4, using a magnet. Beads were then incubated with synaptosomal subfractions for 1 h at room temperature. For preparative isolation of binding partners, the coupled beads were incubated with 2 ml of GFAP-enriched synaptosomal fraction prepared from 10 adult mouse brains. The samples were transferred to a 40 × 10 mm cell culture dish, placed on ice, and exposed to UV light for 15 min using an ultraviolet cross-linker (Amersham Biosciences) with energy setting of 365. Beads were washed successively with PBS containing 1% Triton X-100, and 0.2, 0.5, and 1% SDS. Bound proteins were eluted by boiling the beads in SDS sample buffer (80 mm Tris/HCl, pH 6.8, 10% glycerol, 1% SDS) with 1% dithiothreitol. After removing the beads, samples were subjected to silver staining or to Western blot analysis using horseradish peroxidase-conjugated streptavidin (Perbio) at a 1:20,000 dilution.
Primary cell cultures.
For cultures of astrocytes, brains of 1- to 2-d-old C57BL/6J mice were stripped of meninges, dissected free from brainstem, and cut into small pieces that were subjected to mechanical dissociation by several passages through a Pasteur pipette and successive passage through 200, 150, and 30 μm nylon mesh filters (VWR, Darmstadt, Germany). Poly-l-lysine (0.1 mg/ml) in water was used for coating plastic dishes. Cells were kept for 7 d at 37°C in a water-saturated atmosphere containing 5% CO2 by replacing the medium every 2–3 d. Cells were seeded on plastic 35 mm dishes or 12-well plates (Greiner BioOne, Frickenhausen, Germany) at a density of 105/cm2 in DMEM medium supplemented with 10% FCS, 0.6% glucose, 25 mm NaHCO3, 200 nm glutamine, and antibiotics (50 U/ml penicillin, 50 μg/ml streptomycin, and 50 μg/ml gentamicin).
Cerebellar neurons were prepared as described previously (Chen et al., 1999). After 1 d in culture, cytosine-β-arabinofuranoside was applied to inhibit astrocytic proliferation and the culture was maintained for an additional 6 d. For analysis of the protein expression patterns, cultured astrocytes or cerebellar neurons were lysed with SDS sample buffer with or without 0.5% dithiothreitol and subjected to Western blot analysis.
SDS-polyacrylamide gel electrophoresis and Western blot analysis.
Samples were boiled in SDS sample buffer without (monoclonal β2-ATPase antibody 426 is only reactive under nonreducing conditions) and with 5% mercaptoethanol or 0.5% dithiothreitol and subjected SDS-PAGE using 10 or 12% polyacrylamide gels. Proteins were then transferred onto nitrocellulose membranes (Schleicher and Schüll, Dassel, Germany), which were then incubated for 1 h at room temperature in blocking buffer consisting of PBS, pH 7.4, and 5% skim milk powder, washed three times, and incubated overnight at 4°C with primary antibodies. After washing three times with PBS containing 0.05% Tween 20 (PBST), filters were incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies (Dianova, Hamburg, Germany) in blocking buffer. After five washes with PBST, enhanced chemiluminescence detection was performed using SuperSignal (Perbio).
Immunoaffinity chromatography, immunoprecipitation, and pull-down.
Immunoaffinity purification of basigin was performed as described previously (Heller et al., 2003). In pull-down assays, PrP-Fc, basigin-Fc, or L1-Fc was incubated with purified ATPase or brain homogenate in RIPA buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% NP-40, and 1 mm Na2PO7) for 60 min at room temperature. Protein A magnetic beads (Dynal Biotech) were added and incubated overnight. For immunoprecipitation, polyclonal PrP antibodies were immobilized onto protein G-coupled magnetic beads (Dynal Biotech) by the cross-linker BS3 (Perbio). Beads were incubated overnight with brain homogenate or with astrocytes scraped off the culture dish, all solubilized in RIPA buffer. Alternatively, after preclearing using Protein A/G Agarose Plus (Santa Cruz), solubilized homogenate or astrocytes were incubated with antibodies for 2 h. Protein A/G Agarose Plus was then added and incubated overnight. After washing the beads with RIPA buffer, they were boiled in SDS sample buffer with or without dithiothreitol and subjected to Western blot analysis.
Mass spectrometry.
After SDS-PAGE, the gel was stained with colloidal Coomassie blue, and protein bands were cut out. After successive treatment with dithiothreitol and iodoacetamide, in-gel digestion of proteins by 5 ng/μl trypsin (Promega, Mannheim, Germany) in 50 mm NH4HCO3 was performed overnight at 37°C. Gel pieces were then repeatedly extracted with 50% acetonitrile/5% formic acid, and the combined extracts were dried down in a vacuum concentrator, redissolved in 5% methanol/5% formic acid, desalted on a C18 μZipTip (Millipore), eluted with 1 μl of 60% methanol/5% formic acid, and analyzed by nanoelectrospray mass spectrometry in a QTOF II instrument (Micromass, Manchester, UK). The MS/MS spectra obtained by collision-induced fragmentation of the peptides were evaluated both manually and by the Mascot MS/MS ion search algorithm (Matrix Sciences, London, UK).
Electrophysiological techniques.
Membrane potentials were recorded from cultured astrocytes. Pipettes were pulled from borosilicate glass (GB 150F-8P; Science Products, Hofheim, Germany) with a DMZ-Universal Puller (Zeitz, Augsburg, Germany). Pipette resistances were between 4 and 8 MΩ when filled with intracellular solution (in mm): 140 KCl, 2 MgCl2, 5 EGTA, 0.5 CaCl2, 10 HEPES, pH 7.2 with KOH. The extracellular bath solution contained the following (in mm): 140 NaCl, 2 MgCl2, 2.8 KCl, 1 CaCl2, 10 HEPES, pH 7.2 with NaOH. Membrane potentials were recorded in patch-clamp experiments immediately after establishing the whole-cell configuration in current-clamp mode. Twenty sweeps of 500 ms duration with 3 s sweep interval were averaged. Potentials were recorded with an EPC-9 patch-clamp amplifier (HEKA, Lambrecht, Germany). Data were visualized on a Zeiss (Oberkochen, Germany) Axiovert 135 TV, stored, and processed with PULSE software.
86Rb uptake.
Astrocyte cultures were incubated for 2 h in DMEM-5 medium: serum- and pyruvate-free DMEM (Sigma; D5030) supplemented with 5 mm glucose and 44 mm NaHCO3. The medium was then replaced by 1 ml of DMEM-5 without or with 50 μg of PrP antibodies SAF34 or SAF61 and incubated for 20 min at 37°C. A total of 0.5 ml of DMEM-5 without or with 800 μm l-glutamate or 4 mm ouabain was added followed by the immediate addition of 0.5 ml of DMEM-5 containing 4 μCi of 86RbCl. Cultures were placed on ice, washed three times with 1 ml of cold washing buffer (22 mm mannitol, 150 mm choline chloride, 1.2 mm CaCl2, 1.2 mm MgCl2, 2 mm BaCl2, 5 mm HEPES, pH 7.4), and finally lysed by adding 1 ml of 0.01 m NaOH/0.1% Triton X-100. Aliquots were assayed by liquid scintillation counting in 2 ml of Opti-Phase HiSafe 3 (PerkinElmer, Turku, Finland), and 50 μl aliquots were used for determination of protein concentration using the BCA reagent (Perbio).
