

Abstract
Skeletal muscle tone is modulated in a stereotypical pattern across the sleep–wake cycle. Abnormalities in this modulation contribute to most of the major sleep disorders; therefore, characterizing the neurochemical substrate responsible for transmitting a sleep–wake drive to somatic motoneurons needs to be determined. Glutamate is an excitatory neurotransmitter that modulates motoneuron excitability; however, its role in regulating motoneuron excitability and muscle tone during natural sleep–wake behaviors is unknown. Therefore, we used reverse-microdialysis, electrophysiology, pharmacological, and histological methods to determine how changes in glutamatergic neurotransmission within the trigeminal motor pool contribute to the sleep–wake pattern of masseter muscle tone in behaving rats. We found that blockade of non-NMDA and NMDA glutamate receptors (via CNQX and d-AP-5) on trigeminal motoneurons reduced waking masseter tone to sleeping levels, indicating that masseter tone is maximal during alert waking because motoneurons are activated by an endogenous glutamatergic drive. This wake-related drive is switched off in non-rapid eye movement (NREM) sleep, and this contributes to the suppression of muscle tone during this state. We also show that a functional glutamatergic drive generates the muscle twitches that characterize phasic rapid-eye movement (REM) sleep. However, loss of a waking glutamatergic drive is not sufficient for triggering the motor atonia that characterizes REM sleep because potent activation of either AMPA or NMDA receptors on trigeminal motoneurons was unable to reverse REM atonia. We conclude that an endogenous glutamatergic drive onto somatic motoneurons contributes to the stereotypical pattern of muscle tone during wakefulness, NREM sleep, and phasic REM sleep but not during tonic REM sleep.
Introduction
Skeletal muscle tone is modulated in a stereotypical pattern across the sleep–wake cycle. Jouvet first characterized this pattern by showing that muscle tone is maximal in alert waking, reduced in quiet waking, further reduced in non-rapid eye movement (NREM) sleep, and potently suppressed in rapid eye movement (REM) sleep (Jouvet, 1962, 1967). Since this initial description, considerable attention has been paid to deciphering the mechanisms controlling muscle tone during the sleep cycle because abnormal motor control contributes to the major sleep disorders, including REM sleep behavior disorder, narcolepsy/cataplexy, and obstructive sleep apnea. Although considerable progress has been made in identifying the mechanisms responsible for muscle tone suppression in REM sleep (Kubin et al., 1998; Chase and Morales, 2005), little is known about the neurochemistry of muscle tone regulation in waking and NREM sleep.
Glutamate is the most abundant and potent excitatory neurotransmitter in the mammalian CNS (Collingridge and Lester, 1989; Nakanishi, 1992). It is also the primary transmitter responsible for controlling motoneuron excitability and for mediating motor behaviors such as involuntary and rhythmic movements as well as afferent reflexes (Rekling et al., 2000). Motoneurons express ionotropic glutamate receptors and receive glutamatergic innervation from brainstem regions regulating arousal states and motor control (Rekling et al., 2000). The discharge pattern of neurons in the medial medulla, in which glutamate-containing cells are located (Kaneko et al., 1989), parallels the activity pattern of muscle tone across the sleep cycle, with neuronal activity being maximal in waking, minimal in NREM sleep, and either silent or episodically active in REM sleep (Siegel et al., 1983, 1992). Despite this evidence, the role of glutamate in regulating levels of motoneuron excitability and muscle tone during natural sleep–wake motor behaviors has never been tested.
Therefore, we used reverse-microdialysis, electrophysiology, pharmacological, and histological methods to examine how glutamatergic neurotransmission within the trigeminal motoneuron pool contributes to basal levels of masseter muscle tone during sleep and waking in freely behaving rats. The masseteric motor system was the focus of this study because trigeminal motoneurons innervate masseter and palatal muscles, both of which are affected by and contribute to the pathogenesis of several sleep disorders, including REM sleep behavior disorder, obstructive sleep apnea, cataplexy/narcolepsy, and bruxism (Guilleminault, 1994; Horner, 1996; Kato et al., 2003). In addition, the glutamatergic control of trigeminal motoneurons has been well documented (Chandler, 1989; Kolta, 1997).
This study was designed to determine whether changes in glutamatergic neurotransmission at the trigeminal motor pool are responsible for the sleep–wake pattern of masseter muscle tone. First, we identified an endogenous glutamatergic drive at the trigeminal motor pool during waking and in phasic REM sleep, by antagonizing ionotropic glutamate receptors on trigeminal motoneurons. Then, we determined that the motor suppression of NREM, but not tonic REM, sleep could be restored to waking levels by exogenously replacing the glutamatergic drive that is normally withdrawn in sleep. We conclude that an endogenous glutamatergic drive onto trigeminal motoneurons provides a major neurochemical cue controlling the stereotypical pattern of masseter muscle tone during wakefulness, NREM sleep, and phasic REM sleep but not tonic REM sleep.
Materials and Methods
Animals
Rats were housed individually and maintained on a 12 h light/dark cycle (lights on at 7:00 A.M. and off at 7:00 P.M.), and both food and water were available ad libitum. All procedures and experimental protocols were approved by the University of Toronto animal care committee and were in accordance with the Canadian Council on Animal Care.
Surgical preparation for sleep and microdialysis studies
Studies were performed using 30 male Sprague Dawley rats (average mass, 340 ± 8.2 g). To implant electroencephalogram (EEG) and electromyogram (EMG) electrodes and a microdialysis probe, sterile surgery was performed under anesthesia induced with intraperitoneal ketamine (85 mg/kg) and xylazine (15 mg/kg) and maintained with additional anesthesia given by inhalation (isoflurane, 0.5–2%). Effective depth of anesthesia was determined by the abolishment of the pedal withdrawal and blink reflexes. Body temperature was monitored with a rectal probe (CWE, Ardmore, PA) and maintained at 37 ± 1°C.
Three insulated, multi-stranded stainless steel wire EMG electrodes (Cooner Wire, Chatsworth, CA) were implanted into the left and right masseter muscles. The wires were tunneled subcutaneously to an incision along the dorsal surface of the cranium. Three EMG electrodes were also inserted into the nuchal muscle. Four stainless steel screws (JI Morris Company, Southbridge, MA), attached to insulated 34 gauge wire (Cooner Wire), were implanted in the skull for recording cortical EEG; their coordinates were 2 mm rostral and 2 mm to the left and right of bregma, and 3 mm caudal and 2 mm to the left and right of bregma. An additional reference electrode was also secured onto the skull; its position was 9.4 mm caudal and 0.5 mm lateral to bregma.
