

Abstract
Glutamate transporters are expressed throughout the CNS where their major role is to clear released glutamate from presynaptic terminals. Here, we report a novel function of the transporter in rat pinealocytes. This electrogenic transporter conducted inward current in response to l-glutamate and l- or d-aspartate and depolarized the membrane in patch-clamp experiments. Ca2+ imaging demonstrated that the transporter-mediated depolarization induced a significant Ca2+ influx through voltage-gated Ca2+ channels. The Ca2+ rise finally evoked glutamate exocytosis as detected by carbon-fiber amperometry and by HPLC. In pineal slices with densely packed pinealocytes, glutamate released from the cells effectively activated glutamate transporters in neighboring cells. The Ca2+ signal generated by KCl depolarization or acetylcholine propagated through several cell layers by virtue of the regenerative “glutamate-induced glutamate release.” Therefore, we suggest that glutamate transporters mediate synchronized elevation of l-glutamate and thereby efficiently downregulate melatonin secretion via previously identified inhibitory metabotropic glutamate receptors in the pineal gland.
Introduction
Excitatory amino acid transporters (EAATs) sequester the excitatory amino acids (EAAs) glutamate and aspartate from synaptic clefts in the mammalian CNS (Danbolt, 2001; Tzingounis and Wadiche, 2007). Their major function is to terminate glutamatergic signal transmission preparing postsynaptic glutamate receptors for the next transmission from the presynaptic terminal. Rapid removal of extracellular glutamate is also important for reducing toxic effects of the neurotransmitter (Weiss et al., 1990; Rothstein et al., 1996). So far, five different Na+-dependent glutamate transporters have been cloned: GLAST (glutamate–aspartate transporter) (homolog of EAAT1 cloned in human) (Storck et al., 1992), GLT-1 (EAAT2) (Pines et al., 1992), EAAC1 (EAAT3) (Kanai and Hediger, 1992), EAAT4 (Fairman et al., 1995), and EAAT5 (Arriza et al., 1997). Each subtype is expressed in different cell types and brain regions and facilitates glutamate uptake into neurons or glial cells.
The mammalian pineal gland is one of several brain regions containing high levels of l-glutamate and l- and d-aspartate (Schell et al., 1997). The synaptic-like microvesicles (SLMVs) of pineal cells contain l-glutamate and l-aspartate, which are secreted through regulated exocytosis (Yatsushiro et al., 1997). d-Aspartate is also abundant in the cytoplasm of pinealocytes and is gradually released through a pathway distinct from SLMV-mediated exocytosis (Ishio et al., 1998). After release, the EAAs are resequestered through a GLT-1-type glutamate transporter (Yamada et al., 1997b; Yatsushiro et al., 1997). In addition to their role in glutamate clearance, glutamate transporters can contribute to the membrane potential because they are electrogenic. One stoichiometric uptake cycle transports three Na+, one H+, and one negatively charged glutamate into the cell and one K+ out of the cell, resulting in the net influx of two positive charges (Zerangue and Kavanaugh, 1996; Levy et al., 1998; Owe et al., 2006). Therefore, forward operation of EAATs depolarizes the membrane, an effect most prominent with a negative resting potential, with a high density of the transporters, and with a high membrane input resistance. Furthermore, there is a nonstoichiometric conductance for small anions (such as Cl−) associated with active EAATs that would serve as an additional influence on the membrane potential. Therefore, we addressed whether glutamate transporter currents contribute to the excitability of pinealocytes. Our biophysical and biochemical measurements demonstrate that pineal glutamate transporters can depolarize the membrane enough to activate voltage-gated Ca2+ channels, increase intracellular Ca2+ concentration ([Ca2+]i), and finally induce Ca2+-dependent exocytosis of l-glutamate. We discuss the potential role of this transporter-mediated signaling in terms of melatonin production in the pineal gland based on the fact that aspartate and glutamate inhibit melatonin synthesis (Yamada et al., 1997a; Ishio et al., 1998).
Materials and Methods
Cell culture.