Release of lactate.
Cultured astrocytes were incubated in 1 ml of DMEM-5 medium for 2 h at 37°C. The medium was then replaced by 1 ml of DMEM-5 without or with 200 μm l-glutamate and 25 μg of SAF34, SAF61, or A2D2 antibody or 100 μm NBQX, AP5, pentobarbital, PDC, or pCMBS. After 1 h incubation, cell culture supernatants were collected and assayed for lactate using a lactate determination reagent (Trinity Biotech, Darmstadt, Germany). Cells were lysed in 0.1 m NaOH/0.1% Triton X-100, and protein concentrations were determined.
14C-lactate uptake.
Cultured astrocytes were washed three times with 1 ml of EBSS buffer (in mm: 150 NaCl, 3 KCl, 0.6 MgCl2, 1.7 KH2PO4, 8 K2HPO4, 1 CaCl2, pH 7.4). A total of 1.4 ml of EBSS buffer without or with 50 μg of SAF34, A2D2, L3, or L4 antibodies, 250 μm pCMBS, or 200 μg of synthetic peptide comprising the amino acids 5–22 of mouse basigin was added to the cultures and incubated with the cultured cells for 20 min at 37°C. A total of 0.2 ml of EBSS buffer containing 50 μm l-cycloserine and fluoroacetate was added and incubated for 5 min to inhibit lactate metabolism. A total of 0.2 ml of EBSS buffer without or with 500 μm glutamate and 0.2 ml of EBSS buffer containing 1 μCi of 14C-labeled lactate and 5 mm unlabeled lactate were added and the samples were incubated for 4 min. Alternatively, cultures were incubated in 2 ml of EBSS buffer for 30 min. After replacing the EBSS buffer by 1.6 ml of EBSS buffer supplemented with 50 μm l-cycloserine and fluoroacetate, the cultures were incubated for 5 min. A total of 0.2 ml of EBSS buffer without or with 500 μm glutamate and 2 mm NBQX, AP5, pentobarbital, IEM-1460, PDC, or ouabain, and 0.2 ml of EBSS buffer containing 1 μCi of 14C-labeled lactate, and 5 mm unlabeled lactate were added. The samples were incubated for 4 min. For analysis of uptake kinetics, incubation was done for 0.5, 1, 2, 3, 4, 5, and 6 min.
All samples were placed on ice, washed three times with 1 ml of EBSS buffer and lysed with 0.1 m NaOH. Aliquots were assayed by liquid scintillation counting, and protein concentrations were determined.
Collection of CSF.
CSF was isolated from the cisterna magna compartment of anesthetized 2- to 3-month-old mice. The tissue above the cisterna magna was removed to expose the meninges overlying the cisterna magna under a dissecting microscope. Residual blood or interstitial fluid was removed and a small glass needle was used to penetrate the arachnoid membrane covering the cisterna and to collect 5–10 μl of CSF.
Statistical analysis.
Data were analyzed by Student's t test (two-tailed distribution; two-sample equal variance).
Results
PrP interacts with the α2/β2 Na+/K+-ATPase
To identify interaction partners of PrP, a biochemical cross-linking approach was taken that allows detection of directly bound molecules. PrP-Fc was conjugated to the trifunctional biotin-carrying cross-linker sulfo-SBED and subsequently immobilized to protein A beads. Two different synaptosomal fractions isolated from brains of adult mice, both containing the neuronal marker protein synaptophysin, with one fraction containing high levels of the astrocytic marker GFAP and the other fraction containing no detectable levels of GFAP were prepared by differential centrifugation (Fig. 1A). They were added to the immobilized PrP-Fc/sulfo-SBED conjugate. Bound proteins were cross-linked by exposing the samples to UV light, separated under reducing conditions from the PrP-Fc bait with concomitant transfer of the biotin label to the cross-linked proteins. Biotinylated binding partners were detected by Western blot analysis using peroxidase-conjugated streptavidin. In the synaptosomal fraction with no detectable levels of GFAP, only diffuse background staining but no distinct streptavidin-positive proteins were detectable (Fig. 1B, lane 1). However, in the synaptosomal fraction highly enriched in GFAP, streptavidin-positive protein bands with apparent molecular masses of 90 and 150 kDa, and ranging between 45 and 60 kDa were detected (Fig. 1B, lane 2). For control, the GFAP-enriched synaptosomal fraction was applied to protein A beads carrying the adhesion molecule close homolog of L1 linked to Fc (CHL1-Fc) and coupled to sulfo-SBED or to unconjugated beads. No streptavidin-positive bands were detectable (Fig. 1B, lanes 3 and 4).

Interaction between PrP and α2/β2-ATPase. A, Crude synaptosomal fractions (lanes 1 and 2) containing the synaptosomal marker protein synaptophysin (Synp) show either no detectable or high levels of the astrocyte marker protein GFAP. B, Biotinylated proteins were isolated from a synaptosomal fraction devoid of GFAP (lane 1) or highly enriched in GFAP (lanes 2–6) by chemical cross-linking using magnetic beads conjugated to PrP-Fc (lanes 1, 2, 5, and 6) or CHL1-Fc (lane 3), or unconjugated beads (lane 4). Proteins were subjected to Western blot analysis using peroxidase-conjugated streptavidin (lanes 1–4), to silver staining (lane 5), or to Western blot analysis using a polyclonal antibody to the α2-ATPase (lane 6). The arrowheads indicate the apparent molecular weights of the α2-ATPase. C, PrP-Fc and L1-Fc were incubated with brain homogenate or a purified Na+/K+-ATPase fraction and pulled-down by protein A magnetic beads. A Western blot using a monoclonal antibody against β2-ATPase is shown. D, Immunoprecipitation (IP) using immobilized polyclonal PrP antibody and brain homogenates from wild-type (+/+) and PrP-deficient (−/−) mice. The samples were analyzed by Western blotting using a polyclonal α2-ATPase or a monoclonal β2-ATPase antibody. E, Western blot analysis with polyclonal PrP antibody after immunoprecipitation (IP) using goat polyclonal α2-ATPase or mouse monoclonal β2-ATPase antibody and nonimmune control (ctrl) antibodies from goat or mouse and brain homogenate.