To implant a microdialysis probe into the left trigeminal motor nucleus, a ∼2 mm burr hole was made at 9.4 mm caudal and 1.8 mm lateral to bregma (Paxinos and Watson, 1998). A microdialysis guide probe (CMA/Microdialysis, Solna, Sweden) was then lowered 8.2 mm below the skull surface by stereotaxic manipulation. Dental cement (1234; Lang Dental, Wheeling, IL) secured the probe in place, and, after the cement was dry, EEG and EMG electrodes were connected to pins (Allied Electronics, Bristol, PA) and inserted into a custom-made head plug (Allied Electronics) that was affixed to the skull with dental cement.
After surgery, rats were given an intraperitoneal injection of 0.03 mg/kg buprenorphin and kept warmed by a heating pad. They were also given a dietary supplement (i.e., Nutri-Cal) and soft food for the following 2 d. Rats recovered for at least 7–10 d before experimental testing began.
Experimental procedures for sleep and microdialysis studies
Recording environment.
During experiments, animals were housed in Raturn system (BAS Bioanalytical Systems, West Lafayette, IN), which is a movement-responsive caging system eliminating the need for a commutator or liquid swivel. This caging system was housed inside a sound-attenuated, ventilated, and illuminated (lights on, 110 lux) chamber.
Electrophysiological recordings.
Sleep and muscle activity were recorded by attaching a lightweight cable to a plug on the rat's head, which was connected to a Super-Z head-stage amplifier and BMA-400 alternating current/direct current Bioamplifier (CWE). The EEG was amplified 1000 times and bandpass filtered between 1 and 100 Hz. EMG signals were amplified between 500 and 1000 times and bandpass filtered between 30 Hz and 30 kHz. All electrophysiological signals were digitized at 250 Hz (Spike 2 Software, 1401 Interface; Cambridge Electronic Design, Cambridge, UK) and monitored and stored on a computer.
Microdialysis probe.
A microdialysis probe was used to exogenously perfuse glutamate antagonists and agonists into the trigeminal motor nucleus. The microdialysis probe (CMA/Microdialysis) (34 kDa cutoff; membrane length and diameter, 1 mm × 250 μm) was lowered into the left trigeminal motor nucleus. The microdialysis probe was connected to FEP Teflon tubing (inside diameter, 0.12 mm; Eicom, Moraine, OH), which was connected to a 1 ml gastight syringe via a liquid switch (BAS Bioanalytical Systems). The probe was continually perfused with filtered (0.2 μm nylon; Thermo Fisher Scientific, Waltham, MA) artificial CSF (aCSF) (in mm: 125 NaCl, 5 KCl, 1.25 KH2PO4, 24 NaHCO3, 2.5 CaCl2, 1.25 MgSO2, and 20 d-glucose) at a flow rate of 2 μl/min using a syringe pump (BAS Bioanalytical Systems).
Drug preparation.
All drugs were dissolved in aCSF at the beginning of each experimental day. Glutamatergic antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) [molecular weight (MW), 232.16; Tocris Bioscience, Bristol, UK], a selective non-NMDA receptor antagonist, and d-2-amino-5-phosphonopentanoate (d-AP-5) (MW, 197.13; Tocris Bioscience), a selective NMDA antagonist, were used to block endogenous glutamatergic excitation. Glutamatergic agonists l-glutamic acid (glutamate; MW, 147.13; Tocris Bioscience), AMPA (MW, 186.17; Tocris Bioscience), and NMDA (MW, 147.13; Tocris Bioscience) were used to activate glutamate receptors.
Experimental protocols
Each experiment took 2 d to complete. On the first day at 8:00 A.M. to 10:00 A.M., animals were placed into the recording chamber and given at least 1 h to habituate before they were connected to the electrical tether. They were then given a minimum of 3 h to habituate to this before recordings began. Baseline recordings (without the microdialysis probe in place) were performed on day 1 of experiments, between 1:00 P.M. and 4:00 P.M. The microdialysis probe was inserted between 5:00 P.M. and 7:00 P.M., and aCSF was perfused throughout the night. Probes were inserted the night before experiments began because previous studies demonstrate that probe insertion induces spontaneous neurotransmitter release and local neuronal activation (Di Chiara, 1990; Kodama et al., 1998).
On the second day of experimentation, perfusion of candidate drugs began at 8:00 A.M. to 9:00 A.M. Drug treatments were randomized, and no more than two drug treatments were given to any one animal; an aCSF washout period of at least 2 h followed every drug treatment.
Study 1: antagonism of NMDA and non-NMDA glutamate receptors.
To determine whether an endogenous glutamatergic drive mediates trigeminal motoneuron excitability during natural behavior, NMDA and non-NMDA receptors were antagonized using (1) application of 0.5 mm CNQX and 5.0 mm d-AP-5 in combination, (2) application of 0.5 mm CNQX alone, or (3) 5.0 mm d-AP-5 alone. We used these concentrations because previous in vivo studies demonstrate that they effectively block glutamate neurotransmission onto somatic motoneurons (Steenland et al., 2006). Each drug was applied onto the motor nucleus for 2–4 h; this typically allowed sufficient time for the animal to pass through at least three complete sleep cycles (i.e., wake to NREM sleep to REM sleep). The commencement of drug treatments were not linked to arousal state, that is, drug administration into the motor pool began regardless of the animal's arousal state.
Study 2: agonism of glutamate receptors.
To determine whether addition of glutamate could reverse the sleep-dependent suppression of muscle activity, particularly in REM sleep, NMDA and non-NMDA receptors were activated by applying three separate glutamatergic agonists into the left trigeminal motor pool during both sleep and wakefulness: (1) 25 mm glutamate; (2) 0.1 mm AMPA; or (3) 0.1 mm NMDA. We used 25 mm glutamate because previous studies show that only 10% of glutamate diffuses across the microdialysis membrane (Alessandri et al., 1996). Therefore, based on this observation, it is estimated that only 2.5 mm glutamate will diffuse onto trigeminal motoneurons; this concentration approximates glutamate levels at the mammalian synaptic cleft (Clements et al., 1992), and we found that it activates masseter EMG activity in waking. We used 0.1 mm AMPA and 0.1 mm NMDA because previous in vivo studies show that such doses induce activation of somatic motoneurons (Steenland et al., 2006), and we found that they potently activated masseter muscle tone in waking.
Verification of microdialysis probe location
Two procedures were used to demonstrate that microdialysis probes were both functional and located in the left trigeminal motor pool. At the end of each experiment, 0.1 mm AMPA was perfused into the left trigeminal motor pool, which induced a rapid and potent increase in basal levels of left masseter muscle tone without affecting either the right masseter or neck EMG activity. This result verified that trigeminal motoneurons were viable and able to respond to glutamatergic activation, that microdialysis probes were functional at the end of each experiment, and that probes were located in the trigeminal motor nucleus. We also used postmortem histological analysis to demonstrate that microdialysis probes were physically located in the trigeminal nucleus.