Dissociated pinealocytes were prepared from male Sprague Dawley rats (4–6 weeks old) as described previously (Yamada et al., 1996a). Briefly, pineal glands were removed from rats killed with CO2, according to University of Washington and Okayama University animal guidelines. The glands were dissected into small pieces in cold HBSS (Invitrogen) containing 5 mm d-glucose, 20 mm HEPES, and 1 mg/ml BSA, and treated with 4 mg/ml collagenase (Sigma-Aldrich) in HBSS solution at 37°C for 30–40 min. Cell clusters were dispersed by trituration in a Ca2+-free HBSS containing 1 mm EGTA, 20 mm HEPES, and 10 mg/ml BSA. Isolated cells were plated on glass coverslips and kept at 37°C in a 5% CO2 incubator for 1–2 d in RPMI 1640 culture medium (Invitrogen) containing 10 mm glucose, 10% FBS, 100 μg/ml streptomycin, and 100 IU/ml penicillin before use. For single-cell experiments, pinealocytes were visually identified using differential interference contrast (DIC) optics (see Results).
Preparation and measurement of pineal slices.
A dissected pineal gland was dropped into warmed (39°C) HBSS containing 2% liquefied agar (Difco), sucked into the end of a glass Pasteur pipette (inner diameter, 5.5 mm; VWR Scientific), and cooled rapidly in cold HBSS. The solid agar block containing the gland was sliced using a tissue vibratome (series 1000; Vibratome) in cold HBSS bubbled with 95% O2 and 5% CO2 (Takahashi, 1978). Slices were incubated for at least 30 min at room temperature with gas bubbling and used for experiments during the next 5 h. Pinealocytes in the pineal slices were visualized using infrared DIC video microscopy using a Pixelfly CCD camera (Applied Scientific) on an Eclipse 80i upright microscope (Nikon).
Solutions and drugs.
The standard extracellular saline solution for single-cell measurements contained the following (in mm): 137.5 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.3 adjusted with NaOH. For Ca2+-free extracellular saline solution, CaCl2 was omitted and 100 μm EGTA was added to remove contaminating free Ca2+. For an extracellular saline solution containing high KCl, NaCl was replaced by equimolar KCl. For whole-cell recordings, 10 mm NaCl was replaced by 10 mm tetraethylammonium chloride (TEA-Cl) to block abundant delayed rectifier K+ channels (Aguayo and Weight, 1988). Internal pipette solutions contained the following (in mm): 125 KCl or CsCl, 15 TEA-Cl, 5 HEPES, 1 MgCl2, 4 NaCl, 10 EGTA, pH 7.3 adjusted with KOH or CsOH. For the experiment with impermeable N-methyl-d-glucamine (NMDG) ions, 125 mm KCl was replaced by equimolar NMDG-Cl in the internal solution. Some experiments used SCN−-based internal solution (in mm: 130 KSCN, 10 TEA-Cl, 1 MgCl2, 10 EGTA, 20 HEPES, pH 7.3 adjusted with KOH). EGTA and TEA were included in the internal solution to block Ca2+-activated or other K+ channels. For the measurement of glutamate secretion, the extracellular saline solution contained the following (in mm): 128 NaCl, 1.9 KCl, 1.2 KH2PO4, 2.4 CaCl2, 1.3 MgSO4, 26 NaHCO3, 10 d-glucose, 10 HEPES, pH 7.4 adjusted with NaOH, and the Ca2+-free saline solution included 0.2 mm CaCl2, 1 mm EGTA, and 3.8 mm MgSO4, whereas other components remained same.
Electrical recording and fast application of substrates.
All current measurements were performed using the whole-cell configuration with an EPC-9 patch-clamp amplifier (HEKA Elektronik). When filled with internal solution, patch pipettes had a resistance of 4–6 MΩ. The low-pass filter frequency was 1 kHz, and the sampling rate was 5 kHz.
For rapid application of agonists, we used a double-barreled pipette fabricated from theta glass tubing (outer diameter, 2.0 mm; inner diameter, 1.4 mm; 0.2 mm septum; Harvard Apparatus) and a Piezo-electric element (Noliac) (Dudel et al., 1990). The solution exchange time (20–80%) was 500–800 μs when measured with an open patch pipette during a change between 100% saline and 10% saline solutions. To improve the solution exchange, recorded cells were lifted from the coverslip: After the rupture of the membrane patch, strong negative pressure (∼200 mbar) was maintained for 1–2 min to draw the nucleus to the patch pipette tip. The nucleated cells then could be lifted easily from the bottom and positioned in front of the rapid application system: Single isolated cells, especially ones of round shape, were not yet firmly attached to the substrate 1–2 d after plating. With the lifted cells, the solution exchange was estimated to take 2–3 ms, as judged with K+ currents during a switch of external K+ concentrations from 137.5 to 2.5 mm. In some experiments, rapid actuation of the theta tube perturbed the patched cell, and produced consistent but minor (<1 pA) baseline fluctuations shortly before current activation (see Fig. 1A).
