

Abstract
A current hypothesis proposes that fragile X mental retardation protein (FMRP), an RNA-binding translational regulator, acts downstream of glutamatergic transmission, via metabotropic glutamate receptor (mGluR) Gq-dependent signaling, to modulate protein synthesis critical for trafficking ionotropic glutamate receptors (iGluRs) at synapses. However, direct evidence linking FMRP and mGluR function with iGluR synaptic expression is limited. In this study, we use the Drosophila fragile X model to test this hypothesis at the well characterized glutamatergic neuromuscular junction (NMJ). Two iGluR classes reside at this synapse, each containing common GluRIIC (III), IID and IIE subunits, and variable GluRIIA (A-class) or GluRIIB (B-class) subunits. In Drosophila fragile X mental retardation 1 (dfmr1) null mutants, A-class GluRs accumulate and B-class GluRs are lost, whereas total GluR levels do not change, resulting in a striking change in GluR subclass ratio at individual synapses. The sole Drosophila mGluR, DmGluRA, is also expressed at the NMJ. In dmGluRA null mutants, both iGluR classes increase, resulting in an increase in total synaptic GluR content at individual synapses. Targeted postsynaptic dmGluRA overexpression causes the exact opposite GluR phenotype to the dfmr1 null, confirming postsynaptic GluR subtype-specific regulation. In dfmr1; dmGluRA double null mutants, there is an additive increase in A-class GluRs, and a similar additive impact on B-class GluRs, toward normal levels in the double mutants. These results show that both dFMRP and DmGluRA differentially regulate the abundance of different GluR subclasses in a convergent mechanism within individual postsynaptic domains.
- mental retardation
- fragile X syndrome
- protein synthesis
- synapse
- postsynaptic
- metabotropic glutamate receptor
- synaptic plasticity
Introduction
Fragile X syndrome (FXS) is the leading known genetic cause of both mental retardation and autism spectrum disorders. The disease also produces hyperactivity, hypersensitivity to sensory stimuli, and epileptic seizures (O'Donnell and Warren, 2002; Bagni and Greenough, 2005; Garber et al., 2006). FXS is caused by silencing of the fragile X mental retardation 1 (fmr1) gene, which encodes the RNA-binding, translational regulator fragile X mental retardation protein (FMRP) (Jin and Warren, 2000; Brown et al., 2001; Li et al., 2001). In fmr1 knock-out mice, hippocampal long-term depression (LTD) dependent on activation of the group I class 5 metabotropic glutamate receptor (mGluR) is selectively enhanced (Huber et al., 2002; Koekkoek et al., 2005). This LTD is caused by loss of surface AMPA GluRs, a mechanism requiring protein synthesis (Huber et al., 2000, 2001; Nosyreva and Huber, 2006; Pfeiffer and Huber, 2006). Similarly, cerebral cortex long-term potentiation is reduced concomitant with reduced GluR1 expression (Li et al., 2002; Larson et al., 2005; Zhao et al., 2005). Two major FXS phenotypes in fmr1 knock-out mice, reduced habituation in open-field tests and increased sensitivity to audiogenic seizures can be rescued by the mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) (Yan et al., 2005).
FMRP is localized and translated in postsynaptic terminals, in a mechanism regulated by group I mGluRs, whose activation is a potent stimulus for synaptic protein synthesis (Weiler et al., 1997, 2004; Todd et al., 2003b). Both FMRP synaptic localization and expression are increased by glutamatergic activity, and this regulation is blocked by MPEP (mGluR5 antagonist) and increased by (RS)-3,5-dihydroxyphenylglycine (DHPG) (agonist) (Weiler et al., 1997, 2004; Antar et al., 2004; Hou et al., 2006). mGluR5-dependent LTD is normally completely dependent on new translation, but, in the absence of FMRP, mGluR5 activation does not trigger protein synthesis (Weiler et al., 2004; Aschrafi et al., 2005). The anisomycin (translation inhibitor) response is lost and the DHPG-induced increase of synaptic proteins synthesis is absent in fmr1 knock-out mice (Hou et al., 2006; Nosyreva and Huber, 2006). A primary target of mGluR5 activation is to regulate trafficking of AMPA GluRs (Carroll et al., 1999; Snyder et al., 2001; Xiao et al., 2001; Nosyreva and Huber, 2005). These data suggest a mechanistic connection between mGluR signaling, the translation regulatory function of FMRP, and AMPA GluR expression, a hypothesis termed “the mGluR theory of FXS” (Bear et al., 2004).
We developed a Drosophila FXS model through mutation and overexpression of Drosophila fmr1 (dfmr1) (Zhang et al., 2001; Pan et al., 2004; Zhang and Broadie, 2005). In support of the mGluR theory, feeding dfmr1 mutants with MPEP, as well as class II/III mGluR antagonists, can rescue some mutant phenotypes, including behavioral (courtship learning and memory) and morphological (mushroom body structure) defects (McBride et al., 2005). A great advantage for testing the mGluR theory in Drosophila is that the genome encodes only a single mGluR, DmGluRA (Parmentier et al., 1996; Bogdanik et al., 2004; Mitri et al., 2004). We characterized DmGluRA to show it is synaptically localized and regulates synaptic structure and activity-dependent function at the neuromuscular junction (NMJ) (Bogdanik et al., 2004). This glutamatergic NMJ contains five AMPA-like GluR subunits, GluRIIA–GluRIIE. Three subunits (GluRIIC, GluRIID, and GluRIIE) are essential for all GluRs, whereas the other two subunits define two GluR classes: A-class (GluRIIA) and B-class (GluRIIB), which are differentially regulated, subsynaptically localized, and possess distinct functional properties, including differential conductance and opening kinetics (Schuster et al., 1991; Petersen et al., 1997; DiAntonio et al., 1999; Sigrist et al., 2002; Marrus et al., 2004; Featherstone et al., 2005; M. Qin et al., 2005).
The purpose of this study was to genetically test the mGluR theory of FXS in the Drosophila model, specifically mechanistic links between dFMRP, DmGluRA, and ionotropic GluR expression. In dfmr1 null mutants, A-class GluRs accumulate and B-class GluRs are lost, significantly altering the GluR class ratio in single synapses. In dmGluRA null mutants, both GluR classes increase, showing that the metabotropic receptor negatively regulates both ionotropic receptors. Targeted rescue experiments demonstrate that dfmr1 defects are attributable to a postsynaptic requirement. Postsynaptic overexpression of DmGluRA causes the exact opposite GluR class-specific changes to the dfmr1 null. In dfmr1; dmGluRA double null mutants, there are additive effects on both GluR classes. These results show that DmGluRA glutamatergic signaling and dFMRP regulatory function converge to control the GluR class ratio in the postsynaptic domain, but that the two pathways are at least partially independent.
Materials and Methods
Drosophila genetics.
All Drosophila stocks were maintained at 25°C on standard food under standard conditions. The w1118; dfmr150M null mutant strain was used as the dfmr1 single mutant (Zhang et al., 2001), and w1118 was used as its genetic background control. The P-element imprecise excision dmGluRA112b null mutant was used as the dmGluRA single mutant, and dmGluRA2b (hereafter called 2b), a P-element precise excision line from the same screen, was used as its genetic background control (Bogdanik et al., 2004). For targeted overexpression studies, lines used included w; upstream activating sequence (UAS)–dfmr1 (Zhang et al., 2001; Pan et al., 2004), w; UAS–dmGluRA (Bogdanik et al., 2004), and the tissue-specific Gal4 drivers ELAV (embryonic lethal, abnormal vision, Drosophila)–Gal4 (neural specific) and myosin heavy chain (MHC)–Gal4 (muscle specific). The following strains were generated by standard genetic methods for this study: (1) the dfmr150M; dmGluRA112b double null mutant, (2) the dfmr150M; 2b genetic background control combination, (3) the ELAV–Gal4; dfmr150M, and (4) dfmr150M, UAS–dfmr1.