Histology.
Under deep anesthesia (ketamine at 85 mg/kg and xylazine at 15 mg/kg, i.p.), rats were decapitated, and their brains removed and placed in chilled 4% paraformaldehyde (in 0.1 m PBS) for 24 h. Brains were then cryoprotected in 30% sucrose (in 0.1 m PBS) for 48 h; they were then frozen in dry ice and transversely sectioned in 30 μm slices using a microtome (Leica, Wetzlar, Germany). Brain sections were mounted, dried, and stained with Neutral Red. Tissue sections spanning regions rostral and caudal to the trigeminal motor pool were viewed using a light microscope (Olympus, Tokyo, Japan); the location of probe lesion tracts were plotted on standardized brain maps (Paxinos and Watson, 1998) to verify probe location. These data are summarized in Figure 3B.
Data analysis
Behavioral state.
We identified and classified four behavioral states. Alert wake (AW) was characterized by high-frequency, low-voltage EEG signals coupled with high levels of EMG activity (i.e., chewing, grooming, and drinking) (see Fig. 1A). Quiet wake (QW) was characterized by high-frequency, low-voltage EEG signals but in the absence of overt motor activity. NREM sleep was characterized by high-amplitude, low-frequency EEG signals and minimal EMG activity. REM sleep was characterized by low-amplitude, high-frequency theta-like EEG activity and REM atonia interspersed by periodic muscle twitches. Sleep states were visually identified and analyzed in 5 s epochs using the Sleepscore version 1.01 script (Cambridge Electronic Design).
EMG analysis.
Raw EMG signals were full-wave rectified, integrated, and quantified in arbitrary units. Average EMG activity for left and right masseter and neck muscle activity was quantified in 5 s epochs for each behavioral state. When glutamatergic agents were applied onto the left trigeminal motor pool, EMG data were not analyzed for the first 30 min of perfusion because the latency from the syringe pump to the microdialysis probe was ∼15 min. We analyzed and quantified levels of EMG activity (i.e., left and right masseter and neck muscle) for all behavioral states (i.e., AW, QW, NREM, and REM) occurring across the 2–4 h perfusion period (i.e., aCSF and candidate drugs).
EMG analysis in REM sleep.
REM sleep consists of both tonic and phasic motor events. The stereotypical periods of motor atonia occur during tonic REM sleep and the periodic muscle twitches that punctuate REM atonia occur during phasic REM sleep (i.e., during rapid eye movements) (Aserinsky and Kleitman, 1953; Jouvet, 1967). Because a major goal of this study was to determine the role for glutamatergic neurotransmission in modulating motor activity in REM sleep, we developed an objective method for identifying and quantifying the phasic (i.e., muscle twitches) and tonic (REM atonia) periods of REM sleep. To quantify motor atonia during tonic REM sleep, we determined the 99th percentile of EMG activity during the first 5 s of each REM period because muscle twitches are conspicuously absent during this time (Lu et al., 2005; Brooks and Peever, 2008). The muscle twitches that define phasic REM sleep were classified as motor events that exceeded the 99th percentile of EMG activity during the first 5 s of REM; and conversely, REM sleep atonia was classified as any period in which muscle activity was equal to or less than the 99th percentile of EMG activity during the first 5 s of REM. In each rat, REM atonia and muscle twitches were quantified for each REM episode during baseline conditions and for each drug perfused into the trigeminal motor pool.
EEG spectral analysis.
Spectral analysis was performed using EEG Band Detect version 1.06 in Spike 2. The EEG was windowed using a Hamming function and subjected to a fast Fourier transform to yield the power spectrum. The power within four frequency bands was recorded as absolute power and as a percentage of the total power of the signal that was calculated over each 5 s epoch. The band limits used were as follows: delta, 0.48–4 Hz; theta, 4.25–8 Hz; alpha, 8.25–15 Hz; beta, 15.25–35 Hz.
Statistical analyses
The statistical tests used for analysis are included in Results. Comparisons between treatments for mean basal muscle tone in all behavioral states were made using a two-way repeated-measures (RM) ANOVA with post hoc Tukey's tests to infer statistical significance. Comparisons between treatments for both the amplitude and number of muscle twitches per REM episode were made using paired t tests. All statistical analyses used SigmaStat (SPSS, Chicago, IL) and applied a critical two-tailed α value of p < 0.05. Data are presented as means ± SEM.
Results
Masseter muscle activity follows a stereotypical pattern across the sleep–wake cycle
Because the sleep–wake activity patterns of masseter muscle have not been documented in naturally sleeping rats, our first aim was to characterize how masseter muscle tone changes as a function of sleep–wake state. In 24 rats, we recorded masseter muscle EMG activity across the natural sleep–wake cycle under baseline conditions (i.e., before probe insertion) and compared it with neck muscle activity. A total of 61.8 h of EMG activity were analyzed, with 21.1 h of it spent in AW, 21.8 h in QW, 14.5 h in NREM sleep, and 4.4 h in REM sleep. We found that masseter muscle EMG activity is significantly affected by different sleep–wake states (p < 0.001, one-way RM-ANOVA) (Fig. 1B). Figure 1A is a typical example illustrating that basal levels of masseter muscle tone are highest in AW, reduced in QW, and minimal in NREM sleep; during REM sleep, masseter muscles are atonic except during periods of phasic REM, when flurries of muscle twitches override the background of motor atonia.

Masseter muscle tone exhibits a stereotypical pattern of activity across the natural sleep–wake cycle. A, A typical example showing how different sleep–wake states affect basal levels of masseter and neck muscle tone. Masseter and neck muscle EMG activity are maximal in AW, reduced in QW, and further suppressed in NREM sleep. During tonic REM sleep, masseter and neck muscles are atonic except for flurries of muscle twitches that occur during periods of phasic REM sleep. B, Group data from 24 rats demonstrating that both masseter and neck muscle tone follow a stereotypical pattern of activity across the sleep–wake cycle, with muscle tone being significantly suppressed during both NREM and REM sleep (p < 0.001). Traces were taken during baseline conditions, before a microdialysis probe was inserted into the trigeminal motor pool. Data are expressed as mean percentage changes from alert waking. All values are means ± SEM.