For perforated whole-cell recordings the patch pipettes were front-filled with the normal internal solution and back-filled with the internal solution containing amphotericin B (Horn and Marty, 1988). Recordings started when input resistance became <30 MΩ. All patch-clamp measurements were made at room temperature (22–24°C).
Ca2+ imaging.
Intracellular Ca2+ concentration ([Ca2+]i) was measured using the Ca2+-sensitive fluorescent dye fura-2. The cells were preincubated with 2 μm membrane-permeable fura-2 AM in saline solution for 30 min at room temperature. Fluorescence in single pinealocytes was imaged using an inverted microscope (TE-300; Nikon) fitted with a 175 W xenon arc lamp, a Lambda 10-2 optical filter changer (Sutter), and a Coolsnap fx CCD camera (Photometrics). The excitation wavelengths were 340 and 380 nm, and fluorescence signals were recorded at 510 nm using Metafluor software (Universal Imaging). The sampling rate was 1 Hz and the background fluorescence measured from a cell-free area was subtracted. [Ca2+]i was calculated as described previously (Grynkiewicz et al., 1985; Herrington et al., 1996). For Ca2+ imaging experiments, solutions were exchanged by a local perfusion system that allowed complete exchange of medium within 0.5 s (Koh and Hille, 1997).
For Ca2+ measurements in pineal slices, fura-2 AM concentration and temperature were raised to 4 μm and 37°C, respectively, to facilitate the dye loading into compact tissues (Yuste, 2000). A small flat spatula was used to transfer pineal slices to the loading solution and the recording chamber to minimize damage to the circular agar block supporting the pineal slice. To achieve a relatively fast (<1 s) solution exchange, we designed a new chamber (see supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Subtraction of background fluorescence and calibration of the dye could not be achieved in the slice dense with neighboring cells. Therefore, we present fluorescence ratios instead of calibrated [Ca2+]i values. All Ca2+ imaging experiments were performed at room temperature (22–24°C).
Measurement of exocytosis.
Vesicular exocytosis was measured using carbon fiber amperometry as described previously (Kim et al., 2000). First, SLMVs of cultured pinealocytes were acutely loaded with oxidizable dopamine by fluid-phase endocytosis. This was accomplished by stimulating exocytosis and membrane recycling with a K+-rich solution for 2 min at room temperature in the presence of high concentrations of dopamine (in mm: 67.5 dopamine, 67.5 KCl, 2 NaCl, 2 CaCl2, 1 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.3 adjusted with NaOH). In pinealocytes, the KCl stimulation evokes exocytosis preferentially of SLMVs instead of large dense-core vesicles containing serotonin (Yamada et al., 1996a, 2002). Cells were incubated for 5–10 min in a dopamine-free saline solution before measurements. Vesicular release of the loaded dopamine was monitored as pulses of electric current generated by oxidation of the molecules at the tip of the carbon fiber electrode polarized to +400 mV and gently touching a cell. Carbon fiber microelectrodes were fabricated from 11 μm carbon fibers and polypropylene 10 μl micropipettor tips (Koh and Hille, 1999). Amperometric currents were recorded with an EPC-9 amplifier, filtered at 100 Hz, sampled at 500 Hz, and later analyzed using a macro written in IGOR Pro (WaveMetrics). For amperometric experiments, solutions were exchanged within 0.5 s using a local perfusion system that warmed the solutions to 34–35°C.
Measurement of glutamate and melatonin secretion.
Pinealocytes (106 cells per dish) were preincubated with 2 ml of saline solution for 30 min and then challenged with l- or d-aspartate for 15 min at 37°C. The released glutamate was determined by HPLC with precolumn o-phthalaldehyde derivatization, separation by a reverse-phase Resolve C18 column (4.6 × 150 mm; Waters Ltd.), and fluorescence detection. Melatonin secreted from freshly dissected pineal glands was collected for 6 h at 37°C and measured using an ELISA kit (Alpco Diagnostics). Each measurement used one or two half-glands.