Proteins directly linked to the PrP-Fc/sulfo-SBED conjugate were isolated from the GFAP-enriched synaptosomal fraction and analyzed by silver staining. Intensely stained protein bands of ∼200, 150, 90, 75, and 52 kDa (Fig. 1B, lane 5) were subjected to sequence analysis by mass spectrometry. The 90 kDa band was identified as α2 subunit of the Na+/K+-ATPase, which is predominantly expressed by astrocytes (Peng et al., 1997). Western blot analysis confirmed the presence of the α2-ATPase in the 90 kDa band (Fig. 1B, lane 6). The α2-ATPase has been shown to copurify and, thus, to be associated with the β2 subunit of Na+/K+-ATPase, which is identical with AMOG (Antonicek et al., 1987; Gloor et al., 1990) and regulates the pump activity of the α2 subunit. To analyze whether PrP interacts with the β2-ATPase, we used a pull-down approach in which PrP-Fc was exposed to detergent-solubilized adult mouse brain homogenate or Na+/K+-ATPases purified from bovine brain. PrP-Fc, but not L1-Fc as control, pulled down β2-ATPase from brain homogenate and from a purified Na+/K+-ATPase preparation (Fig. 1C).
To further substantiate the interaction of PrP with the α2/β2-ATPase complex, coimmunoprecipitations using polyclonal PrP antibody and detergent extracts from adult mouse brain homogenates of either wild-type or PrP-deficient mice were carried out. Western blot analysis of the immunoprecipitates showed coisolation of PrP with both the α2- and β2-ATPase subunits from wild-type mice, but not from PrP-deficient mice (Fig. 1D). Immunoprecipitation using antibodies against β2-ATPase or α2-ATPase resulted in coimmunoprecipitation of PrP from wild-type brain homogenates, but not by control antibodies (Fig. 1E). The combined results indicate that PrP interacts with the α2/β2-ATPase.
The α2/β2 Na+/K+-ATPase interacts with basigin
The β2-ATPase, which is identical with the oligomannosidic glycan carrying AMOG, interacts with the immunoglobulin superfamily adhesion molecule basigin, which is a lectin for oligomannosidic glycans (Heller et al., 2003). Western blot analysis of the eluate from a basigin monoclonal antibody column (Heller et al., 2003) showed that β2-ATPase copurifies with basigin (Fig. 2A), both of which have almost the same apparent molecular weight of ∼45 kDa. On specific enzymatic removal of oligomannosidic glycans from β2-ATPase containing eluate with endoglycosidase H, the apparent molecular weight of β2-ATPase shifted from ∼45 to ∼30 kDa, whereas basigin was not affected in its apparent molecular weight (Fig. 2B). In a lectin blot of the β2-ATPase containing eluate with Galanthus nivalis agglutinin (GNA), which recognizes oligomannosidic glycans, only one glycoprotein of ∼45 kDa, which corresponds to the β2-ATPase, was detectable within a large number of brain glycoproteins reactive with GNA (Fig. 2B). After endoglycosidase H treatment of the β2-ATPase-containing eluate, no GNA-positive bands were seen (Fig. 2B). These results show that β2-ATPase is the predominant oligomannose-carrying glycoprotein in adult mouse brain and that β2-ATPase copurifies with basigin, indicating a tight association between these two molecules.

Interaction between basigin and α2/β2-ATPase. A, Brain homogenate (H) and an eluate (E) obtained from a monoclonal basigin antibody column were subjected to Western blot analysis using polyclonal basigin and a monoclonal β2-ATPase antibody. B, The eluate was treated without (lanes 1, 3, and 5) or with endoglycosidase H (lanes 2, 4, and 6) and subjected to Western blot analysis using a monoclonal β2-ATPase (lanes 1 and 2) or a polyclonal basigin (lanes 3 and 4) antibody and to a lectin blot using GNA (lanes 5 and 6). As reference for the lectin staining, brain homogenate was applied (lane 7). C, Basigin-Fc (bsg-Fc) and L1-Fc were incubated with brain homogenate or purified Na+/K+-ATPases and pulled-down by protein A magnetic beads. A Western blot with polyclonal antibody against α2-ATPase is shown. D, Immunoprecipitation using brain homogenate and a goat polyclonal basigin (bsg) or α2-ATPase antibody or a rat monoclonal β2-ATPase and the corresponding nonimmune control (ctrl) antibodies from goat or rat. Western blot (WB) analysis of the immunoprecipitates was performed using a polyclonal α2-ATPase or basigin antibody.
To investigate whether basigin is also associated with the α2-ATPase, we performed a pull-down assay using basigin-Fc or L1-Fc as control and brain homogenate or Na+/K+-ATPase purified from bovine brain. Basigin-Fc, but not L1-Fc, pulled-down α2-ATPase from detergent-solubilized mouse brain homogenate and from purified Na+/K+-ATPase (Fig. 2C). Furthermore, the α2-ATPase was detected by Western blot analysis in the immunoprecipitate with a monoclonal antibody to basigin. Immunoprecipitation with antibodies against α2-ATPase or β2-ATPase and subsequent Western blot analysis using a polyclonal basigin antibody showed that basigin coimmunoprecipitates both with α2- and β2-ATPase (Fig. 2D). The combined results indicate that basigin interacts with the α2/β2-ATPase.
PrP and basigin are expressed by cultured astrocytes and interact with the α2/β2-ATPase
To characterize a functional interaction between the associated molecules, we resorted to investigate glial cell cultures. It has been shown that the α2- and β2-ATPase are expressed by glial cells, in particular by astrocytes (Peng et al., 1997). Predominantly or even exclusively expressed in astrocytes are also the monocarboxylate transporters MCT1 and MCT4 (Debernardi et al., 2003), which interact with basigin and are regulated in their activity by basigin. Because α2/β2-ATPase interacts with basigin and PrP, PrP may be involved in the regulation of the glial α2/β2-ATPase pump activity and transport of lactate. Furthermore, α2-ATPase is involved in regulating the MCT-mediated release of lactate from cultured astrocytes (Pellerin et al., 1998). PrP may thus regulate lactate release from astrocytes via Na+/K+-ATPase and the basigin-associated MCT1 and/or MCT4 transporters. We therefore tested whether PrP influences ATPase pump activity and release of lactate in primary cultures of astrocytes isolated from early postnatal mouse brain. Beforehand, we checked whether PrP and basigin are expressed by these cells. Western blot analysis showed high levels of PrP and basigin as well as α2-ATPase and β2-ATPase as expected (Fig. 3A). Only MCT1, but not MCT4, was detectable in our astrocyte cultures (data not shown). Cerebellar neurons expressed low levels of basigin and PrP, and no detectable levels of α2- or β2-ATPase (Fig. 3A). To verify that the astrocyte cultures did not contain neurons, we checked for L1 as a neuronal marker and found that it was not detectable (Fig. 3A).

Expression and interaction of PrP and basigin with α2/β2-ATPase in cultured astrocytes. A, Astrocytes (A) and cerebellar neurons (N) from primary cell cultures were analyzed by Western blotting using polyclonal antibodies to basigin (bsg), PrP, α2-ATPase or L1, and monoclonal β2-ATPase antibody. B, Immunoprecipitation (IP) using immobilized polyclonal PrP antibody and lysate of cultured astrocytes from wild-type (+/+) and PrP-deficient (−/−) mice. The samples were analyzed by Western blotting using a polyclonal α2-ATPase or a monoclonal β2-ATPase antibody. C, Western blot analysis with polyclonal PrP antibody after immunoprecipitation using goat polyclonal α2-ATPase or mouse monoclonal β2-ATPase antibody and nonimmune control (ctrl) antibodies from goat or mouse and brain homogenate. D, Immunostaining of cultured astrocytes using monoclonal PrP antibody SAF61 or monoclonal basigin antibody and polyclonal α2-ATPase antibody.