Microdialysis probes were located in the left trigeminal motor nucleus
We found that inserting a probe into the left trigeminal nucleus caused an immediate 293 ± 42% increase in left masseter activity (p = 0.014, one-way RM-ANOVA) (Fig. 2A,B), this effect lasted for 99 ± 50 s before returning to preinsertion values (p = 0.997). This intervention had no affect on either right masseter or neck muscle activities (right masseter, p = 0.271; neck, p = 0.537) (Fig. 2A), demonstrating that probe insertion only activates motoneurons in the left trigeminal motor pool. We also found that microdialysis of 0.1 mm AMPA into the left trigeminal motor pool during the last 15 min of each experiment significantly increased left masseter muscle activity (p = 0.012, paired t test) (Fig. 3C,D) without altering levels of either the right masseter (p = 0.574) (Fig. 3C,D) or neck muscle activity (p = 0.431; data not shown). This procedure verified that probes were located within the left trigeminal nucleus and that microdialysis probes were functional, and that motoneurons were viable and able to respond to compounds that manipulate glutamatergic transmission. In 28 rats, we confirmed by postmortem histology that microdialysis probes were located within or immediately adjacent to the left trigeminal motor nucleus (Fig. 3A,B). However, in two rats, left masseter EMG activity was unaffected by either probe insertion or AMPA application; histology confirmed that probes were located outside of the trigeminal motor nucleus (Fig. 3B). These two animals were not included in this study.

Insertion of a microdialysis probe into the trigeminal motor pool has transient affects on left masseter muscle tone. A, A representative EMG trace showing that insertion of a microdialysis probe into the left trigeminal motor pool (during wakefulness) induced a transient activation of left masseter tone (LM); this intervention never affected right masseter (RM) EMG activity. B, Group data demonstrating that left masseter muscle tone significantly increased above baseline levels during probe insertion (p = 0.014); however, this affect only endured for 99 ± 50 s before it returned to baseline levels (p = 0.997). C, Group data demonstrate that the stereotypical pattern of left masseter muscle tone across the sleep cycle is unaffected by placing a probe in the trigeminal nucleus. Data are expressed as mean percentage changes from alert waking. All values are means ± SEM; *p < 0.05. A.U., Arbitrary units.

Microdialysis probes were located in the left trigeminal motor nucleus. A, a is a photograph depicting a lesion made by a microdialysis probe in the trigeminal motor nucleus. b and c are sections that immediately flank the rostral and caudal borders of the trigeminal nucleus; there was no lesion in either area, demonstrating that the probe was located exclusively in the motor pool. Scale bars, 1 mm. B, Black filled circles represent the location of the lesions in the left trigeminal nuclei in the 28 rats used in these studies. Triangles represent the location of lesions not in the trigeminal motor nucleus; data from these two rats were not used in the study. C, A typical recording showing that perfusion of 0.1 mm AMPA into the left motor pool increases left masseter tone (LM) without affecting muscle activity in the right masseter (RM). D, Group data showing that AMPA perfusion induced a potent increase in left (p = 0.012) but not right masseter tone (p = 0.574). All values are means ± SEM; *p < 0.05. A.U., Arbitrary units.
Basal levels of masseter muscle tone are unaffected by placing a microdialysis probe into the trigeminal motor nucleus
To demonstrate that inserting a microdialysis probe into the trigeminal motor pool had no affect on the sleep–wake pattern of masseter muscle activity, we compared levels of masseter muscle EMG activity before and 1 h after probe placement (with aCSF flowing at 2 μl/min). We found no difference between basal levels of masseter muscle tone before compared with after probe insertion (p > 0.252 for all states, two-way RM-ANOVA) (Fig. 2C) and conclude that the sleep–wake pattern of masseter muscle tone is unaffected by probe placement.
Glutamatergic receptor agonists/antagonists affect masseter muscle tone but not sleep–wake behavior
Because the trigeminal motor pool is located in close proximity to pontine regions that regulate sleep (e.g., locus ceruleus and sublaterodorsal nucleus), we wanted to verify that manipulating glutamatergic neurotransmission in the trigeminal nucleus did not affect sleep–wake behaviors. We found that, under baseline conditions, rats spent 32% of the time in AW, 33% in QW, 24% in NREM, and 7% in REM sleep (with the remaining time being spent in transition states). During application of glutamate, AMPA, NMDA, CNQX/d-AP-5, CNQX, or d-AP-5 rats spent an average of 31% of the recording period in AW, 30% in QW, 27% in NREM, and 9% in REM sleep. There was no significant difference in the amount of time spent in each state when baseline and agonist/antagonist treatment were compared (p = 0.450, two-way RM-ANOVA). There was also no change in the ratio of high to low frequencies in EEG powers (i.e., percentage beta/percentage delta) before and after drug treatment (p = 0.922, two-way RM-ANOVA), indicating that these interventions did not substantially affect sleep–wake regulation.
Antagonism of glutamate receptors on trigeminal motoneurons reveals the presence of an endogenous glutamatergic drive during wakefulness and phasic REM sleep
Combined non-NMDA and NMDA receptor antagonism
To determine how endogenous glutamate release contributes to mediating basal levels of masseter muscle tone during different waking and sleeping states, we antagonized non-NMDA and NMDA receptors on trigeminal motoneurons using 0.5 mm CNQX and 5.0 mm d-AP-5 while monitoring masseter muscle tone across the sleep–wake cycle in six rats. Although antagonists were continuously applied into the left trigeminal motor pool for 3.1 ± 0.15 h, neither right masseter muscle activity (p = 0.921, two-way RM-ANOVA) (Fig. 4) nor neck muscle activity (p = 0.199, two-way RM-ANOVA; data not shown) changed (relative to baseline) during any behavioral state.

Antagonism of non-NMDA and NMDA receptors on trigeminal motoneurons in the left motor pool suppresses left but not right masseter tone. A, A typical example demonstrating that application of CNQX and d-AP-5 into the left trigeminal motor pool suppresses left masseter muscle tone during alert waking and REM sleep, without affecting right masseter muscle activity. B–D, Group data (n = 6) comparing right masseter muscle tone before (i.e., baseline) and after perfusion of CNQX and d-AP-5 into the left trigeminal motor pool. Although non-NMDA and NMDA receptor antagonism suppressed left masseter tone (see Fig. 5) during alert and quiet waking and reduced muscle twitch activity during REM sleep, this intervention had no affect on right masseter muscle tone during any behavioral state. These observations demonstrate that manipulations in the left motor pool do not affect motoneurons in the right trigeminal motor pool. All values are means ± SEM; A.U., arbitrary units.