Immunohistochemistry.
Wandering third-instar larvae were dissected in Ca2+-free standard saline containing 2 mm l-glutamate, followed by ice-cold methanol fixation for 5 min. The monoclonal mouse antibody against GluRIIA (8B4D2, used at 1:20) was obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). The polyclonal rabbit antibodies against GluRIIB (used at 1:2000) (Marrus et al., 2004) and GluRIII/IIC (used at 1:2000) (Marrus et al., 2004) were a generous gift from Dr. Aaron DiAntonio (Washington University, St. Louis, MO). The specificity of these GluR subunit-specific antibodies has been rigorously demonstrated in Western blot analyses and genetic tests in subunit-specific null mutants (Yoshihara and Littleton, 2002; Marrus et al., 2004; Liebl et al., 2005; G. Qin et al., 2005). The monoclonal mouse antibody against NC82/bruchpilot (used as 1:200) was obtained from the Developmental Studies Hybridoma Bank (Wagh et al., 2006). All primary antibodies were visualized using fluorescent dye-conjugated secondary antibodies, including Alexa Fluor 488 goat anti-mouse IgG (1:200; Invitrogen, Carlsbad, CA), Alexa Fluor 488, 546, and 633 goat anti-rabbit IgG (1:200; Invitrogen), and cyanine 5- and Texas Red-conjugated anti-horseradish peroxidase (1:200; Jackson ImmunoResearch, West Grove, PA). All fluorescent images were collected using a Zeiss (Oberkochen, Germany) LSM 510 meta laser scanning confocal microscope and image-collection software. All image processing was done with Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA).
Fluorescent intensity quantification.
For any given experiment, animals of control and mutant genotypes were simultaneously processed, together in the same tube, and imaged using identical confocal settings. A neuronal membrane specific probe, anti-HRP, was used in all experiments to label the NMJ synapse and as an internal fluorescence staining control. All images used in fluorescent intensity quantification were three-dimensional projections from complete Z-stacks through the entire NMJ on the lateral, longitudinal muscle 4 in abdominal segment A3. Data from the two paired hemisegments were averaged for each animal, to produce each single data point. All images were analyzed using LSM 5 Confocal Image Examiner software in the “histogram” display mode. Synaptic regions were user defined with the closed free shape curve drawing tools, defined by the boundary of HRP staining. For quantification, multiple synaptic regions were defined, and the fluorescence intensity was calculated as an average of all regions. The software output reports fluorescence intensity for each fluorescent channel and the calculated area for each region. Thus, all reports are density measurements. In the direct ratio tests, green channel (GluRIIA) intensity was divided by red channel (GluRIIB) intensity to determine GluRIIA/GluRIIB ratio in the same synaptic regions. Statistical analysis was done using InStat 3 software (GraphPad Software, San Diego, CA). Significance levels in figures were represented as 0.001 < p < 0.05 (*); 0.0001 < p < 0.001 (**); p < 0.0001 (***). All error bars represent SEM, appropriate for comparison of the mean of means distribution.
Results
dFMRP differentially regulates GluR classes
Subunit-specific antibodies were used to examine three GluR populations at the NMJ: A-class receptors only (anti-GluRIIA), B-class receptors only (anti-GluRIIB), and the total GluR population (anti-GluRIIC) (Yoshihara and Littleton, 2002; Marrus et al., 2004; Liebl et al., 2005; G. Qin et al., 2005). Fluorescence intensity was compared with an internal fluorescent signal marking the NMJ synaptic terminal: anti-HRP labels a carbohydrate epitope in the neuronal membrane of NMJ boutons (Katz et al., 1988; Wang et al., 1994; Sun and Salvaterra, 1995). The dfmr1 null and w1118 genetic background control [wild-type (WT)] animals were processed simultaneously and imaged using identical microscope settings. All quantification was done on type IB synaptic boutons in the muscle 4 NMJ in segment A3. Fluorescence intensity was calculated at each NMJ terminal and then averaged between the two A3 hemisegments to generate each data point for each genotype. Representative images and quantification results are shown in Figure 1.

dFMRP differentially regulates A- and B-class GluRs. Third-instar NMJs were probed using subunit-specific anti-GluRs (green: IIA, IIB, IIC) and anti-HRP (red) antibodies. The HRP antibody recognized a neural membrane epitope and thus labels the NMJ synapse and acts as an internal fluorescence control for intensity quantification. Type IB boutons on muscle 4 in abdominal segment A3 were used for all quantified analysis. A–C, Representative images of GluRIIA, IIB, and IIC expression in WT (w1118) and dfmr1 null mutant (dfmr150M). D–F, Fluorescence intensity quantification of all three GluR subunits and the HRP internal standard. D, The GluRIIA subunit expression is significantly increased in dfmr1 mutants (n = 15 for both WT and dfmr1). E, The GluRIIB subunit expression is significantly decreased in dfmr1 mutants (n = 10 for both WT and dfmr1). F, The GluRIIC subunit expression is comparable in WT and dfmr1 mutants (n = 10 for both WT and dfmr1). **0.0001 < p < 0.001; ***p < 0.0001.
Synaptic A-class GluRs were very significantly increased in dfmr1 null mutants compared with matched controls (Fig. 1A). Average fluorescence intensity in NMJ synaptic boutons was increased by 30% in mutant terminals relative to controls (WT, 96.3 ± 5.03; dfmr1, 124.62 ± 4.84; n = 15 for each genotype; p < 0.0001) (Fig. 1D). There was no difference in HRP fluorescent intensities between mutants and controls (Fig. 1D). In contrast, B-class GluRs were very significantly decreased in dfmr1 null mutants compared with matched controls (Fig. 1B). GluRIIB levels were decreased to a similar level (23%) in dfmr1 null terminals relative to controls (WT, 129.3 ± 5.67; dfmr1, 99.62 ± 3.81; n = 10 for each genotype; p = 0.0004) (Fig. 1E). There was no difference in HRP fluorescent intensities between mutants and controls (Fig. 1E). Total GluR content at the same synaptic terminals was assayed using an antibody against the common GluRIIC subunit (Fig. 1C). Because the two GluR classes displayed similar opposing abundance changes, as might be predicted, there was no significant difference in total GluR abundance between dfmr1 and control synapses (WT, 108.94 ± 2.95; dfmr1, 105.34 ± 7.83; n = 10 for each genotype; p = 0.76) (Fig. 1F). These data show that the two GluR classes are differentially modulated in opposite directions in the absence of dFMRP function, with A-class GluRs increasing, B-class GluRs decreasing, but total GluR levels remaining normal.
All GluR classes decreased by dfmr1 overexpression in postsynaptic muscle
Neuronal overexpression of the dFMRP protein results in synaptic structural and functional defects that are primarily the inverse of dfmr1 null mutant phenotypes, in both the NMJ and in the CNS (Zhang et al., 2001; Pan et al., 2004). However, roles of dFMRP in the postsynaptic muscle have not been investigated. To begin to assay whether GluR phenotypes in dfmr1 null mutants arise through postsynaptic dysfunction, as predicted, and to determine whether GluR phenotypes are also inversely correlated in loss and gain-of-function conditions, GluR levels were next quantified in dFMRP overexpression (OE) mutants. The UAS-dfmr1 transgenic line was crossed with neuronal specific (ELAV-Gal4) and muscle specific (MHC–Gal4) drivers to induce dFMRP overexpression in either presynaptic (in neuron, NOE) or postsynaptic (in muscle, MOE) compartments (Fig. 2). As above, fluorescence intensities for all three GluR populations were compared relative to the internal HRP fluorescence control. Overexpression of dfmr1 in the muscle had no significant impact on the presynaptic HRP fluorescence in any of these experiments.