Western blot analysis of immunoprecipitates of PrP from lysates of cultured astrocytes using antibodies against α2- and β2-ATPase showed coimmunoprecipitation of PrP with both ATPase subunits from wild-type astrocytes, but not from PrP-deficient astrocytes (Fig. 3B). Western blot analysis of immunoprecipitates of α2- and β2-ATPase from detergent lysates of wild-type astrocytes showed coimmunoprecipitation with basigin (Fig. 3C). In contrast, PrP did not coimmunoprecipitate with basigin, and vice versa, basigin did not coimmunoprecipitate with PrP under the conditions of this study (data not shown).
For indirect immunofluorescence analysis, cultured astrocytes were incubated with monoclonal antibodies against PrP or basigin, and, after fixation, an antibody against α2-ATPase was applied. PrP as well as basigin colocalized with α2-ATPase (Fig. 3D).
The combined observations indicate that basigin and PrP are associated with the α2/β2-ATPase not only in brain tissue but also in cultured astrocytes.
Astroglial Na+/K+-ATPase pump activity is regulated by PrP in cultured astrocytes
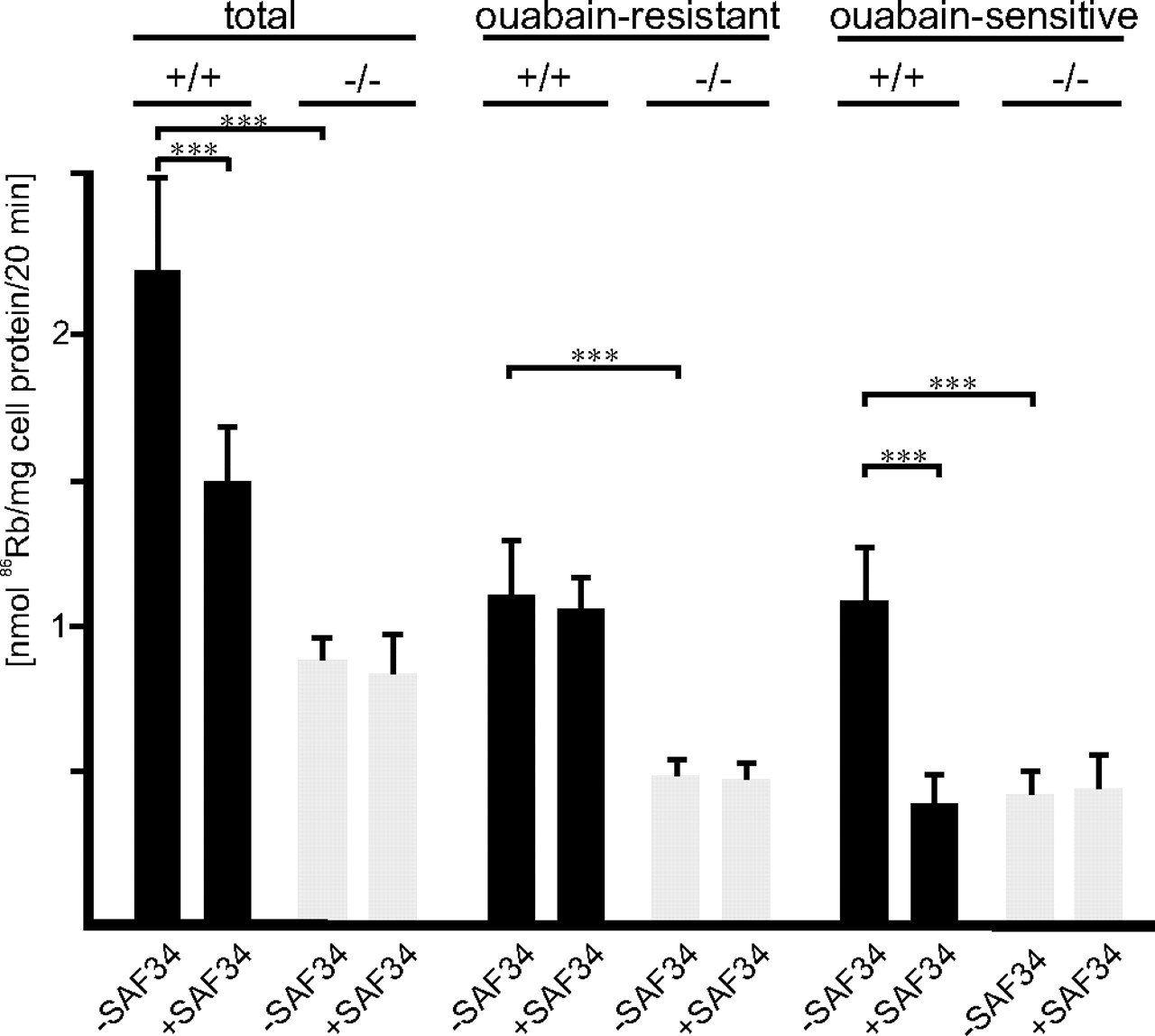
We next investigated whether the interaction between PrP and astroglial α2/β2-ATPase regulates Na+/K+ pump activity in cultured astrocytes. Uptake of the K+ analog 86Rb by astrocytes from PrP-deficient and wild-type mice was measured in the absence and presence of ouabain to determine total and ouabain-resistant and thus Na+/K+ pump-independent 86Rb uptake, respectively. Na+/K+-ATPase-dependent pump activity was then calculated as ouabain-sensitive 86Rb uptake from the difference between total and ouabain-resistant 86Rb uptake. In parallel, we investigated whether the monoclonal PrP antibodies SAF61 or SAF34 directed against two different domains of PrP interfered with Na+/K+ pump activity. Antibody SAF61 is directed against a membrane proximal epitope of PrP, and antibody SAF34 against an epitope in the amino-terminal octarepeat domain of PrP.