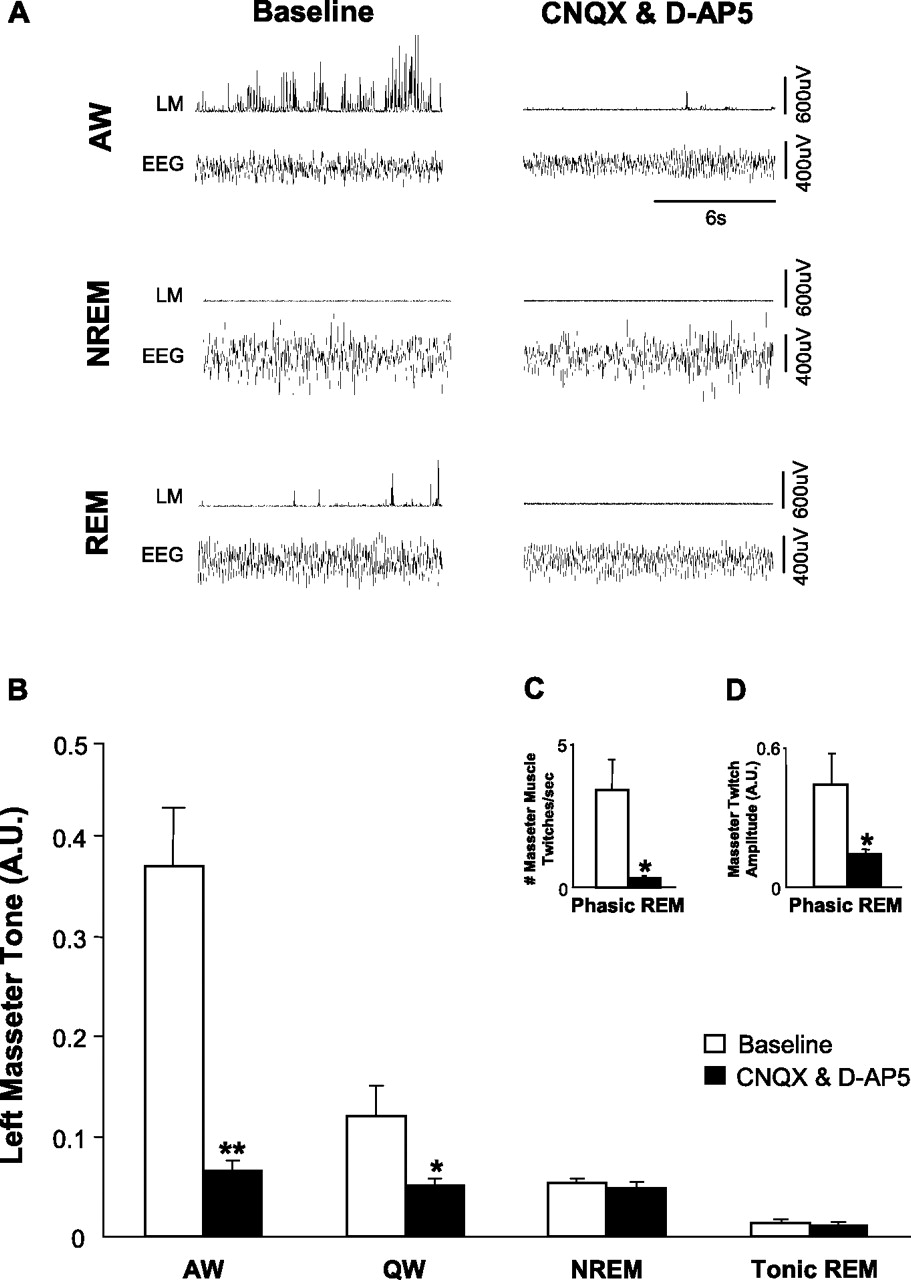
Antagonism of non-NMDA and NMDA receptors resulted a significant suppression of left masseter muscle tone during both alert and quiet waking (p = 0.005, two-way RM-ANOVA) (Fig. 5). Compared with left masseter muscle activity under baseline conditions, application of both CNQX and d-AP-5 markedly reduced basal levels of left masseter activity during AW by 79% (p < 0.001) and during QW by 58% (p = 0.03) (Fig. 5B). This reduction was so potent that it reduced waking masseter muscle tone to levels that were no longer significantly different from those during NREM sleep under baseline conditions (p = 0.172) (Fig. 6A), indicating that withdrawal of a wake-related glutamatergic drive contributes to the suppression of motor tone in NREM sleep. However, antagonism of non-NMDA and NMDA receptors did not suppress waking levels of muscle tone to atonic REM sleep values (p < 0.001) (Fig. 6A), indicating that withdrawal of glutamatergic inputs is not sufficient for inducing REM sleep atonia.

An endogenous glutamatergic drive onto trigeminal motoneurons is functional during wakefulness and REM sleep. A, A typical example showing that antagonism of non-NMDA and NDMA receptors on trigeminal motoneurons in the left motor pool reduces basal tone of left masseter muscle during alert waking; this intervention also abolishes the muscle twitches of phasic REM sleep. B, Group data demonstrating that application of CNQX and d-AP-5 caused a significant reduction in left masseter activity during alert and quiet waking but had no affect on basal masseter tone during either NREM or tonic REM sleep. C, D, Antagonism of non-NDMA and NMDA receptors decreased the number of muscle twitches in the left masseter muscle during phasic REM sleep and decreased the amplitude of the remaining muscle twitches. All values are means ± SEM; *p < 0.05 and **p < 0.001; A.U., arbitrary units.

Withdrawal of a wake-related glutamatergic drive onto motoneurons contributes to the suppression of basal masseter muscle tone during NREM but not tonic REM sleep. A, Perfusion of CNQX and d-AP-5 into the trigeminal motor pool reduced waking masseter muscle tone to NREM sleep levels during baseline conditions (downward arrow; p < 0.001); however, this intervention was unable to reduce waking motor tone to tonic REM sleep levels during baseline conditions (i.e., significant difference between baseline tonic REM and AW during CNQX/d-AP-5, p < 0.05). B, Glutamate application during NREM sleep increased masseter muscle tone to waking levels during baseline conditions (p < 0.001); however, it did not increase muscle tone during tonic REM sleep (p = 0.916). All values are means ± SEM; A.U., arbitrary units.
Glutamate receptor antagonism had no effect on basal masseter tone during either NREM (p = 0.879) or tonic REM (p = 0.939) sleep. However, this intervention significantly reduced the number of muscle twitches during phasic REM sleep by 90% of baseline conditions (p = 0.03, paired t test) (Fig. 5C); it also reduced the mean amplitude of the remaining muscle twitches by 67% of baseline levels (p < 0.001) (Fig. 5D).
NMDA and non-NMDA receptor antagonism
Having demonstrated that there is an endogenous glutamatergic drive on trigeminal motoneurons during wakefulness and phasic REM sleep, we wanted to determine which of the two major ionotropic glutamate receptors (i.e., non-NMDA or NMDA) mediates this excitatory drive. To do this, we selectively blocked non-NMDA or NMDA receptors by applying either 0.5 mm CNQX (n = 6) or 5.0 mm d-AP-5 (n = 5) into the left trigeminal motor pool while recording left masseter muscle EMG activity.