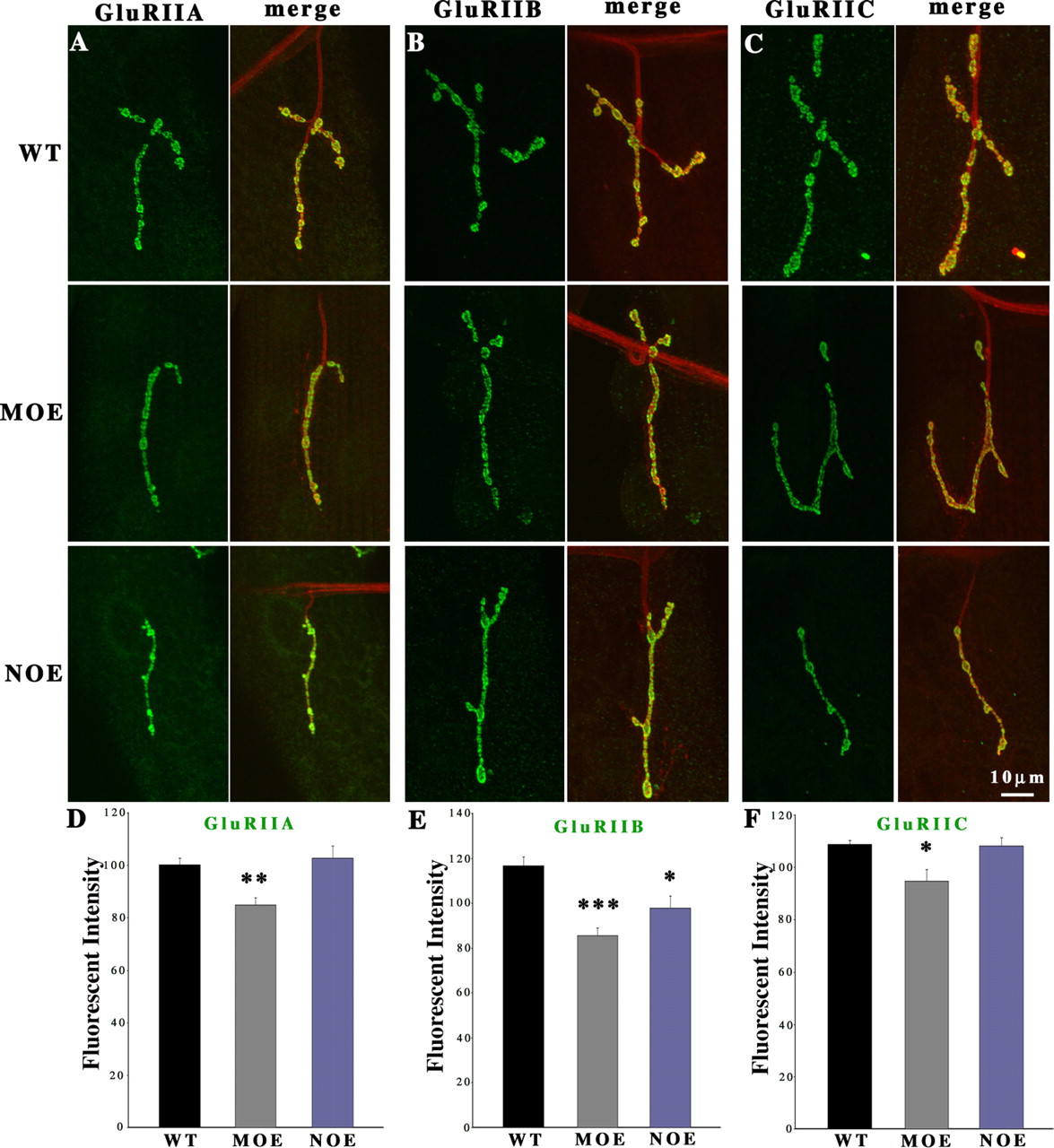
Total GluR abundance is decreased by dfmr1 overexpression in muscle. Third-instar NMJs were probed using subunit-specific anti-GluRs (green) and anti-HRP (red) antibodies. Type IB boutons on muscle 4 in A3 were used for all imaging analysis. A–C, Representative images of GluRIIA, IIB, and IIC expression in dFMRP muscle overexpression (MOE) and neuronal overexpression (NOE). D–F, Fluorescence intensity quantification for each GluR subunit. D, GluRIIA is decreased in dfmr1 MOE mutants but unchanged in dfmr1 NOE mutants (n = 11 for all genotypes). E, GluRIIB is decreased in both dfmr1 MOE and NOE mutants (n = 13 for all genotypes). F, The GluRIIC subunit is decreased in dfmr1 MOE mutants but not changed in dmfr1 NOE mutants (n = 10 for all genotypes). *0.001 < p < 0.05; **0.0001 < p < 0.001; ***p < 0.0001.
As predicted, dFMRP acts in the postsynaptic muscle to regulate the postsynaptic expression of A-class GluRs, and there is an inverse relationship between the increase observed in the dfmr1 null (Fig. 1A,D) and the decrease caused by dfmr1 muscle overexpression (MOE) in the postsynaptic cell (Fig. 2A,D). GluRIIA levels were decreased 15% compared with controls, a highly significant (p = 0.0008) reduction. Furthermore, as expected, neural overexpression (NOE) of dfmr1 in the presynaptic compartment caused no change in the abundance of the postsynaptic A-class GluRs (Fig. 2A,D). In contrast, the situation with the B-class GluRs was more complex. Overexpression of dfmr1 in the muscle caused a 27% decrease in GluRIIB expression (Fig. 2B,E), which resembled the decrease observed in the dfmr1 null synapses (Fig. 1B,E). Moreover, presynaptic dfmr1 overexpression also caused a smaller (15%), but still significant (p = 0.008), decrease in GluRIIB expression (Fig. 2B,E). The change in the total GluR population was primarily consistent with the combined changes of the two subclasses. The total GluR level also significantly (p < 0.01) decreased by overexpression of dfmr1 in the muscle (Fig. 2C,F). Neural overexpression of dfmr1 in the presynaptic terminal had no distinguishable impact on total GluR abundance in the postsynaptic domain (Fig. 2C,F). These data support a primary role for dFMRP within the postsynaptic cell regulating the postsynaptic abundance of GluRs but also show that presynaptic dFMRP levels can impact postsynaptic GluRIIB.
All ionotropic GluRs increase in the absence of metabotropic GluR signaling
Mammals possess three classes of metabotropic GluRs (mGluRs) and eight specific mGluR types. Activation of different mGluR classes differentially regulates synaptic AMPA GluR expression (Snyder et al., 2001; Nosyreva and Huber, 2005). Recently, mounting evidence has suggested a connection between type I mGluR (mGluR5) activation and the translational regulatory function of FMRP to promote downregulation of synaptic AMPA GluR1 (Hou et al., 2006; Nosyreva and Huber, 2006). The situation in Drosophila is much simpler. The Drosophila genome encodes only a single mGluR, DmGluRA. We showed previously that this receptor is localized to the glutamatergic NMJ and that dmGluRA null mutants display strikingly elevated short-term synaptic facilitation and augmentation (Bogdanik et al., 2004) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In that previous work, we identified a P-element deletion excision line, 112b, as a null mutant (hereafter called dmGluRA) and a companion precise excision line, 2b, as a genetic background control. Here, these lines were used to investigate the role of mGluR signaling in the regulation of ionotropic GluR abundance in the NMJ synaptic terminal.