Data analysis.
Analysis was performed with IGOR Pro (WaveMetrics). Concentration–response curves were fitted with the Hill equation: f(c) =1/[1 + (EC50/c)a], where c denotes substrate concentration, EC50 represents the half-maximal effective concentration, and a is the Hill coefficient. All numerical values are given as mean ± SEM. The number of measured cells is indicated by n in the text. Statistical difference of two groups was evaluated by Student's t test. Probabilities of *p ≤ 0.01 or **p ≤ 0.001 were considered significant.
Results
General morphology and electrical properties of pinealocytes
Pinealocytes, the principal cell type in the pineal gland, were cultured for 1–2 d and visually identified by tiny spots on the cell surface (Yatsushiro et al., 2000), structures that may correspond to extensive surface membrane blebs observed by electron microscopy (Aguayo and Weight, 1988). In agreement with previous reports (Aguayo and Weight, 1988; Castellano et al., 1989), the pinealocytes selected this way exhibited both TEA (10 mm)-sensitive delayed rectifier and 4-AP (4-aminopyridine) (5 mm)-sensitive transient outward K+ currents in dissociated preparations and in slices (for the transient K+ currents, see Fig. 8A). Average membrane capacitance measured in the whole-cell patch configuration was 10.1 ± 0.4 pF (n = 141).
The pineal glutamate transporter conducts inward ionic currents
We begin by describing aspartate- and glutamate-induced currents in patched pinealocytes (Fig. 1). Many of the experiments use l-aspartate because it is a poor agonist for ionotropic glutamate receptors (iGluRs) yet is well transported by EAATs. Rapid application of l-aspartate to a lifted whole cell using a piezo-driven solution exchange system (see Materials and Methods) evoked small inward currents with an average peak current density of 1.1 ± 0.1 pA/pF (n = 18) at a holding potential of −80 mV (Fig. 1A,B). With a Cs+-based internal solution, the current was transient, declining to the basal level with a time constant of 4.0 ± 0.3 ms. Currents activated by l-glutamate showed similar amplitudes and time courses. l-Glutamate is the endogenous agonist for iGluRs and is also well transported by EAATs. Typical iGluRs would show linear, outwardly, or doubly rectifying I–V relationships reversing near 0 mV, with significant outward currents above +40 mV for most subtypes (Koh et al., 1995; Dingledine et al., 1999). In contrast, for both l-aspartate and l-glutamate, the pinealocyte currents were primarily inward, did not reverse at 0 mV, and did not show large outward current at more positive potentials (Fig. 1A, current–voltage relationships). Such characteristics have been reported instead for currents in endogenous and cloned EAATs (Brew and Attwell, 1987; Eliasof and Werblin, 1993; Wadiche et al., 1995b; Bergles et al., 1997, 2002; Grewer et al., 2000). d-Aspartate also effectively induced current, whereas d-glutamate failed to do so (Fig. 1B). Dose–response curves for peak currents show that the three active amino acids evoke currents at high micromolar concentrations (Fig. 1C–E). Half-maximal effective concentrations (EC50) of 70–306 μm are comparable with those obtained with the GLT-1-type glutamate transporter of brain (Danbolt, 2001; Bergles et al., 2002) but higher than with the EAAC1 subtype (Grewer et al., 2000).

Glutamate-transporter mediated inward currents in cultured pinealocytes. A, Top, Family of currents activated by rapid application of 1 mm l-aspartate (left) or 1 mm l-glutamate (right) at membrane potentials between −80 and +40 mV in 20 mV steps. Solutions were applied to the lifted cell with exchange within a few milliseconds. Each trace is the mean of four records. All recordings were from the same cell with CsCl-based internal solution. Bottom, Current–voltage (I–V) relationships of peak currents evoked by l-aspartate (□) or l-glutamate (■) fitted with third-order polynomials. B, Average current density at V = −80 mV with several agonists of glutamate transporters and iGluRs applied at 1 mm, except AMPA and KA at 0.5 mm. Measured peak currents were divided by the capacitance of each cell. For l-, d-aspartate and l-glutamate, only cells showing current were included in the analysis. The NMDA application included 10 μm glycine, and external Mg2+ ions were omitted. C–E, Concentration dependence of currents activated by three excitatory amino acids normalized to the value with 10 mm in each cell. The averages were fitted with the Hill equation (see Materials and Methods). Half-maximal effective concentrations (Hill coefficient) are 89 μm (1.1), 73 μm (1.2), 306 μm (0.7) for l-aspartate, l-glutamate, and d-aspartate, respectively. Shown is the mean of three to four cells at −80 mV with CsCl-based internal solution. Error bars indicate SEM.