Total 86Rb uptake by PrP-deficient astrocytes was reduced to 40 ± 11% when compared with wild-type astrocytes (Fig. 4). In wild-type astrocytes, antibody SAF34 reduced total 86Rb uptake to 72 ± 14% when compared with the uptake in the absence of antibodies (Fig. 4), whereas antibody SAF61 did not affect total 86Rb uptake (data not shown). In contrast, total 86Rb uptake of PrP-deficient astrocytes was not affected in the presence of either antibody (Fig. 4), indicating that PrP is involved in K+ uptake into astrocytes. Ouabain-sensitive, Na+/K+-ATPase-dependent 86Rb uptake by PrP-deficient astrocytes was reduced to 37 ± 14% when compared with wild-type astrocytes (Fig. 4). In the presence of antibody SAF34, wild-type astrocytes showed a reduction of ouabain-sensitive 86Rb uptake to 29 ± 14% (Fig. 4) relative to that of wild-type astrocytes in the absence of antibody, indicating that ATPase pump activity depends on PrP and its octarepeat. Interestingly, ouabain-resistant 86Rb uptake into PrP-deficient astrocytes was reduced to 56 ± 13% relative to that of wild-type astrocytes (Fig. 4), suggesting that the lack of PrP leads to an impairment of a Na+/K+-ATPase-independent K+-transport or K+-uptake systems in astrocytes. The ouabain-resistant 86Rb uptake by wild-type astrocytes was not affected by antibody SAF34, indicating that the octarepeat of PrP is not involved in Na+/K+-ATPase-independent K+ uptake. The combined observations indicate that absence of PrP in PrP-deficient astrocytes or the binding of an antibody to the octarepeat domain within PrP leads to a reduction in astroglial Na+/K+-ATPase pump activity, suggesting that PrP is involved in the regulation of this pump. However, PrP is also involved in pump activity-independent K+ uptake, although not via its octarepeat.

PrP regulates Na+/K+ pump activity in cultured astrocytes. After preincubating wild-type (+/+) or PrP-deficient (−/−) astrocytes in the absence or presence of the PrP antibody SAF43, ouabain-resistant, ouabain-sensitive, and total 86Rb uptake were determined. Mean values ± SD are from at least three independent experiments. ***p < 0.0001.
Reduced Na+/K+ pump activity in PrP-deficient astrocytes does not affect the membrane potential
Reduced pump activity may lead to alterations of the membrane potential or could result from an altered membrane potential. We therefore measured the membrane potential of cultured astrocytes from wild-type and PrP-deficient mice. As reported for astrocytes in tissue slices (Matthias et al., 2003), we also observed two subpopulations of wild-type astrocytes with different membrane potential. One population had a membrane potential of 75.8 ± 6.8 mV, whereas the other population had a potential of 30.2 ± 11.2 mV. PrP-deficient astrocytes also consisted of two subpopulations with membrane potentials of 77.2 ± 7.7 and 31.1 ± 11.1 mV, respectively. Membrane potentials are thus not different in PrP-deficient versus wild-type astrocytes. These results exclude the possibility that reduced pump activity in PrP-deficient astrocytes alters the membrane potential and that a reduced membrane potential leads to a reduction in pump activity.
PrP regulates release of lactate from astrocytes
Because PrP regulates astroglial Na+/K+-ATPase activity, we investigated whether PrP also influences the release of lactate from astrocytes. This release is mediated by monocarboxylate transporters and is regulated by basigin (Wilson et al., 2005), which like PrP also interacts with the astroglial Na+/K+-ATPase. We therefore determined the release of lactate from wild-type and PrP-deficient astrocytes. PrP-deficient astrocytes showed ∼1.6-fold higher levels of lactate in the culture supernatant in comparison with wild-type astrocytes (Fig. 5A). Next, we tested whether the monoclonal PrP antibodies would interfere with the release of lactate. Levels of lactate in the presence of SAF61 antibody were comparable with those obtained in the absence of antibody (Fig. 5A). However, in the presence of antibody SAF34, lactate levels in cell culture supernatant were ∼1.9-fold higher in comparison with levels obtained in the absence of antibody (Fig. 5A), indicating that the octarepeat of PrP is involved in the release of lactate. The elevated release of lactate from astrocytes of PrP-deficient mice was not influenced by either antibody (Fig. 5A).

PrP regulates lactate release from cultured astrocytes. A, Astrocytes from wild-type (+/+) or PrP-deficient (−/−) mice were incubated in the absence (−ab) or presence of monoclonal PrP antibodies SAF61 or SAF43. B, Wild-type astrocytes (+/+) preincubated in the absence or presence of SAF43 or monoclonal basigin antibody A2D2 or PrP-deficient astrocytes (−/−) were incubated in the absence (−) or presence (+) of glutamate (glu). A, B, After incubation, lactate levels were determined in the cell culture supernatants. Mean values ± SD are from at least three independent experiments. ***p < 0.00001. C, CSFs taken from the cisterna magna of wild-type (n = 17) and PrP-deficient mice (n = 15) were assayed for lactate levels. Mean values ± SD are shown. ***p < 0.00001.
Because the release of lactate by cultured astrocytes is triggered by glutamate (Pellerin and Magistretti, 1994), levels of lactate were analyzed in the culture supernatants also after application of glutamate. Release of lactate from wild-type astrocytes was ∼1.5-fold higher after application of glutamate than in the absence of glutamate (Fig. 5B). Glutamate did not affect the enhanced release of lactate from PrP-deficient astrocytes (Fig. 5B). Also, the enhanced release from astrocytes of wild-type mice is not affected by antibody SAF34 (Fig. 5B). These observations indicate that the release of lactate is insensitive to stimulation by glutamate, when astrocytes are deficient in PrP or on ablation of its function by an antibody that binds to the octarepeat of PrP.
To further confirm that basigin is involved in regulating the release of lactate from astrocytes, we tested whether a monoclonal antibody against basigin would interfere with this release. In the presence of this antibody, levels of lactate were ∼1.5-fold higher compared with those obtained in the absence of antibody (Fig. 5B). Elevated levels of lactate induced by basigin antibody were not further elevated by glutamate (Fig. 5B). Thus, binding of basigin antibody leads to a glutamate-insensitive increase in the release of lactate. These results indicate that basigin is involved in regulation of glutamate-mediated release of lactate.
Because release of lactate is dysregulated in PrP-deficient astrocytes, we tested whether this release would be also altered in vivo by analyzing lactate levels in the CSF of PrP-deficient mice. CSF was collected from the cisterna magna of wild-type and PrP-deficient mice. Lactate levels in the CSF of wild-type mice were 1.11 ± 0.32 mm, whereas those of PrP-deficient mice were 2.03 ± 0.45 mm and thus significantly higher (Fig. 5C).
PrP and basigin regulate glutamate-dependent lactate uptake in cultured astrocytes
We investigated whether not only release but also uptake of lactate into astrocytes depended on PrP and basigin. Because our cultured astrocytes expressed only the lactate transporter MCT1 and lactate transport via MCT1 is bidirectional and not saturated at high lactate concentrations, we analyzed the uptake of radioactively labeled lactate via MCT1 into cultured astrocytes in the presence of high extracellular lactate concentrations. However, metabolism of lactate via the tricarboxylic acid cycle results in an inwardly directed diffusional transporter-independent flux of lactate (Dienel and Hertz, 2001). To measure only transporter-mediated lactate uptake, the contribution of fast diffusional metabolism-driven lactate uptake was excluded by inhibiting the metabolism of lactate using fluoroacetate, a potent inhibitor of the tricarboxylic acid cycle. In addition, cycloserine was added as an inhibitor of alanine aminotransferase, which catalyzes the conversion of pyruvate to alanine, thus excluding metabolism of lactate via pyruvate to alanine.