The three different GluR populations were examined in the dmGluRA null mutant compared with its 2b genetic background control (Fig. 3). All three tested GluR subunits were significantly increased in dmGluRA mutants: GluRIIA was increased 15% (WT, 107.5 ± 4.2; dmGluRA, 123.5 ± 3.2; n = 13 for each genotype; p = 0.006) (Fig. 3A,D), GluRIIB was increased 11% (WT, 98.21 ± 3.24; dmGluRA, 109.12 ± 3.27; n = 16 for each genotype; p = 0.03) (Fig. 3B,E), and GluRIIC was increased 15% (WT, 109.08 ± 4.38; dmGluRA, 124.7 ± 3.42; n = 13 for each genotype; p = 0.0096) (Fig. 3C,F). The HRP internal fluorescence standard displayed no change between the dmGluRA null and the 2b genetic background control in any of the experimental trials (Fig. 3). These data show that loss of all mGluR signaling results in the upregulation of total ionotropic GluR abundance in the postsynaptic domain and that A- and B-class GluRs are similarly suppressed by DmGluRA glutamatergic signaling.

Total GluR abundance is increased in the absence of mGluR signaling. Third-instar NMJs were probed using subunit-specific anti-GluRs (green) and anti-HRP (red) antibodies. Type IB boutons on muscle 4 in A3 were used for all imaging analysis. A–C, Representative images of GluRIIA, IIB, and IIC expression in WT (2b, P-element precise excision line) and dmGluRA null mutant (dmGluRA112b, imprecise deletion line). D–F, Fluorescence intensity quantification of all three GluR subunits and the HRP internal standard. All three tested GluR subunits (GluRIIA, GluRIIB, and GluRIIC) were increased in dmGluRA mutants, whereas HRP showed no significant change in any trial (n = 13 for both genotypes in GluRIIA and GluRIIC tests, n = 16 for both genotypes in GluRIIB test). *0.001 < p < 0.05.
Postsynaptic DmGluRA overexpression induces GluR class-specific changes
To determine whether the regulatory function of DmGluRA on ionotropic GluRs reflects presynaptic or postsynaptic signaling, GluR levels were next examined in targeted DmGluRA overexpression (OE) mutants. As above, the UAS–DmGluRA transgenic line (Bogdanik et al., 2004) was driven by neural ELAV–Gal4 or muscle MHC—Gal4 to induce DmGluRA overexpression in either presynaptic (NOE) or postsynaptic (MOE) cells. The results are displayed in Figure 4.

DmGluRA postsynaptic overexpression causes the opposite GluR class changes to the dfmr1 null. NMJs probed using subunit-specific anti-GluRs (green) and anti-HRP (red) antibodies. Type IB boutons on muscle 4 in A3 were used for all imaging analysis. Representative images of GluRIIA (A) and GluRIIB (B) with DmGluRA muscle overexpression (MOE) and neuronal overexpression (NOE). C, D, Fluorescence intensity quantification for each GluR class. C, GluRIIA is decreased with both DmGluRA MOE and NOE (n = 15 for all genotypes). D, GluRIIB is increased with MOE but decreased with NOE mutants (n = 12 for all genotypes). *0.001 < p < 0.05.
Postsynaptic overexpression of DmGluRA changes GluR class abundance in exactly the opposite direction of the dfmr1 null; A-class GluRs are decreased and B-class GluRs are increased (compare Figs. 1, 4). GluRIIA expression was decreased 10% in DmGluRA muscle overexpression (Fig. 4A,C) (n = 15; p = 0.003), and GluRIIB was similarly increased by 10% (Fig. 4B,D) (n = 12; p = 0.05). As in the dfmr1 null with opposing GluR class changes, there are no change in total GluR based on the common GluRIIC subunit. In addition, however, presynaptic overexpression of DmGluRA downregulates both A- and B-class GluRs, which is an opposite phenotype to the dmGluRA null (compare Figs. 3, 4). The expression of GluRIIA was decreased 10% (Fig. 4A,C) (n = 15; p = 0.015), and GluRIIB was decreased 20% (Fig. 4B,D) (n = 12; p = 0.006). These results show that presynaptic DmGluRA function indirectly regulates postsynaptic GluR abundance, presumably via a homeostatic mechanism (Paradis et al., 2001; Fong et al., 2002; Wierenga et al., 2005).
dFMRP regulates the ratio of A- to B-class GluRs in single postsynaptic domains
We next queried where dFMRP and DmGluRA regulate A- and B-class within individual synaptic boutons. The two GluR classes occupy primarily overlapping expression patterns in distinct postsynaptic membrane domains directly apposing presynaptic active zones (Fig. 5). There are ∼20 of these punctate GluR postsynaptic domains within each type IB NMJ bouton. It has been reported, however, that the two GluR classes can also spatially segregate in nonoverlapping puncta in a minority of cases (Marrus et al., 2004). By staining for the two class-specific subunits simultaneously, the distribution and fluorescence intensities of A- and B-class GluRs were directly compared in both dfmr1 and dmGluRA mutants (Fig. 5).

Ratio of A- to B-class GluRs is dramatically changed in dfmr1 mutants. Third-instar NMJs were probed using subunit-specific anti-GluRIIA (green), anti-GluRIIB (red), and anti-HRP (purple) antibodies. Type IB boutons on muscle 4 in A3 were used for all imaging analysis. A, B, Representative images of WT (w1118) and dfmr1 mutant NMJs at a high magnification showing individual punctate GluR postsynaptic domains. GluRIIA is highly increased and GluRIIB is highly decreased within overlapping punctate domains in dfmr1 mutants. There was no detectable spatial shift in GluR classes relative to each other in dfmr1 mutants compared with control.
As might be predicted, the GluRIIA/GluRIIB ratio at individual synaptic boutons was dramatically altered by the absence of dFMRP. The IIA/IIB ratio is approximately equal in control synapses (1.07 ± 0.03), with the two GluR subclasses showing a similar distribution in the punctate postsynaptic domains (Fig. 5A). In quite evident contrast, the dfmr1 null mutant displayed a coincident strong elevation in GluRIIA and downregulation in GluRIIB, dramatically altering the GluR presentation within single synaptic boutons and in individual postsynaptic punctate domains (Fig. 5B). As a result, the IIA/IIB ratio was strongly shifted toward the A-class receptor (1.53 ± 0.08; p < 0.0001; n = 9 for each genotype). Comparing highly magnified images of dfmr1 null and control boutons, there was no detectable shift in the distribution pattern of the two GluR classes. Specifically, there was no evidence of the appearance/disappearance of new postsynaptic puncta or a differential change in the ratio of IIA/IIB within subclasses of postsynaptic puncta (Fig. 5). Therefore, the strongly skewed IIA/IIB receptor ratio represents an increase of A-class GluR and decrease of B-class GluR within the same postsynaptic receptor domains in the absence of dFMRP function.
The IIA/IIB ratio was similarly examined in the dmGluRA null mutants. As might be predicted, there was no apparent difference between the WT (2b) control and dmGluRA, with the mutant showing a coincident increase in both A- and B-class GluRs without apparent change in ratio or distribution (data not shown). Quantification of the IIA/IIB receptor ratio showed no significant change between the mutant and control (p = 0.14; n = 11 for each genotype). These results show that DmGluRA signaling negatively regulates A- and B-class GluRs in common within single synaptic boutons, whereas dFMRP bidirectionally regulates the two receptor classes to determine the IIA/IIB expression ratio within single postsynaptic punctate domains.