Although some properties of these currents, such as the rapid activation and decrease, are reminiscent of currents in iGluRs, the general features are more consistent with mediation by glutamate transporters. In accordance, several agonists of iGluRs such as NMDA (n = 13), AMPA (n = 6), and kainate (KA) (n = 7) failed to activate any current (Fig. 1B). In contrast, the same AMPA and kainate solutions (both 500 μm) routinely evoked ∼100 pA of current mediated by AMPA-type glutamate receptors in pancreatic islet cells (J.-H. Cho and D.-S. Koh, unpublished observations). In addition, several antagonists of iGluRs failed to reduce the currents evoked by l-aspartate. The aspartate-evoked currents were not significantly reduced by a high concentration of 6-cyano-2,3-dihydroxy-7-nitro-quinoxaline (CNQX) (100 μm), a specific blocker of AMPA-type glutamate receptors (94 ± 24% of control at V = −80 mV; n = 3; p = 0.16), or by 50 μm d-2-amino-5-phosphonopentanoate (d-AP5), a specific blocker of NMDA-type glutamate receptors (103 ± 26%; n = 3; p = 0.52).
We tested the l-aspartate-evoked currents for two known ionic features of EAAT currents: sensitivity to intracellular cations and sensitivity to intracellular anions. First, we consider the cations and models for glutamate transporters. The transport cycle of GLT-1 (Levy et al., 1998; Bergles et al., 2002) and EAAC1 (Grewer et al., 2000) transporters has been described in terms of a complex kinetic model with multiple states of different orientation and different loading with Na+, H+, K+, and glutamate. At rest, the transporter is “outwardly facing.” On addition of glutamate or aspartate the transporter picks up three Na+, one H+, and one glutamate. This complex makes a transition to cytoplasmically facing states that successively unload the substrates, pick up one cytoplasmic K+, and returns to the outwardly facing condition to complete the cycle. This final return step is supposed to be slow and rate-limiting for the transport cycle. Without intracellular K+, the transporter cannot complete the second one-half of the transport cycle and becomes stuck in an inwardly facing state. The half-cycle it completes would transport one aliquot of Na3H-glutamate per transporter (+3 charges) inwardly. According to the Bergles model, Cs+ ion catalyzes the return step poorly, seven times slower than K+. This fits with the transient l-aspartate-evoked currents we see with a Cs+-based pipette solution; they decayed almost completely within 15 ms (n = 36) (Figs. 1A, 2A,B) at −80 mV. However, with K+ ion in the pipette, there was a considerable steady-state (S-S) current (n = 22) (Figs. 2C,D, 3), presumably as the transporter continued to cycle. This difference was apparent at all potentials. Similar residual S-S currents were observed with the EAAT substrates l-glutamate and d-aspartate (data not shown). As was also seen with heterologously expressed EAAC1 and GLT-1 transporters (Grewer et al., 2000; Bergles et al., 2002), the evoked peak and S-S currents had quite different sensitivities to l-aspartate (Fig. 2E,F). S-S currents were near maximal with 0.01 mm l-aspartate, whereas peak current was much increased in 1 mm l-aspartate.

Steady inward currents require intracellular K+ ions. Currents were activated by 1 mm l-aspartate with CsCl (A)- or KCl (C)-based internal solutions at V = −80 mV (different cells; means of 2–3 records). The dashed lines represent the basal current level. B, D, I–V relationships of peak (■) and S-S currents (□; mean between 350 and 650 ms) in the same cells as A and C. The lines are fitted third-order polynomials. E, Currents were activated by 0.01 and 1 mm l-aspartate with KCl-based internal solution at V = −80 mV in the same cell. Traces are means of two to three records. The arrow indicates the peak current. F, Average current density evoked by 0.01 mm l-aspartate (n = 3) and 1 mm l-aspartate (n = 9). Error bars indicate SEM.