To test whether metabolism-driven uptake of lactate was indeed inhibited and whether the remaining uptake of lactate showed a linear transport rate, which is indicative for facilitated transporter-mediated lactate uptake, the kinetics of lactate uptake after preincubation and in the presence of the inhibitors was determined by measuring radiolabeled lactate uptake after 0.5, 1, 2, 3, 4, 5, and 6 min. First-order linear regression analysis of lactate uptake showed a linear correlation (r2 = 0.99) between uptake and time (Fig. 6A), indicating that the contribution of metabolism-driven uptake was abolished and that transporter-mediated facilitated lactate uptake could be measured, when metabolism of lactate was blocked. To analyze whether the transporter-mediated lactate uptake is stimulated by application of glutamate, the kinetics of lactate uptake into astrocytes in the presence of glutamate was determined. In the presence of glutamate, uptake of lactate showed a linear correlation (r2 = 0.99) with time and was ∼1.5-fold higher than in the absence of glutamate (Fig. 6A), indicating that transporter-mediated lactate uptake is enhanced by glutamate.
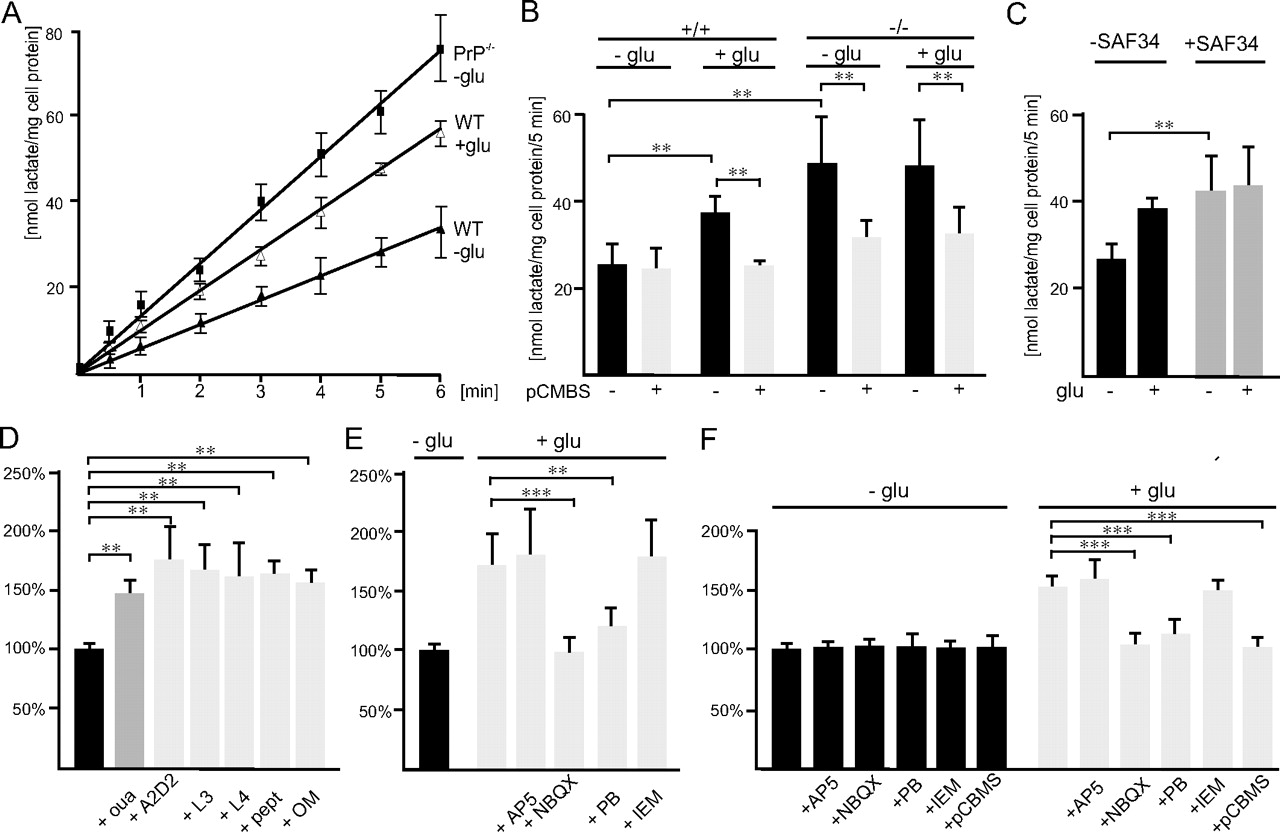
Lactate transport via MCT1 is regulated by PrP and GluR2-containing AMPA receptors in cultured astrocytes. A, Astrocytes from wild-type (WT) and PrP-deficient (−/−) mice were preincubated with fluoroacetate and cycloserine to inhibit the metabolism of lactate. Uptake of 14C-labeled lactate was then determined after 0.5, 1, 2, 3, 4, 5, and 6 min in the absence or presence of glutamate (glu). B, Astrocytes from wild-type (+/+) and PrP-deficient (−/−) mice were pretreated in medium without or with pCMBS. Uptake of 14C-labeled lactate was determined in the absence or presence of glutamate (glu). C, Uptake of 14C-labeled lactate into astrocytes from wild-type mice incubated in the absence or presence of SAF34 without or with glutamate (glu). D, Astrocytes from wild-type mice were incubated in the absence or presence of ouabain (oua), of the monoclonal basigin antibody A2D2 directed to the carbohydrate recognition domain of basigin, of the oligomannose-specific antibodies L3 and L4, of the synthetic peptide comprising the oligomannose binding site of basigin (pept), or of purified oligomannosidic glycans (OM). E, Astrocytes from wild-type mice were incubated in the absence or presence of glutamate without and with AP5, NBQX, pentobarbital (PB), or IEM-1460. B, C, Mean values ± SD of the uptake rates are shown and derived from at least three independent experiments. D, E, Uptake rates are given in percentage relative to the uptake rate in the absence of glutamate. F, Astrocytes from wild-type mice were incubated in the absence or presence of glutamate and AP5, NBQX, pentobarbital (PB), IEM-1460, or pCMBS. Mean values ± SD are from at least three independent experiments. **p < 0.0001; ***p < 0.00001.
To test whether the glutamate-stimulated uptake of lactate is mediated by MCT1, astrocytes were pretreated with pCMBS, a specific inhibitor of lactate transport via MCT1 by covalent modification of its ancillary protein basigin (Wilson et al., 2005). In the absence of glutamate, uptake of lactate was not altered by pretreatment with pCMBS (Fig. 6B), indicating that basal uptake of lactate is not mediated by MCT1. However, in the presence of glutamate, the elevated uptake of lactate was reduced by pCMBS to basal levels observed in the absence of glutamate (Fig. 6B). These findings indicate that the uptake of lactate via MCT1 is glutamate dependent.
We then investigated whether the uptake of lactate via MCT1 is also regulated by PrP and determined the uptake of lactate in PrP-deficient astrocytes in the absence and presence of glutamate and on pretreatment with pCMBS. Uptake of lactate in PrP-deficient astrocytes was approximately twofold higher when compared with wild-type astrocytes and was not further elevated by glutamate (Fig. 6B). Uptake was reduced to ∼35% after pCMBS pretreatment (Fig. 6B). This finding indicates that in PrP-deficient astrocytes uptake of lactate via MCT1 is dysregulated.