The postsynaptic regulatory function of dFMRP
Transynaptic signaling strongly modifies synapse assembly at the Drosophila NMJ. The level of glutamate release from presynaptic terminals can alter the abundance of postsynaptic GluRs (Featherstone et al., 2002; Chen and Featherstone, 2005), and, conversely, GluR overexpression can feedback to alter active zone assembly and function (Sigrist et al., 2002). Because dFMRP function is known to regulate presynaptic glutamate release (Zhang et al., 2001) and increase postsynaptic A-class GluR abundance (Fig. 1), it is critical to examine possible transynaptic mechanisms for these changes. To further determine whether dFMRP regulates GluRs by presynaptic or postsynaptic mechanisms, two experiments were performed to examine presynaptic consequences of dFMRP loss.
Presynaptic active zones were assayed with a probe for NC82/bruchpilot, an ELKS/CAST (active zone-associated protein) protein at the active zone (Fig. 6) (Kittel et al., 2006; Wagh et al., 2006). Each NC82-positive punctum was counted as one active zone within a synaptic bouton area, which was defined by presynaptic membrane HRP staining. Compared with control, dfmr1 null mutants display significantly more active zones per bouton (WT, 18.8 ± 0.75, n = 164; dfmr1, 23.1 ± 1.2, n = 152; p = 0.003) (Fig. 6A,C). However, dfmr1 mutants possess significantly larger synaptic boutons than controls (WT, 6 ± 0.25, n = 164; dfmr1, 7.7 ± 0.46, n = 152; p = 0.0009). When active zone density is considered (number per area, square micrometers), there was no difference in dfmr1 mutants, which had identical density to controls (WT, 3.2 ± 0.04, n = 152; dfmr1, 3.2 ± 0.06, n = 164; p = 0.73) (Fig. 6D). In dmGluRA mutants, there was no change in either active zone number or density (Fig. 6B–D) (n = 103, WT; n = 114, dmGluRA). Note that, in all GluR measurements, fluorescent density per synaptic area was assayed, and therefore the changes in GluR class abundance occur over an area in which active zone density was unchanged.

Presynaptic active zone density normal in dfmr1 and dmGluRA mutants. Third-instar NMJs probed using anti-NC82 (green) and anti-HRP (red) antibodies. Type IB boutons on muscle 4 in A3 were used for all imaging analysis. Bouton area was defined by the boundary of HRP staining. A, Representative images of WT (w1118) and dfmr1 mutant. B, Representative images of WT (2b) and dmGluRA (112b) mutant. C, Quantification of the number of active zones per bouton. The active zone number per bouton is increased in dfmr1 mutants, but there is no difference between WT and dmGluRA mutants. D, Quantification of active zone density per area (square micrometer). There is no difference in active zone density in both dfmr1 and dmGluRA mutants. *0.001 < p < 0.05.
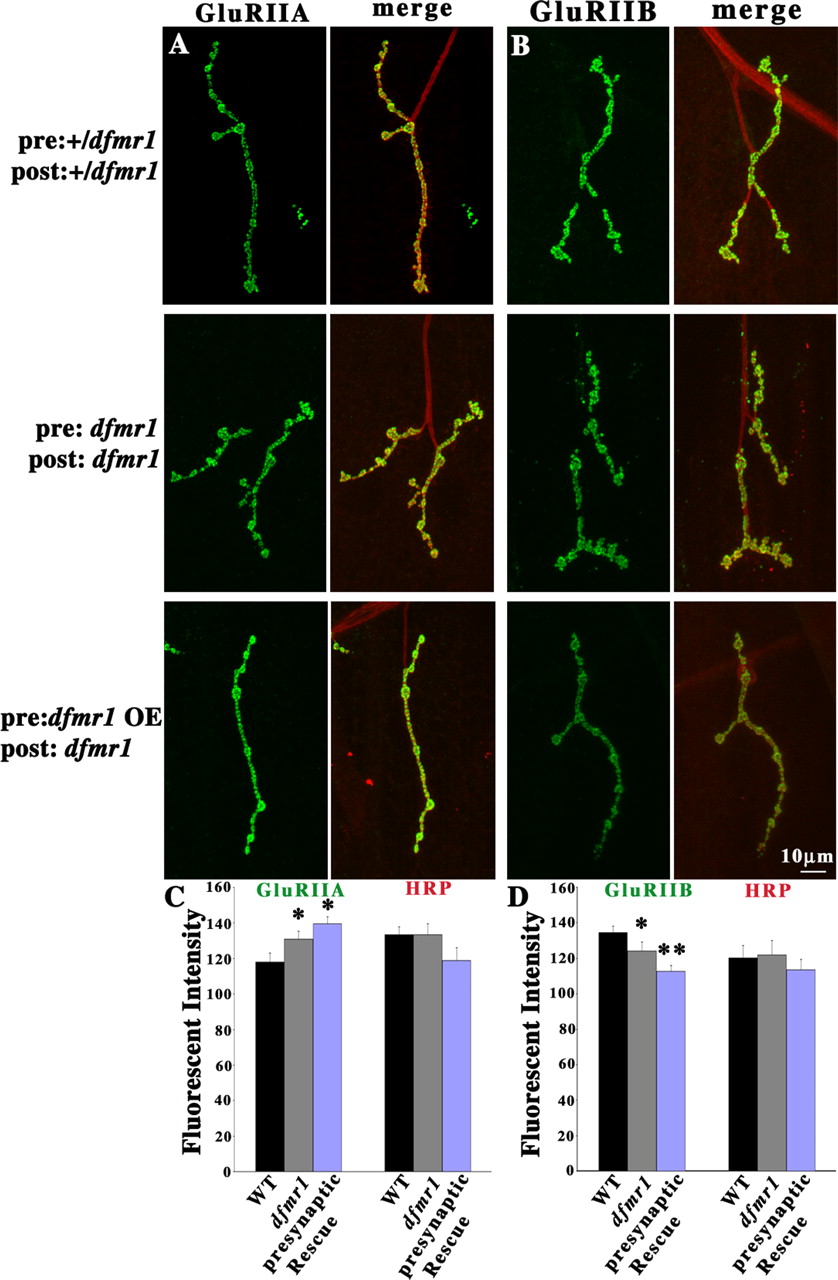
To directly test a possible presynaptic dFMRP role, targeted presynaptic dFMRP expression was driven in the dfmr1 null to assess the impact on the GluR phenotypes. The neural ELAV–Gal4 driver was crossed into the dfmr1 null background with a recombinant line of UAS–dfmr1. This combination strongly expressed dFMRP in the presynaptic neurons, whereas dFMRP was undetectable in the postsynaptic muscles (data not shown). The two controls were ELAV–Gal4 in the heterozygous background (+/dfmr1) and the dfmr1 null (null in both presynaptic and postsynaptic cells). The presynaptic-only expression of dFMRP failed to rescue either GluRIIA or GluRIIB phenotypes (Fig. 7). The A-class GluRs were increased in this single-side rescue (+/dfmr1, 118 ± 4.88; dfmr1, 130.8 ± 4.56; presynaptic rescue, 139.5 ± 3.87; n = 9 for all genotypes; p = 0.002 between +/dfmr1 and presynaptic rescue) (Fig. 7A,C). The B-class GluRs were decreased in single-side rescue (+/dfmr1, 134.63 ± 3.65; dfmr1, 124.16 ± 5; presynaptic rescue, 112.56 ± 3.58; n = 10 for all genotypes; p = 0.004 between +/dfmr1 and presynaptic rescue) (Fig. 7B,D). No significant changes were detected on GluRIIC levels in either dfmr1 null and presynaptic rescue conditions (data not shown).