Sensitivity of current to intracellular anions and pharmacology. A, l-Aspartate (1 mm) evoked a large current of >50 pA with an SCN−-rich intracellular solution. The current was reduced by 300 μm DHK or TBOA present continuously. Traces from one cell at V = −80 mV. Full recovery of the current from DHK and TBOA was tested (data not shown). B, Average current density evoked by 1 mm l-aspartate with KCl (from Fig. 2F)- or KSCN (n = 11)-containing internal solutions at V = −80 mV. S-S current was averaged between 35 and 65 ms. C, Block of 1 mm l-aspartate-evoked currents by 300 μm DHK (n = 3) and TBOA (n = 3). Peak and S-S currents with KSCN-based internal solution were analyzed as in B and compared with those of control. V = −80 mV. Error bars indicate SEM.
Transporting capacity of the pineal glutamate transporter
Using the currents mediated by transporters we calculated a rough estimate of the number of functional transporters and their substrate turnover numbers in pinealocytes. We assume that current transients that decline to the baseline (e.g., peak currents with the internal Cs+ solution) correspond to operation of a single half-cycle of inward glutamate transport that transfers a net three positive charges into the cell per transporter. Because Cs+ was suggested to support the full-cycle, albeit with a low cycling rate (∼6/s) (Bergles et al., 2002), we used an internal solution containing impermeant NMDG-Cl to block the outward transport completely. Average peak current activated by 1 mm l-aspartate at −80 mV was 0.9 ± 0.1 pA/pF (n = 3), and the current declined to the baseline with a time constant of 3.9 ± 0.3 ms. Therefore, the half cycle takes ∼5 ms, so the turnover number for the full cycle can be no more than 200/s. On addition of substrate at −80 mV, we saw inward current transients that moved ∼5 × 10−15 coulombs of charge per picofarad of membrane (the integral of the current transient is 5.2 fC/pF for l-aspartate). This corresponds to 31,000 electronic charges/pF crossing the cell membrane before all transporters become arrested in the inward facing configuration, or 310 charges/μm2 (assuming 100 μm2/pF for biological membranes). So we need at least 100 transporters/μm2. This would be an underestimate if at −80 mV adding glutamate does not engage all the transporters in a complete half-cycle of transport. Indeed, it is probable that the three transported Na+ ions have already been loaded before glutamate is delivered. This preloading of Na+ reduces charge moved after glutamate delivery to 2.5 instead of 3 (Wadiche et al., 1995a), so the estimated number of transporters would rise to 120/μm2. Furthermore, some fraction of transporters might not bind Na+ (Wadiche et al., 1995a).
What is the substrate turnover number? If the 0.5 pA/pF S-S current seen at −80 mV (with K+ internal) is carried by 120 transporters passing a stoichiometric net charge of +2 per full cycle, then each transporter would transport ∼130 glutamates/s. This rough estimate compares with that for GLT-1 (100/s) and EAAC1 or EAAT1 (66 and 17/s) (Wadiche and Kavanaugh, 1998; Bergles et al., 2002; Grewer and Rauen, 2005). The turnover rate is known to depend on intracellular Na+ and glutamate concentrations (Wadiche et al., 2006). Intracellular Na+ and glutamate reduce turnover compared with K+ alone. Using SBFI (sodium-binding benzofuran isophthalate) as a fluorescent indicator of Na+ in pinealocytes, we detected no cytoplasmic Na+ increase (n = 17) on activation of glutamate transporter by 1 mm l-aspartate as long as 30 s. In contrast, the Na+ ionophore monensin (20 μm) elicited significant increases of cytoplasmic Na+.
Together, pineal glutamate transporters would clear EAAs accumulated in the interstitial space in the pineal gland efficiently.