Antagonism of non-NMDA receptors caused a potent suppression of left masseter muscle EMG activity during both waking and phasic REM sleep. Compared with baseline conditions, CNQX application reduced basal levels of left masseter activity during waking by 82% (p < 0.001, two-way RM-ANOVA) (Fig. 7A). Basal levels of masseter tone were not affected during either NREM (p = 0.939) or tonic REM (p = 0.985) sleep. However, non-NMDA receptor antagonism significantly reduced the number of muscle twitches during phasic REM sleep by 86% of baseline conditions (p = 0.036, paired t test) (Fig. 7B). The amplitude of muscle twitches that persisted during CNQX application were significantly reduced by 85% of baseline levels (p < 0.001) (Fig. 7C).

The functional glutamatergic drive onto trigeminal motoneurons during waking and phasic REM is mediated primarily by non-NMDA receptors. A, Group data (n = 6) demonstrating that antagonism of non-NMDA receptors by perfusion of 0.5 mm CNQX into the left trigeminal motor pool significantly decreased left masseter muscle tone during waking but had no effect on levels of masseter tone during either NREM (p = 0.939) or tonic REM (p = 0.985) sleep. B, C, This same intervention significantly decreased the number of muscle twitches during phasic REM sleep and the magnitude of the remaining muscle twitches. D, Group data (n = 5) demonstrating that antagonism of NMDA receptors by perfusion of 5.0 mm d-AP-5 into the left trigeminal motor pool significantly decreased left masseter muscle tone during waking by 49% but had no effect on levels of masseter tone during either NREM (p = 0.896) or tonic REM (p = 0.974) sleep. E, F, This same intervention did not decrease the number of muscle twitches during REM sleep (p = 0.231) or the amplitude of muscle twitches (p = 0.197). All values are means ± SEM; *p < 0.05 and **p < 0.001; A.U., arbitrary units.
The reduction in masseter muscle activity during blockade of non-NMDA receptors was comparable with that observed when both non-NMDA and NMDA receptors were both inactivated. We found that simultaneous application of both CNQX and d-AP-5 decreased left masseter muscle activity during AW by 79% and during phasic REM sleep by 90%; application of CNQX alone decreased muscle tone by the same magnitude, decreasing it by 81% in AW and by 86% in phasic REM sleep. There was no significant difference between the magnitude of masseter tone suppression during combined antagonism versus non-NMDA antagonism alone (two-way ANOVA, p = 0.828), suggesting that non-NMDA receptors mediate glutamatergic drive onto trigeminal motoneurons.
However, we found that antagonism of NMDA receptors alone also decreased left masseter EMG activity but only during waking. Application of d-AP-5 into the left trigeminal nucleus reduced left masseter tone during waking by 49% (p = 0.034, two-way RM ANOVA) (Fig. 7D); however, this intervention had no affect on masseter tone during either NREM (p = 0.896) or tonic REM (p = 0.974) sleep, nor did it affect muscle twitches during phasic REM sleep. Application of d-AP-5 did not affect (relative to baseline) either the number (p = 0.109, paired t test) (Fig. 7E) or amplitude (p = 0.197) (Fig. 7F) of muscle twitches during REM sleep.
Glutamate receptor agonism potently activates trigeminal motoneurons during all behavioral states except tonic REM sleep
Glutamate application
Having demonstrated that glutamate exerts an endogenous excitatory drive on trigeminal motoneurons during waking and phasic REM sleep, but not during either NREM or tonic REM sleep, we wanted to determine whether exogenous application of glutamate during sleep could restore basal levels of masseter tone to waking levels. We did this by applying 25 mm glutamate onto trigeminal motoneurons while measuring levels of masseter tone during sleep and waking. In six rats, glutamate was microdialyzed into the left trigeminal motor pool for 3.0 ± 0.21 h, over which time it had a potent excitatory effect on trigeminal motoneurons resulting in an increase in left masseter tone (Fig. 8A).

Exogenous glutamate application at the trigeminal motor pool potently increases masseter muscle tone during all behavioral states except tonic REM sleep. A, A typical example showing that perfusion of glutamate into the left trigeminal motor pool potently increases left masseter muscle (LM) tone during both alert waking and during NREM sleep. Remarkably, glutamate application is unable to overcome the atonia of tonic REM sleep; however, it does increase the number of muscle twitches during phasic REM. B, Group data (n = 6) demonstrating that exogenous glutamate application significantly increased left masseter muscle tone in waking and NREM sleep (p < 0.002), without changing levels of motor tone during tonic REM sleep (p = 0.916). C, D, Perfusion of glutamate into the left trigeminal motor pool also increased the number of muscle twitches during phasic REM sleep, but there was no change in the amplitude of muscle twitches after glutamate application (p = 0.099). All values are means ± SEM; *p < 0.05 and **p < 0.002; A.U., arbitrary units.
During glutamate perfusion, masseter EMG activity increased (relative to baseline) by 128% in AW (p < 0.001, two-way RM ANOVA) (Fig. 8B), by 332% in QW (p < 0.001), and by 556% in NREM sleep (p = 0.002). The excitatory actions of glutamate were maintained with equipotency (i.e., receptors did not desensitize to the stimulus) across the application period; basal levels of masseter muscle tone were the same at the beginning and end of the 3 h application period (p = 0.199, paired t test, first 30 s vs last 30 s NREM sleep).
Glutamate application during NREM sleep increased masseter muscle activity during this state to levels that were not significantly different from those during waking under baseline conditions (p = 0.713) (Fig. 6B), suggesting that sleeping levels of muscle tone can be restored to waking levels by replacing the glutamate that is naturally withdrawn from trigeminal motoneurons during NREM sleep. However, this same excitatory stimulus had no effect on masseter tone during tonic REM sleep (p = 0.916) (Fig. 8B). Figure 9A depicts a typical example illustrating that the stimulatory effects of glutamate during NREM sleep are rapidly abolished during entry into REM sleep.

The excitatory effects of glutamate are rapidly lost during entry into REM sleep but immediately regained in post-REM waking. A, A typical example showing that glutamate perfusion into the left trigeminal motor pool causes a potent activation of left masseter muscle (LM) activity during NREM sleep, but this excitatory effect is immediately abolished during entrance into REM sleep. Note that right masseter muscle (RM) tone is unaffected by glutamate application. B, Group data demonstrating that the excitatory effects of glutamate on trigeminal motoneurons are present in NREM sleep (glutamate vs baseline, p < 0.001) but rapidly lost on entry into tonic REM sleep (glutamate vs baseline, p = 0.916) and immediately regained during post-REM waking (glutamate vs baseline, p < 0.001). All values are means ± SEM; A.U., arbitrary units.