Targeted presynaptic dFMRP expression in dfmr1 null mutants. Third-instar NMJs probed using subunit-specific anti-GluRs (green) and anti-HRP (red) antibodies. Type IB boutons on muscle 4 in A3 were used for all imaging analysis. Representative images of GluRIIA (A) and GluRIIB (B) in heterozygous mutant (+/dfmr1), dfmr1 null, and presynaptic-only rescue of dFMRP expression. C, D, Fluorescence intensity quantification of each GluR subunit and the HRP internal standard. C, GluRIIA is significantly increased in dfmr1 mutants and is not rescued by presynaptic expression of dFMRP (n = 9 for all genotypes). D, GluRIIB is significantly decreased in dfmr1 mutants and is not rescued by presynaptic expression of dFMRP (n = 10 for all genotypes). *0.001 < p < 0.05; **0.0001 < p < 0.001.
DmGluRA and dFMRP functions converge to regulate synaptic GluR expression
The above results demonstrate that both dFMRP and DmGluRA function to regulate GluR abundance in the postsynaptic compartment. Loss of function of these two genes increases A-class GluRs, whereas B-class GluRs are downregulated in the absence of dFMRP and upregulated in the absence of DmGluRA. The best possible test for interactions between these two proteins is to generate double null mutants, to determine whether GluR phenotypes may cancel out, be additive, or show a synergistic interaction. If the two proteins function in the same signaling pathway, the prediction is that an epistatic interaction will be observed, with the downstream function epistatic to the upstream function. In this case, dFMRP translation regulation function is clearly predicted to be downstream of DmGluRA glutamatergic signaling. If the two proteins act in pathways that converge on GluR regulation, then mutant phenotypes are predicted to be additive, without any epistasis. To test these two possibilities, we generated a dfmr1; dmGluRA double null mutant. We crossed dfmr1 into both the 2b (dmGluRA background control) and dmGluRA null mutant backgrounds. There was no significant difference between the original single dfmr1 mutant and dfmr1; 2b in any assays (data not shown); therefore, genetic background is not a complicating factor. All three GluR populations were assayed in all mutant combinations (Fig. 8).

GluR expression in dfmr1; dmGluRA double null mutants. Third-instar NMJs were probed using subunit-specific anti-GluRs (green) and anti-HRP (red) antibodies. Type IB boutons on muscle 4 in A3 were used for all imaging analysis. A–C, Representative images of GluRIIA, IIB, and IIC expression in WT (2b), dfmr1; 2b single mutants, and dfmr1; dmGluRA double mutant NMJs. D–F, Fluorescence intensity quantification of each GluR subunit and the HRP internal standard in independent trials. D, GluRIIA is significantly increased in dfmr1 single mutants, and the dfmr1; dmGluRA double mutants display an additive, highly significant level of GluRIIA expression (n = 27 for dfmr1; 2b and dfmr1; dmGluRA). E, GluRIIB is very significantly decreased in dfmr1 single mutants, and the dfmr1; dmGluRA double mutants display a reduced, less significant change in GluRIIB expression, indicating an additive interaction (n = 18 for dfmr1; 2b and dfmr1; dmGluRA). F, GluRIIC showed no significant change in dfmr1 single or dfmr1; dmGluRA double mutants (n = 13 for dfmr1; 2b and dfmr1; dmGluRA). In all independent trials, there was no significant change of HRP levels among the three genotypes. *0.001 < p < 0.05; **0.0001 < p < 0.001.
For both GluR classes, the dfmr1; dmGluRA double null mutant showed the additive phenotypes of dfmr1 and dmGluRA single null mutants. For GluRIIA, the dfmr1; 2b single mutant displayed a significant increase and the double mutant displayed a significantly larger, additional elevation in A-class receptor expression (dfmr1; dmGluRA, 114.24 ± 3.24; n = 27; p = 0.0004) (Fig. 8A,D). For GluRIIB, the dfmr1; dmGluRA double mutant similarly showed an additive phenotype. The dfmr1 single mutant displayed a 20% decrease in GluRIIB level compared with WT (2b) controls [dfmr1; 2b, 91.92 ± 23.16; WT (2b), 112.29 ± 3.32; n = 18, p = 0.0007] (Fig. 8B,E). However, the dmGluRA single mutant displayed an apposing 11% increase in GluRIIB (Fig. 3B,E). The dfmr1; dmGluRA double mutant displayed an intermediate, additive phenotype, with a 12% increase in B-class GluRs relative to the dfmr1 single mutant (dfmr1; 2b, 91.92 ± 23.16; dfmr1; DmGluRA, 102.74 ± 2.88; n = 18 for both; p = 0.016) (Fig. 8B,E). Thus, the double mutant displayed only an 8% decrease in GluRIIB compared with the control, a significant difference (p = 0.05) compared with the dfmr1 single mutant. For GluRIIC, there were no significant changes between any of the genotypes [WT (2b), 109.03 ± 5.67, n = 8; dfmr1; 2b, 114.54 ± 4.46; dfmr1; dmGluRA, 118.93 ± 4.11; n = 13 for both mutants; p = 0.48 between WT and dfmr1; p = 0.45 between dfmr1 and dfmr1; dmGluRA) (Fig. 8C,F). Similarly, the ratio of GluRIIA/GluRIIB subunits in dfmr1; 2b single mutants and dfmr1; dmGluRA double mutants was not significantly different (dfmr1; 2b, 1.51 ± 0.05; dfmr1; dmGluRA, 1.42 ± 0.06; n = 9 for each genotype; p = 0.24). These data show an additive interaction between DmGluRA signaling and dFMRP function, which suggests convergent mechanisms on GluR regulation.
Discussion
The finding of elevated group I mGluR5-dependent hippocampal LTD in the fmr1 knock-out mouse (Huber et al., 2002) has elicited a great deal of attention and excitement. This type of LTD is caused by loss of surface expression of AMPA GluRs, in a mechanism requiring protein synthesis (Huber et al., 2000, 2001; Nosyreva and Huber, 2006; Pfeiffer and Huber, 2006). Because synaptic protein translation is regulated by FMRP (Weiler et al., 1997, 2004; Todd et al., 2003a; Antar et al., 2004; Aschrafi et al., 2005; Qin et al., 2005), these observations suggest a mechanistic connection between mGluR signaling and FMRP translation regulation in the control of GluR expression at the synapse and indicate that this pathway may be a critical regulator of functional synaptic plasticity. This idea has been formally expressed as the mGluR theory of FXS (Bear et al., 2004). Our study directly investigates this hypothesized connection between mGluR signaling, FMRP regulatory function, and the synaptic expression of GluRs using the Drosophila FXS model. In Drosophila, there is a single FMR1 protein [dFMRP (Wan et al., 2000; Zhang et al., 2001)] and a single mGluR [DmGluRA (Parmentier et al., 1996; Bogdanik et al., 2004)]. dFMRP structure, expression, and regulative functions closely resemble mammalian FMRP (Wan et al., 2000; Zhang et al., 2001; Dockendorff et al., 2002; Morales et al., 2002; Lee et al., 2003; Schenck et al., 2003; Ling et al., 2004; Reeve et al., 2005; Zhang et al., 2005). In contrast, DmGluRA is more homologous to mammalian group II/III mGluRs (Parmentier et al., 1996), not the group I mGluRs implicated in the FMRP mechanism. However, these mGluR class distinctions may mean little in Drosophila, with its single mGluR. The mammalian group I mGluR antagonist MPEP rescues morphological and behavioral phenotypes in dfmr1 null mutants (McBride et al., 2005), and DmGluRA modulates synaptic architecture (Bogdanik et al., 2004), which is a known function of mammalian group I mGluRs (Vanderklish and Edelman, 2002). These findings suggest that DmGluRA likely also occupies the group I mGluR niche in Drosophila. In any case, DmGluRA is the only mGluR capable of mediating glutamatergic signaling in the Drosophila system.