To ensure that only transporter-mediated uptake of lactate into PrP-deficient astrocytes and not metabolism of lactate is altered in the absence of PrP, the rate of lactate uptake into PrP-deficient astrocytes was determined. Uptake of lactate into PrP-deficient astrocytes was found to be linear with time and approximately twofold higher at 0.5, 1, 2, 3, 4, 5, and 6 min when compared with the uptake kinetics of wild-type astrocytes (Fig. 6A). The time course of lactate uptake in the presence of glutamate was nearly identical with that observed in the absence of glutamate (data not shown). These results show that enhanced lactate uptake by PrP-deficient astrocytes is indeed transporter mediated and that the elevated MCT1-mediated transport in the absence of PrP is glutamate independent.
Next, we analyzed the effect of antibody SAF34 on the uptake of lactate in wild-type astrocytes. In its presence, uptake of lactate increased approximately twofold and was not further increased by glutamate (Fig. 6C), indicating that uptake of lactate is dysregulated by binding of the antibody to the octarepeat domain of PrP.
To verify that ATPase pump activity is involved in regulation of lactate uptake, astrocytes were incubated in the presence of a low concentration of ouabain (1 μm) allowing specific inhibition of α2-ATPase activity. Uptake of lactate was increased ∼1.5-fold (Fig. 6D) by inhibition of α2-ATPase activity, suggesting that the Na+/K+ pump regulates lactate transport.
Basigin-mediated lactate uptake depends on oligomannosidic glycans
We previously showed that the monoclonal antibody against basigin binds to the oligomannose recognition domain within basigin (Heller et al., 2003). Because this antibody affects the release of lactate, we investigated whether binding of basigin to oligomannosidic glycans is involved in regulation of lactate uptake in astrocytes via basigin-associated MCT1. We therefore determined uptake of lactate in the presence of this monoclonal antibody. In addition, we tested antibodies against oligomannosidic glycans (Schmitz et al., 1993), a synthetic peptide that comprises the carbohydrate binding site within basigin (Heller et al., 2003) and purified oligomannosidic glycans. Interestingly, in all cases, uptake of lactate was increased ∼1.5-fold (Fig. 6D) and was not further elevated by application of glutamate (data not shown). These results indicate that interference with the interaction between basigin and oligomannose-carrying proteins, most likely the β2-ATPase, leads to glutamate-independent activation of lactate transport.
Basigin-dependent lactate transport depends on the AMPA receptor subunit GluR2
We could show that glutamate induces transport of lactate in cultured astrocytes via a basigin-associated lactate transporter. We next analyzed whether glutamate transporters are involved in this process by using PDC, an inhibitor of glutamate transporters. Inhibition of glutamate uptake by PDC did not affect the glutamate-dependent increase of lactate uptake (data not shown). We then tested the involvement of glutamate receptors by using the NMDA receptor antagonist AP5 and the AMPA receptor antagonist NBQX. AP5 also did not affect the glutamate-induced lactate uptake, whereas NBQX inhibited the glutamate-induced increase in lactate uptake (Fig. 6E), but not the uptake in the absence of glutamate (data not shown). These results indicate that stimulation of AMPA receptors triggers the uptake of lactate.
To further characterize the regulation of lactate transport by AMPA receptors, uptake of lactate by astrocytes was measured in the presence of the inhibitors IEM-1460 or pentobarbital, which inhibit Ca2+-permeable GluR2-lacking AMPA receptors or Ca2+-impermeable GluR2-containing AMPA receptors, respectively (Essin et al., 2002). IEM-1460 did not affect the glutamate-induced increase in lactate uptake, whereas pentobarbital inhibited this increase (Fig. 6E), indicating that GluR2-containing AMPA receptors are involved in triggering lactate transport. Pentobarbital did not alter uptake of lactate in the absence of glutamate (data not shown).
We also tested whether the release of lactate showed the same characteristics as its uptake. Indeed, glutamate-induced release of lactate was not inhibited by AP5 or IEM-1460, but was inhibited by NBQX and pentobarbital as well as by pCMBS (Fig. 6F). These observations indicate that the glutamate-induced release of lactate via monocarboxylate transporters is regulated by GluR2-containing AMPA receptors.
PrP interacts with the AMPA receptor subunit GluR2
Because we could show that PrP regulates the transport of lactate, we investigated whether PrP interacts with GluR2, which needs to be activated for glutamate-induced transport of lactate via MCT1. A polyclonal PrP antibody coimmunoprecipitated GluR2 from brain homogenates of wild-type mice but not from those of PrP-deficient mice as shown in Western blot analysis of the immunoprecipitate (Fig. 7A). Furthermore, GluR2 was coimmunoprecipitated with PrP from lysates of cultured wild-type astrocytes by a polyclonal PrP antibody, but not by a control antibody (Fig. 7B). However, GluR2 was not found to coimmunoprecipitate with α2/β2-ATPase or with basigin (data not shown). Indirect immunofluorescence using antibody SAF61 and a polyclonal GluR2 antibody showed colocalization of PrP and GluR2 in cultured astrocytes (Fig. 7C). These results indicate that PrP and GluR2 are associated with each other.

AMPA receptor subunit GluR2 is associated with PrP. A, Immunoprecipitation (IP) using immobilized polyclonal PrP antibody and brain homogenates from wild-type (WT) and PrP-deficient mice (−/−) and Western blot analysis of the immunoprecipitates using polyclonal GluR2 antibody. B, Immunoprecipitation from lysate of cultured astrocytes (astro) using goat polyclonal PrP antibody or nonimmune goat control antibody (crtl) and Western blot analysis with polyclonal GluR2 antibody. C, Immunostaining of cultured astrocytes using monoclonal PrP antibody and polyclonal GluR2 antibody.
Discussion
In this study, we obtained indications that PrP, the AMPA receptor subunit GluR2, the astroglial α2/β2-ATPase, the adhesion molecule basigin, and the monocarboxylate transporter MCT1 functionally interact or form a functional complex in the plasma membrane of astrocytes (Fig. 8). Biochemical cross-linking, pull-down, coimmunoprecipitation or coimmunofluorescence experiments showed that PrP interacts directly with the α2/β2-ATPase. The β2 subunit of this enzyme, which is identical with AMOG, is a prominent carrier of oligomannosidic glycans, which mediate its binding to the immunoglobulin superfamily adhesion molecule basigin (Heller et al., 2003), which thus is an oligomannose binding lectin. Here, we observed that the β2 subunit is the only oligomannose-carrying protein that copurifies with basigin from mouse brain tissue, indicating a tight interaction between these molecules. Because PrP does not coimmunoprecipitate with basigin, it is likely that they do not directly interact with each other. However, coimmunoprecipitation and coimmunofluorescence experiments suggest a direct interaction of PrP with GluR2. A direct interaction of GluR2 with either α2/β2-ATPase or basigin is unlikely because we have not found them to coimmunoprecipitate. The tight and direct interaction between basigin and MCT1 is well documented (Kirk et al., 2000), and basigin is required for the trafficking and catalytic activity of MCT1 (Wilson et al., 2005). MCT1 mediates the proton-coupled bidirectional lactate flux in astrocytes and thus functions as a lactate transporter.