To demonstrate that the mechanism responsible for blocking the excitatory effects of glutamate were rapid, reversible, and specific to REM sleep, we analyzed levels of masseter EMG activity in the 30 s immediately preceding and after each REM period. In the 64 REM periods analyzed (mean REM duration, 65.4 ± 13.5 s), we found that glutamate increased masseter tone (relative to baseline levels) in all pre-REM (i.e., NREM; p < 0.001) and post-REM (i.e., waking; p < 0.001) periods (Fig. 9B). However, there was rapid and complete loss of the excitatory effects of glutamate during periods of tonic REM sleep that resulted in the persistence of motor atonia despite the continued presence of glutamate at the trigeminal motor pool (Fig. 9A,B). This nullifying effect was immediately reversed during entrance into post-REM waking, therefore demonstrating that the excitatory actions of glutamate are rapidly regained during entry into waking.
Although exogenously applied glutamate had no effect on trigeminal motoneuron activity during periods of tonic REM sleep, it significantly affected masseter muscle activity during periods of phasic REM sleep. Compared with baseline levels, the number of muscle twitches increased by 200% during phasic REM (p = 0.035, paired t test) (Fig. 8C), with the amplitude of muscle twitches being unaffected by glutamate perfusion (p = 0.099) (Fig. 8D).
AMPA and NMDA application
To demonstrate that the loss of the excitatory effects of glutamate during tonic REM sleep was not attributable to an insufficient dosage of glutamate, we also activated trigeminal motoneurons with doses of non-NMDA and NMDA receptor agonists that have been demonstrated previously to potently excite motoneurons (Steenland et al., 2006). We applied either 0.1 mm AMPA or 0.1 mm NMDA into the left trigeminal motor pool during both sleep and wakefulness.
Compared with baseline, AMPA increased left masseter EMG activity by 167% in AW (n = 6; p < 0.001, two-way RM-ANOVA) (Fig. 10A) and by 934% in NREM sleep (p = 0.002). The excitatory effects of AMPA were rendered completely ineffective during tonic REM sleep (p = 0.879). The number of phasic muscle twitches was significantly increased with AMPA application (p < 0.001, paired t test) (Fig. 10B), but the amplitude of phasic activity was unchanged (p = 0.978) (Fig. 10C). There was a rapid loss of the excitatory actions of AMPA at the transition of NREM to REM sleep (p = 0.012) that resulted in the persistence of atonia, but the stimulatory effects of AMPA were immediately reinstated during entrance into post-REM waking (p = 0.032).

AMPA and NMDA at the trigeminal motor pool increase masseter muscle tone during waking and NREM sleep but not during tonic REM sleep. A, Group data (n = 6) demonstrating that AMPA application significantly increased left masseter muscle tone in waking and NREM sleep, without changing levels of motor tone during tonic REM sleep (p = 0.879). B, C, During phasic REM sleep, AMPA application increased the number of muscle twitches without affecting twitch amplitude (p = 0.978). D, NMDA application also increased masseter muscle tone in waking and NREM sleep, without changing levels of muscle tone during tonic REM sleep (p = 0.895). E, F, During phasic REM sleep, NMDA application did not increase either the number (p = 0.192) or amplitude of muscle twitches (p = 0.688). All values are means ± SEM; **p < 0.003; A.U., arbitrary units.
Activation of NMDA receptors on trigeminal motoneurons also increased masseter tone during AW by 95% (n = 6; p < 0.001, two-way RM-ANOVA) (Fig. 10D) and in NREM sleep by 267% (p = 0.003) but not during tonic REM sleep (p = 0.895). There was no significant change in either the amount of phasic activity during REM sleep (p = 0.192, paired t test) (Fig. 10E) or the amplitude of muscle twitches (p = 0.688) (Fig. 10F) during NMDA application. There was a rapid loss of the excitatory effects of NMDA as soon as tonic REM sleep began (p = 0.024), and these effects were rapidly regained during post-REM waking (p = 0.022).
Discussion
Skeletal muscle tone is regulated across the sleep–wake cycle: it is maximal during alert waking, suppressed at sleep onset and during NREM sleep, and minimal or absent during REM sleep except for periodic muscle twitches. Here we provide the first evidence that an endogenous excitatory glutamatergic drive onto motoneurons is a contributing factor controlling the stereotypical pattern of muscle tone during wakefulness, NREM sleep, and phasic REM sleep but not during tonic REM sleep.
An endogenous glutamatergic drive mediates somatic motoneuron excitability and basal muscle tone during alert and quiet waking
We demonstrate that a functional glutamatergic drive contributes to motoneuron activity during natural motor behaviors. We show that antagonism of ionotropic glutamate receptors on trigeminal motoneurons significantly suppresses masseter tone during waking, indicating that a glutamatergic drive plays an important role in controlling motoneuron excitability and motor tone during waking behaviors. This waking glutamatergic drive is transduced by both non-NMDA and NMDA receptors. However, non-NMDA receptors play the predominant role because their blockade reduced waking masseter tone by 82%, whereas blockade of NMDA receptors only reduced masseter tone by 49%. This observation confirms previous findings that non-NMDA receptors transduce the majority of the excitatory effects of glutamate on motoneurons (Funk et al., 1993; Del Negro and Chandler, 1998).
Additional excitatory neuromodulators also contribute to waking levels of muscle tone because non-NMDA and NMDA antagonism did not eliminate basal muscle tone. Possible sources of excitatory wake-related drives to motoneurons include inputs from orexinergic (hypocretinergic), serotonergic, and noradrenergic cell groups, which not only project to and facilitate motoneuron excitation (Peever et al., 2003; Yamuy et al., 2004; Fenik et al., 2005; Lee et al., 2005), but also discharge maximally during wakefulness (McGinty and Harper, 1976; Horvath et al., 1999; Mileykovskiy et al., 2005).
Withdrawal of a waking glutamatergic drive contributes to the suppression of muscle tone in NREM sleep
We report that withdrawal of a wake-related glutamatergic drive during NREM sleep is, at least in part, responsible for the reduction in basal muscle tone observed during this state. We found that antagonism of ionotropic glutamate receptors on trigeminal motoneurons had no effect on muscle tone during NREM sleep, indicating that there is negligible glutamatergic excitation during this state. We also found that glutamate receptor antagonism reduced waking muscle tone to NREM sleep levels, thus suggesting that withdrawal of this excitatory drive contributes to muscle tone suppression in NREM sleep.
Although withdrawal of glutamatergic excitation may be the primary neurochemical responsible for reducing motor tone in NREM sleep, other transmitters may also regulate motoneuron activity during this state. Indeed, we recently identified the presence of functional excitatory noradrenergic and inhibitory glycinergic drives that contribute to muscle activity during NREM sleep (Mir et al., 2006; Brooks and Peever, 2008); similar NREM drives have also been identified in the hypoglossal motor pool (Morrison et al., 2003; Chan et al., 2006).