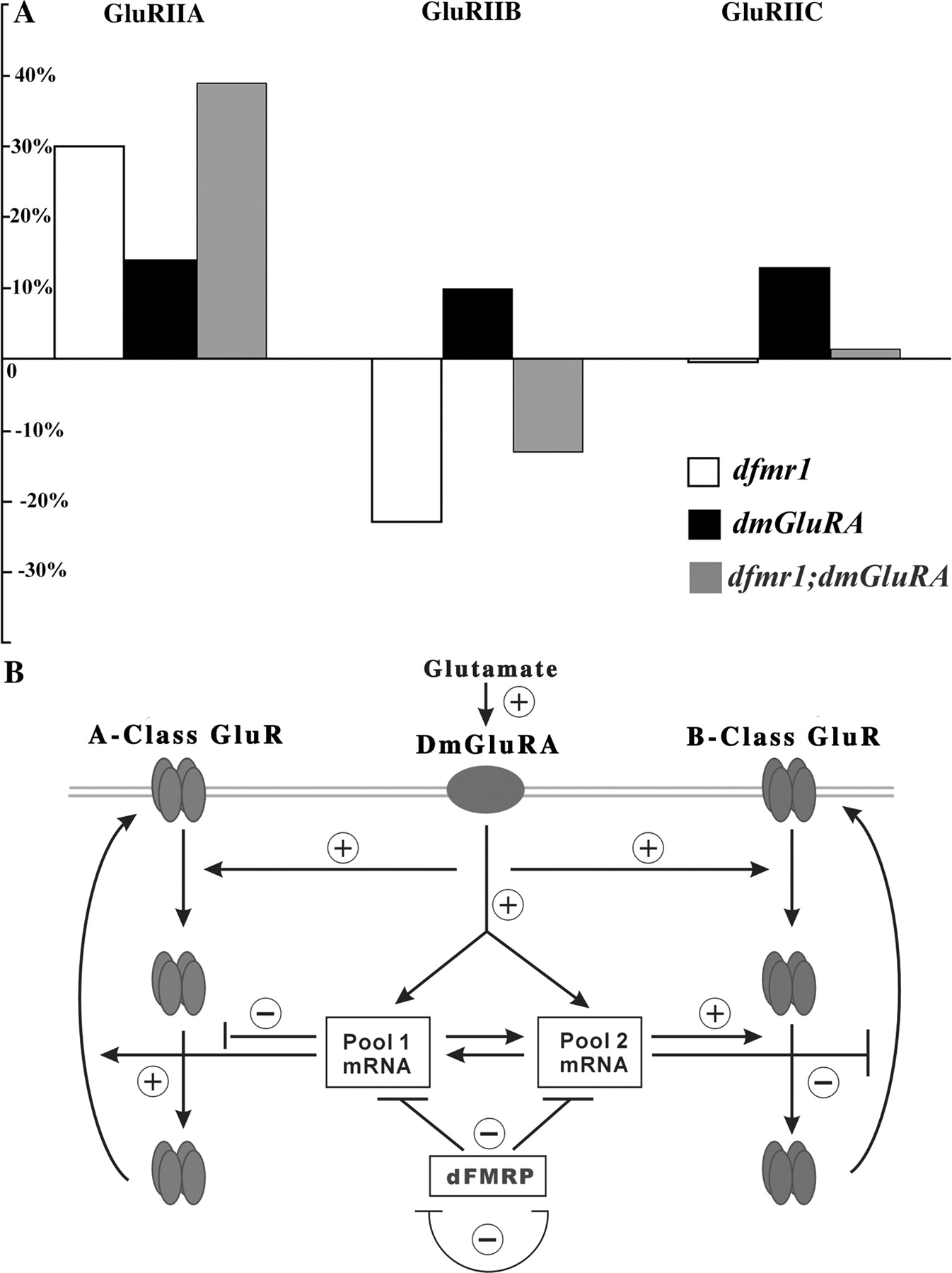
We showed previously that both dfmr1 and dmGluRA mutants have strong defects in glutamatergic synaptic function at the Drosophila NMJ (Zhang et al., 2001; Bogdanik et al., 2004). Neurotransmission at this synapse is mediated by A- and B-class AMPA-type GluRs, which have distinctive functional properties and subsynaptic distributions and are regulated by distinct mechanisms (Schuster et al., 1991; Petersen et al., 1997; Sigrist et al., 2002; Marrus et al., 2004; Featherstone et al., 2005; G. Qin et al., 2005). Here, we show in dfmr1 null mutants that A-class GluRs accumulate and B-class GluRs are lost (Fig. 9A). The total GluR content does not change, but rather there is a striking shift in the GluR class ratio within single postsynaptic domains. This subclass-specific regulation of GluRs is a novel finding. In dmGluRA null mutants, we show that both GluR classes, and therefore the total GluR population, are significantly increased (Fig. 9A). This is a novel finding for DmGluRA but consistent with findings in mammals showing that GluR1 AMPA receptors are decreased in synaptic terminals when mGluR activity is induced (Snyder et al., 2001; Xiao et al., 2001). Moreover, we show that postsynaptic overexpression of DmGluRA induces exactly opposite changes of A- and B-class GluRs compared with dfmr1 null mutants. By testing active zone density and targeted presynaptic rescue of dFMRP in the dfmr1 null, we show that the regulatory function of dFMRP on the GluR classes is a postsynaptic mechanism. Finally, we show in dfmr1; dmGluRA double null mutants that both GluR class phenotypes are additive (Fig. 9A); A-class GluRs increase further with the additive increases of dfmr1 and dmGluRA single mutants, and B-class GluRs tend toward normal levels, with the additive downregulation in the dfmr1 single mutant and upregulation in the dmGluRA single mutant. These results suggest that DmGluRA signaling and dFMRP function converge to regulate the synaptic expression of these two GluR classes but that independent pathways of DmGluRA signaling and dFMRP function also exist.

mGluR and dFMRP pathways converge in the regulation of GluRs. A, Comparison of dfmr1 and dmGluRA single mutants and dfmr1; dmGluRA double mutants in the expression of the three GluR subunits. The WT control is set as zero, and each bar represents percentage change in expression. B, Model relating mGluR signaling and FMRP translation regulatory function controlling synaptic expression of ionotropic GluR classes. DmGluRA negatively regulates both A- and B-class GluRs. dFMRP negatively regulates A-class GluRs and positively regulates B-class GluRs. Genetic tests (A) show that the regulation of these two pathways is additive and, therefore, that the pathways converge on the regulation of these GluR classes. The mGluR theory of FXS postulates that these pathways both regulate local postsynaptic mRNA translation: activated by mGluR signaling and inhibited by dFMRP. Some newly synthesized proteins (Pool 1 mRNA) promote synaptic expression of A-class GluRs, whereas others (Pool 2 mRNA) depress synaptic expression of B-classes GluRs. Thus, the postsynaptic ratio of A- to B-class GluRs is independently regulated.
Presynaptic and postsynaptic functions: overlapping and independent mechanisms
This study suggests that dFMRP and DmGluRA perform in both overlapping and independent pathways in the regulation of postsynaptic GluR classes (Fig. 9B). Targeted presynaptic expression of dFMRP in the dfmr1 null fails to provide any rescue of class-specific GluR misregulation, showing that the dFMRP requirement is in the postsynaptic compartment. Consistently, targeted postsynaptic overexpression of DmGluRA causes the opposite class-specific GluR misregulation of the dfmr1 null, suggesting an intersection of DmGluRA signaling and dFMRP function in the postsynaptic compartment (Fig. 9B). In the dfmr1 null, quantal size is increased (Zhang et al., 2001), a hallmark postsynaptic defect. A mechanistic cause suggested by this study is the elevated A-class GluR level, consistent with former reports that GluRIIA overexpression increases quantal size. Moreover, GluRIIA overexpression increases active zone number per bouton, based on the NC82/bruchpilot probe, but does not alter active zone density (Sigrist et al., 2002), which is identical to the phenotype reported here for dfmr1 mutants. These results support the conclusion that both dFMRP and DmGluRA function in the postsynaptic domain in class-specific GluR regulation (Fig. 9B) and that this mechanism may feedback to alter presynaptic properties.