Anion sensitivity and pharmacology of the pineal glutamate transporter
Next, we consider the anion sensitivity of EAAT currents. The stoichiometric transport cycle of EAATs is accompanied by a nonstoichiometric anion conductance with the selectivity sequence, SCN− > ClO4− > NO3− > I− > Br− > Cl− > F− ≫ gluconate (Wadiche et al., 1995b; Wadiche and Kavanaugh, 1998; Danbolt, 2001; Bergles et al., 2002). In the glutamate transporter models, the nonstoichiometric conductance is associated with specific states of the active transporter (Grewer et al., 2000; Otis and Kavanaugh, 2000; Bergles et al., 2002). In the presence of transported substrates, even if nonstoichiometric current is not obvious when Cl− is the major anion, it should become more obvious when the solutions contain NO3− or SCN−. Correspondingly, switching from a KCl-based internal solution to a KSCN-based one, increased the l-aspartate-evoked peak current density (8.9 ± 1.5 pA/pF; n = 11) and the S-S current in pinealocytes approximately eightfold (Fig. 3A,B).
If the evoked currents are carried by EAATs, they should be sensitive to the competitive EAAT inhibitors, dihydrokainate (DHK) and dl-threo-β-benzyloxyaspartate (TBOA). SCN−-mediated currents were evoked by 1 mm l-aspartate in the absence and presence of 300 μm DHK (Fig. 3A,C). The peak response with KSCN was significantly blocked by DHK, but the S-S response was slightly increased. Exactly the same paradoxical effect has been reported for heterologously expressed EAAC1 and GLT-1 (Grewer et al., 2000; Bergles et al., 2002). The effect has been explained and modeled: DHK occupies the transporters at rest and prevents the initial synchronous conversion to a high-conducting state (peak current) on application of amino acid substrate; however, within a few milliseconds the DHK molecules dissociate and are competitively replaced by substrate, so the transporter returns to full steady-state operation. The evoked steady-state current appears larger than in control because the DHK had also blocked a small standing inward anion leak current at rest (already enhanced by SCN−); subsequent binding of the amino acid substrate restores this missing component of inward current as well. The lack of DHK block of S-S currents evoked by 1 mm l-aspartate might appear inconsistent with previous results obtained with GLT-1 (EAAT2) overexpressed in oocytes (Arriza et al., 1994; Wadiche et al., 1995a) where currents (presumably S-S current attributable to slow agonist application to oocytes) evoked by low (100 μm) l-glutamate were significantly blocked by DHK and kainate. However, DHK and kainate are competitive blockers so their blocking action depends on the relative concentrations of agonist and antagonist. In line with this explanation, with low concentrations of l-glutamate (100 μm) the S-S currents of the pineal glutamate transporters were significantly blocked by 500 μm DHK (relative current compared with control, 0.15 ± 0.08; n = 3) and kainate (0.21 ± 0.03; n = 5). TBOA (300 μm) depressed both peak and S-S currents (Fig. 3A,C). The block by DHK and TBOA was fully reversible (data not shown).
Glutamate transporter-mediated currents depolarize the membrane and increase intracellular Ca2+
Pinealocytes express L-type Ca2+ channels that are responsible for exocytosis of SLMVs containing l-glutamate and l-aspartate (Yatsushiro et al., 1997). Can inward transporter currents evoked by these transmitters depolarize the membrane potential enough to activate voltage-dependent Ca2+ channels and consequently increase intracellular Ca2+ concentration ([Ca2+]i)? To address this question, we first measured the membrane potential of pinealocyte in perforated whole-cell configuration. In the current-clamp mode with no current injection, the resting membrane potential was −45 ± 3 mV (n = 7). Because pinealocytes have a high input resistance (see below), we included only those cells with seal resistance >1 GΩ in the analysis for a reliable estimation of the membrane potential. Significant fluctuation of the resting membrane potential was observed, reflecting a high input resistance and a small K+ leakage conductance responsible underlying the resting potential. Then the cells were perfused with 1 mm l-aspartate via a local application system (exchange time, <0.5 s) (see Materials and Methods) (local solution exchange was used because we could not lift perforated whole cells). The neurotransmitter depolarized the membrane by 9 ± 2 mV (n = 3) (Fig. 4A). In the same cells, the evoked transporter current was 4 ± 1 pA (n = 3; at −60 mV) using voltage-clamp mode (Fig. 4C). Hence the membrane depolarization during l-aspartate application was induced by S-S transporter currents and pinealocytes have a high input resistance. An extracellular solution containing 50 mm KCl depolarized the membrane by 32 ± 7 mV as expected from the Nernst equation (n = 3) (Fig. 4B), and the K+ inward current elicited at −60 mV was 43 ± 19 pA (n = 3) (Fig. 4D).