Glutamatergic control of muscle tone during phasic and tonic REM sleep
Motor control in REM sleep is unique compared with either waking or NREM sleep because it is characterized by flurries of periodic muscle twitches that occur on a background of motor atonia. We demonstrate that an endogenous glutamatergic drive is sufficient for generating muscle twitches in REM sleep. We show that antagonism of non-NDMA receptors on trigeminal motoneurons abolishes muscle twitches without affecting REM atonia. However, antagonism of NMDA receptors had no affect on twitch activity during REM sleep; this may be related to the fact that voltage-dependent NMDA receptors are rendered inactive in REM sleep when motoneurons are hyperpolarized (Chase and Morales, 1983; Mayer et al., 1984). These findings suggest that muscle twitches are mediated by glutamate release and confirms intracellular studies in cats showing that trigeminal motoneurons receive non-NMDA-mediated glutamatergic excitation during phasic REM sleep (Soja et al., 1995).
Two recent studies suggest that inspiratory genioglossus muscle activity is suppressed during REM sleep because noradrenergic excitation of hypoglossal motoneurons is withdrawn during this state (Fenik et al., 2005; Chan et al., 2006). If loss of an excitatory noradrenergic drive at nonrespiratory motor pools is also responsible for suppressing basal muscle tone during REM sleep, then pharmacologically activating trigeminal motoneurons should reverse REM atonia. Our results show that, despite potent glutamatergic excitation of trigeminal motoneurons during REM sleep, we were unable to override REM atonia. We conclude that reduced excitation of motoneurons does not trigger REM sleep atonia; however, this mechanism may underlie the suppression of inspiratory activity during REM sleep. This contention is supported by the demonstration that norepinephrine release is decreased within the hypoglossal motor pool during stimulation-induced REM atonia (Lai et al., 2001).
The loss of glutamatergic excitation during tonic REM sleep is in striking contrast to the excitatory response that glutamate evokes on muscle tone during all other behavioral states. We show that the excitatory effects of glutamate are rapidly lost on entrance into REM sleep and immediately regained on exiting it. Previous findings also report that the excitatory actions that neuromodulators (e.g., norepinephrine) have on trigeminal (Mir et al., 2006) and hypoglossal motoneuron activity are also nullified during REM sleep (Chan et al., 2006). We therefore conclude that motor atonia is mediated by a powerful REM-specific inhibitory mechanism (Chase et al., 1989; Kodama et al., 2003). This contention is strengthened by the fact that glutamatergic activation during REM sleep failed to increase muscle twitch amplitude; this lack of effect probably occurs because motoneurons are maximally hyperpolarized during periods of phasic REM sleep (Chase et al., 1989), which obviates the excitatory actions of glutamate on motoneurons, thereby limiting muscle twitch magnitude.
Intracellular studies show that somatic motoneurons are bombarded by inhibitory glycinergic and GABAergic potentials during natural REM sleep (Nakamura et al., 1978; Soja et al., 1987), and microdialysis studies demonstrate that glycine and GABA release onto motoneurons is increased during pharmacologically induced muscle atonia (Kodama et al., 2003). However, neither glycinergic nor GABAergic inhibition are sufficient to induce motor suppression in REM because antagonism of glycine and GABAA receptors at either the trigeminal or hypoglossal motor pools does not reverse REM atonia (Morrison et al., 2003; Brooks and Peever, 2008). This indicates that another inhibitory mechanism must be responsible for REM sleep atonia; the source of inhibition remains unknown.
Determining the neural substrate responsible for inducing REM atonia is of major clinical significance. For example, if a REM-specific inhibitory mechanism could be pharmacologically reproduced, it could serve as powerful therapeutic tool to suppress the pathological movements associated with motor disorders such as Parkinson's disease. Indeed, De Cock et al. (2007) recently demonstrated that parkinsonian symptoms such as tremor, rigidity, and bradykinesia are present in both waking and NREM sleep but abolished during REM sleep in Parkinson's patients (De Cock et al., 2007). This observation is consistent with our findings that glutamate-induced motor excitation is blocked only in REM sleep.
What is the source of the endogenous glutamatergic drive to motoneurons?
The major implication of this study is that a functional glutamatergic drive onto motoneurons regulates levels of muscle tone during wakefulness and phasic REM sleep. A possible source of this drive could be the medial reticular formation, a brainstem region that plays a pivotal role in coupling arousal state and motor activity (Siegel et al., 1991). Glutamate-containing cells in this region project to and facilitate motoneuron activity (Davidson et al., 2007); most cells in this region also discharge maximally in REM sleep, moderately in waking, and minimally in NREM sleep (Siegel et al., 1983, 1992). These cells could therefore provide the source of endogenous drive onto motoneurons during both REM sleep and waking; the inactivity of these cells during NREM could also explain the absence of a glutamatergic drive during this state.
Another potential and very potent source of a waking glutamatergic drive could be from the dopaminergic, noradrenergic, and serotonergic cell groups that promote arousal and facilitate motor activity (Wu et al., 1999; Jacobs et al., 2002; Lu et al., 2006). These monoaminergic cells not only exhibit a wake–active discharge pattern, project to and excite motoneurons, but they also synthesize and corelease glutamate (for example, 86% of noradrenergic cells in the locus ceruleus coexpress glutamate) (Liu et al., 1995). Therefore, in addition to providing an endogenous waking drive (e.g., from norepinephrine), monoaminergic cells may also be a major source of glutamatergic input to motoneurons during wakefulness (Fung et al., 1994; Bouryi and Lewis, 2003). In fact, several lines of evidence demonstrate that motor activation produced by monoaminergic stimulation is primarily mediated by glutamate, with monoamine activity playing a minor role (Bouryi and Lewis, 2003; Trudeau, 2004). Therefore, it is possible that monoaminergic cell groups mediate their excitatory actions on motoneurons primarily by glutamatergic excitation; this is consistent with our findings that a prevailing glutamatergic drive is responsible for controlling levels of motor tone during waking. That glutamate plays the dominate role in regulating basal motor tone may explain why pharmacological strategies aimed at enhancing monoaminergic neurotransmission (e.g., serotonin reuptake inhibitors) have limited success at increasing motor tone during sleep and cataplexy (Berry et al., 1999; Sonka et al., 2006; Horner, 2007).
Footnotes
-
This work was supported by grants from the Canadian Institutes of Health Research and Natural Sciences and Engineering Research Council of Canada (J.P.). J.S. was supported by the Medical Research Service of the Department of Veterans Affairs, National Institutes of Health Grant NS14610, and United States Public Health Service Grant HL41370. We are grateful to Dr. Richard Horner and Patti Brooks for their helpful comments. We also thank Saba Mir and Jahan Salma for their technical assistance.
- Correspondence should be addressed to Dr. John Peever, Department of Cell and Systems Biology, University of Toronto, 25 Harbord Street, Toronto, Ontario, Canada M5S 3G5. john.peever{at}utoronto.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}