In addition to the postsynaptic mechanism, there appears to also be presynaptic roles of both dFMRP and DmGluRA that can impact the postsynaptic GluR domains. We have shown previously that both proteins are expressed in the presynaptic neuron of the Drosophila NMJ (Zhang et al., 2001; Bogdanik et al., 2004). Single null mutants show differential misregulation, with the dfmr1 null displaying the class-specific change reflecting its postsynaptic function but the dmGluRA null increasing both GluR classes in common. This must reflect a presynaptic function for DmGluRA. Consistently, presynaptic overexpression of DmGluRA depresses the level of both A- and B-class GluRs, the opposite phenotype as the dmGluRA null. Likewise, presynaptic overexpression of dFMRP also reduces B-class GluR expression, although it does not change the abundance of A-class receptors. Presumably, these presynaptic roles reflect the know functions of dFMRP and DmGluRA in regulating presynaptic glutamate release properties, which we have shown previously (Zhang et al., 2001; Bogdanik et al., 2004), and therefore the GluR changes reflect transynaptic signaling in a homeostatic mechanism.
By strict genetic criteria, the prediction for the interaction of two proteins within a common regulatory pathway is that the mutant phenotype for the gene product downstream in the pathway should be epistatic to that of the gene product upstream in the pathway. Clearly, the FMRP translation regulatory activity should be downstream of mGluR surface glutamate reception. Such a strict epistatic relationship is not observed for DmGluRA and dFMRP in the control of GluR expression. Rather, the null mutant phenotypes are obviously additive in double mutants (Fig. 9A). The A-class GluR goes up in both single mutants and goes up further in the double mutant. The B-class GluR goes down in dfmr1 and up in dmGluRA and shows an intermediate, additive level in the double mutant. Such additive phenotypes show that dFMRP and DmGluRA have overlapping functions but can be operating in the independent pathways (Fig. 9B). Together, our results suggest that dFMRP and DmGluRA pathways converge on the regulation of GluR synaptic expression and that this involves both presynaptic and postsynaptic interactions.
GluR classes are regulated by independent mechanisms
Regulating GluR class composition in the postsynaptic domain is an important mechanism controlling neurotransmission strength and synaptic plasticity properties. The subunit composition of mammalian NMDA and AMPA receptors are both known to be regulated in this manner (Sheng et al., 1994; Washburn et al., 1997). Similarly at the Drosophila NMJ, the independent regulation of GluR classes is critical, because each receptor class has distinct functional properties, e.g., the A-class specifically is negatively regulated by protein kinase A phosphorylation, is modulated by atypical protein kinase C, is important in retrograde signaling, and mediates larger, slower-decaying transmission events with a smaller single channel conductance (Petersen et al., 1997; Davis et al., 1998; DiAntonio et al., 1999; Ruiz-Canada et al., 2004; Chen et al., 2005). The molecular mechanisms for controlling each GluR therefore must be distinct and, indeed, distinct mechanisms have been identified. For example, the PDZ [postsynaptic density 95 (PSD-95)/Discs Large (DLG)/zona occludens-1]-domain scaffold DLG, a PSD-95 homolog, is involved in the localization of many synaptic proteins (Lahey et al., 1994; Budnik et al., 1996; Guan et al., 1996; Tejedor et al., 1997; Thomas et al., 1997, 2000; Mathew et al., 2002; Roche et al., 2002) but plays a specific role in B-class GluR regulation: GluRIIB abundance correlates with DLG level, but GluRIIA localization is unaffected in dlg mutants (Chen et al., 2005). Similarly, the Rho-type guanine nucleotide exchange factor dPix (the Drosophila homolog of the Pak interacting exchange factor) (Werner and Manseau, 1997; Hakeda-Suzuki et al., 2002), its interacting Drosophila p-21 activated kinase (dPak), a serine threonine kinase activated by GTPases Rac and cell division cycle 42 (Harden et al., 1996; Newsome et al., 2000; Mentzel and Raabe, 2005), and a dPak binding partner, the adaptor Dreadlocks (Dock; Nck homolog) (Rao and Zipursky, 1998; Buday et al., 2002; Ang et al., 2003; Rao, 2005), are all required to facilitate synaptic expression of A-class GluRs, but GluRIIB is reportedly not affected in mutants of this pathway (Parnas et al., 2001; Albin and Davis, 2004). Trafficking mechanisms likely involve GluR tethering to the cytoskeleton. The actin-interacting Coracle (mammalian brain 4.1 protein) binds only GluRIIA to specifically regulate its abundance, with no role in B-class GluR tethering (Chen et al., 2005). Thus, separable mechanisms for A- and B-class GluR regulation clearly exist.
FMRP/dFMRP is an RNA-binding protein and a regulator of protein translation at the synapse (Weiler et al., 1997, 2004; Zhang et al., 2001). Although FMRP/dFMRP is best defined as a negative regulator of translation, it may also positively regulate the translation of a distinct set of synaptic mRNAs (Todd et al., 2003a; Khandjian et al., 2004; Stefani et al., 2004; Weiler et al., 2004; Bagni and Greenough, 2005; Garber et al., 2006). Presumably, these translation regulation mechanisms underlie the differential, and opposing, regulation of A- and B-class GluRs by dFMRP. Some newly synthesized proteins may promote synaptic expression of A-class GluRs, whereas others promote the diminution of B-classes GluRs. One protein whose synaptic translation is regulated by both mGluR signaling and FMRP function is PSD-95, implicated in both NMDA and AMPA GluR synaptic expression (Todd et al., 2003a; Muddashetty et al., 2007). Thus, altered regulation of the Drosophila PSD-95 homolog DLG is an attractive candidate mechanism for GluR phenotypes discovered here. In addition, the synaptic cytoskeleton affects synaptic expression of both NMDA and AMPA GluRs (Rosenmund and Westbrook, 1993; Shen et al., 2000; Zhou et al., 2001; Chen et al., 2005). Notably, a key function of FMRP/dFMRP is regulating microtubule and actin filament dynamics via regulating expression of key cytoskeleton-binding proteins, such as Futsch/microtubule-associated protein 1b (Zhang et al., 2001; Lee et al., 2003; Schenck et al., 2003; Lu et al., 2004; Antar et al., 2006). The future goal of our studies will be to test these candidate downstream factors as regulators of GluR synaptic expression in Drosophila, downstream of both DmGluRA signaling and dFMRP translational control.
Footnotes
-
This work was supported by National Institutes of Health Grants HD40654 and GM54544 (K.S.B.). We thank Dr. Aaron DiAntonio for the generous gifts of GluRIIB and GluRIIC (III) antibodies. We thank the Iowa Developmental Studies Hybridoma Bank for providing the GluRIIA and NC82 antibodies. We also thank members of Broadie laboratory for discussion, especially Drs. Charles Tessier and Cheryl Gatto for comments on this manuscript and Dr. Jeffrey Rohrbough for imaging advice.
- Correspondence should be addressed to Prof. Kendal S. Broadie, 6270 MRB III, 465 21st Avenue South, Nashville, TN 37232. kendal.broadie{at}vanderbilt.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}