Schematic presentation of the functional interactions of GluR2, PrP, α2/β2-ATPase, basigin, and MCT. At the cell surface of astrocytes, PrP directly interacts with the α2 and β2 subunits of Na+/K+-ATPase and is associated with the GluR2 subunit of AMPA receptors. The oligomannosidic glycans carried by the β2-ATPase subunit interact with the adhesion molecule basigin, which also interacts with the α2-ATPase subunit. Basigin is tightly associated with the lactate transporter MCT1. In the absence of glutamate, MCT1 is in an inactive state and does not transport lactate. In the presence of glutamate, MCT1 is transformed to an active state, leading to an increase of release or uptake of lactate depending on the lactate concentration inside and outside of the astrocyte. Lack of PrP or ablation of PrP function by binding of an antibody (position A) reduces α2-ATPase pump activity and activates the basigin/MCT1-associated lactate transport. A similar activation is observed by disturbing the interaction of basigin with oligomannosidic glycans (position B) or by inhibiting α2-ATPase by low concentrations of ouabain (position C). Modification of basigin by pCMBS or application of AMPA receptor inhibitors blocks the basigin/MCT1-associated glutamate-dependent lactate transport. The functional interplay of GluR2, PrP, α2-ATPase, β2-ATPase, basigin, and MCT1 tightly regulates the transport of lactate in astrocytes.
Lactate is released predominantly from astrocytes and this release is enhanced during neuronal activity (e.g., in the presence of the excitatory neurotransmitter glutamate) (Pellerin and Magistretti, 1994; Pellerin et al., 1998). Lactate released from astrocytes is taken up by neurons and represents an important energy source or substrate for neurons (Pellerin and Magistretti, 1994; Pellerin et al., 2005), at least during high neuronal activity or under ischemic or hypoxic conditions (Chih et al., 2001; Dienel and Hertz, 2001). Neuronal activity leads to a twofold to threefold increase of lactate levels and under ischemic conditions extracellular lactate levels rise up to >20 mm, indicating that lactate plays a crucial role in the metabolism of the brain. In this study, we observed a 1.5- to 2-fold increase in lactate transport of astrocytes by application of glutamate. Furthermore, we showed that this glutamate-triggered transport of lactate is completely abolished by treatment of astrocytes by pCBMS, which specifically inhibits lactate transport via the basigin-associated monocarboxylate transporter MCT1. This result clearly shows that the glutamate-dependent transport of lactate in cultured astrocytes is mediated by MCT1. Inhibition of glutamate-dependent lactate transport by NBQX and pentobarbital, but not by IEM-1460, showed that lactate transport via MCT1 is triggered by GluR2-containing AMPA receptors. Basal lactate influx or efflux in the absence of glutamate does not appear to be mediated by MCT1, because it is not inhibited by pCMBS.
We obtained indications in cultured astrocytes that the functional interplay of GluR2, PrP, α2/β2-ATPase, and basigin regulates the transport of lactate via MCT1 (Fig. 8). This interplay maintains MCT1 in an inactive state in the absence of glutamate and facilitates the transition of MCT1 from an inactive to an active state in the presence of glutamate. Disturbance of the interplay at different sites within this molecular complex leads to a dysregulation of lactate transport that manifests itself in a nonphysiological activation of lactate transport. Binding of a monoclonal PrP antibody to the octarepeat of PrP leads to dysregulation of MCT1-mediated lactate transport, resulting in a glutamate-independent elevation of lactate transport. A monoclonal PrP antibody that binds to the first α-helix did not interfere with the transport of lactate. Thus, the octarepeat domain plays a crucial role in the regulation of lactate transport. Disturbing the oligomannose-mediated interaction of basigin with β2-ATPase by several inhibitors or competitors of the lectin-binding site in basigin also leads to a dysregulated and thus elevated glutamate-independent lactate transport. Furthermore, a similar dysregulated lactate transport was observed on specific inhibition of α2-ATPase activity by low concentrations of ouabain, suggesting that the Na+/K+ pump is involved in the transport of lactate. Because Na+/K+ pump activity is reduced in PrP-deficient astrocytes or by application of a PrP antibody against the octarepeat to wild-type astrocytes, it is likely that PrP regulates α2/β2-ATPase pump activity and thereby indirectly influences the transport of lactate via MCT1. This notion is supported by the finding that the reduced pump activity in PrP-deficient astrocytes did not alter the membrane potential. This is plausible, because the α2/β2-ATPase is not involved in maintaining the membrane potential of astrocytes by stabilization of Na+ and K+ gradients (Calabresi et al., 1995). Our observations favor a scheme in which the AMPA glutamate receptor is the initial and upstream trigger of a chain reaction that continues via its interaction with PrP, which in turn interacts with the α2/β2-ATPase that docks into basigin, which is associated with the lactate transporter MCT1. Other regulatory mechanisms are not excluded in our scheme, such as, for instance, a more indirect interaction of the AMPA receptor with MCT1 or an indirect regulation of MCT1 via Na+/K+-ATPase activity.
Absence of PrP in PrP-deficient astrocytes dysregulates the transport of lactate and leads to a glutamate-independent increase in lactate transport in vitro. Indication that PrP regulates the release of lactate also in vivo derives from the observation that lactate levels in the CSF of PrP-deficient mice are elevated approximately twofold in comparison with levels observed in the CSF of wild-type mice. Increased lactate levels were also found in the CSF of Creutzfeldt–Jakob-diseased patients (Awerbuch et al., 1988). In animals or humans suffering from prion disease, a conformationally altered PrP leads to a loss of normal PrP function. Because our present results imply that PrP regulates lactate transport by astrocytes, it is tempting to assume that loss of PrP function in astrocytes of prion-diseased brains may result in elevated levels of extracellular lactate and in lactate-induced acidosis and ensuing neuronal damage. Thus, reduction of PrP functions in the homoeostatic network of astrocyte–neuron interactions may play an aggravating role in the stressed or diseased brain and cause deficits in cellular communication as a function of altered cell surface recognition and concomitantly disturbed cellular metabolism.
Footnotes
-
This work was supported by Bundesministerium für Bildung und Forschung (01KO 0512, GZ GF/GFKO01068105). M.S. is New Jersey Professor of Spinal Cord Research. We are grateful to Ute Bork and Ute Eicke-Kohlmorgen for excellent technical assistance, Prof. U. Schumacher for help in collecting CSFs, Dr. Alexander Dityatev for helpful discussions, and Eva Kronberg for animal care.
- Correspondence should be addressed to Melitta Schachner, Zentrum für Molekulare Neurobiologie, Universität Hamburg, Martinistrasse 52, D-20246 Hamburg, Germany. melitta.schachner{at}zmnh.uni-hamburg.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}