Depolarization of the membrane potential by glutamate transporter-mediated currents. A, B, Membrane depolarizations evoked by 1 mm l-aspartate and 50 mm KCl (current-clamp mode). C, D, Membrane currents evoked by l-aspartate and KCl (voltage-clamp mode at V = −60 mV). All recordings were from the same cell in the perforated whole-cell configuration.
Next, we tested whether the small depolarization induced by the transporter is enough to activate voltage-gated Ca2+ channels and can increase [Ca2+]i (Fig. 5). The average resting [Ca2+]i measured with Ca2+-sensitive dye fura-2 was 101 ± 4 nm (n = 114). On application of 1 mm l-aspartate using the same local perfusion system as for measurement of the membrane potential, [Ca2+]i increased to a peak within a few seconds and then declined slowly (Fig. 5A–D). Most cells responded to l-aspartate (102 of 118 cells; 373 ± 41 nm Ca2+ increase in responding cells), l-glutamate (29 of 33; 360 ± 41 nm), and d-aspartate (11 of 13; 328 ± 17 nm), but no cells responded to the iGluR agonists d-glutamate (0 of 13), NMDA (0 of 11), AMPA (0 of 26), or kainate (0 of 13) (Fig. 5B,E). This agonist profile is identical to that for the glutamate transporter-mediated currents in Figure 1B.

[Ca2+]i rise induced by glutamate transporters. Ratiometric calcium traces as agonists at 1 mm (except AMPA and kainate, 0.5 mm) were applied for 30 s using a local perfusion system (exchange within 0.5 s). A, B, Three transporter substrates evoked Ca2+ responses. d-Glutamate and two specific agonists of iGluR did not. C, D, TBOA (300 μm) and nimodipine (5 μm) reduced the [Ca2+]i rise induced by 1 mm l-aspartate. DHK at the same concentration were less effective (shown in F). E, Summary of experiments like A and B. F, Summary of experiments like C and D, normalized to l-aspartate alone; DHK (300 μm; n = 21), TBOA (n = 30), nimodipine (n = 14), 50 μm d-AP5 (n = 10), or 10 μm CNQX (n = 13). G, Dose–response relationship for l-aspartate evoked Ca2+ responses (n = 38–54 for each concentration). EC50 and Hill coefficient are 12 μm and 1.5. Error bars indicate SEM.
Pharmacological experiments confirmed that the Ca2+ rise evoked by l-aspartate required glutamate transporters. The Ca2+ rise was blocked reversibly by TBOA and, to a lesser extent, by DHK (Fig. 5C,F) consistent with the transporter current measurements (Fig. 3). The l-aspartate-evoked Ca2+ response was not sensitive to the antagonists of NMDA- and AMPA-type receptors, d-AP5 and CNQX (Fig. 5F). Efficient block by 5 μm nimodipine identified the major pathway of Ca2+ influx as an L-type Ca2+ channel (Fig. 5D,F), suggesting that the glutamate transporter itself has little Ca2+ permeability, as previously reported (Danbolt, 2001). The same concentration of nimodipine also blocked the Ca2+ rise induced by l-glutamate (n = 9) (data not shown). The Ca2+ rise was not attributable to activation of endogenous mGluRs triggering Ca2+ release from internal Ca2+ stores because l-glutamate evoked the Ca2+ response only in the presence of external Ca2+ (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). In addition, even in cells treated with thapsigargin to deplete the internal Ca2+ stores, the Ca2+ rise by l-aspartate was comparable with the control [compare supplemental Fig. 1B (available at www.jneurosci.org as supplemental material), Fig. 5A]. This result again indicates that Ca2+ influx through depolarization-activated Ca2+ channels was the major source for cytoplasmic Ca2+ rise evoked by l-aspartate.
Interestingly, the Ca2+ rise was significant even at low concentrations of l-aspartate (Fig. 5G) with an EC50 (12 μm) about eight times lower than that for l-aspartate-evoked peak currents (Fig. 1C). This result implies that S-S inward currents activated at low concentrations of EAAT substrates (Fig. 2E,F) depolarize the membrane enough to activate voltage-gated Ca2+ channels significantly.
The Ca2+ rises evoke glutamate release via exocytosis
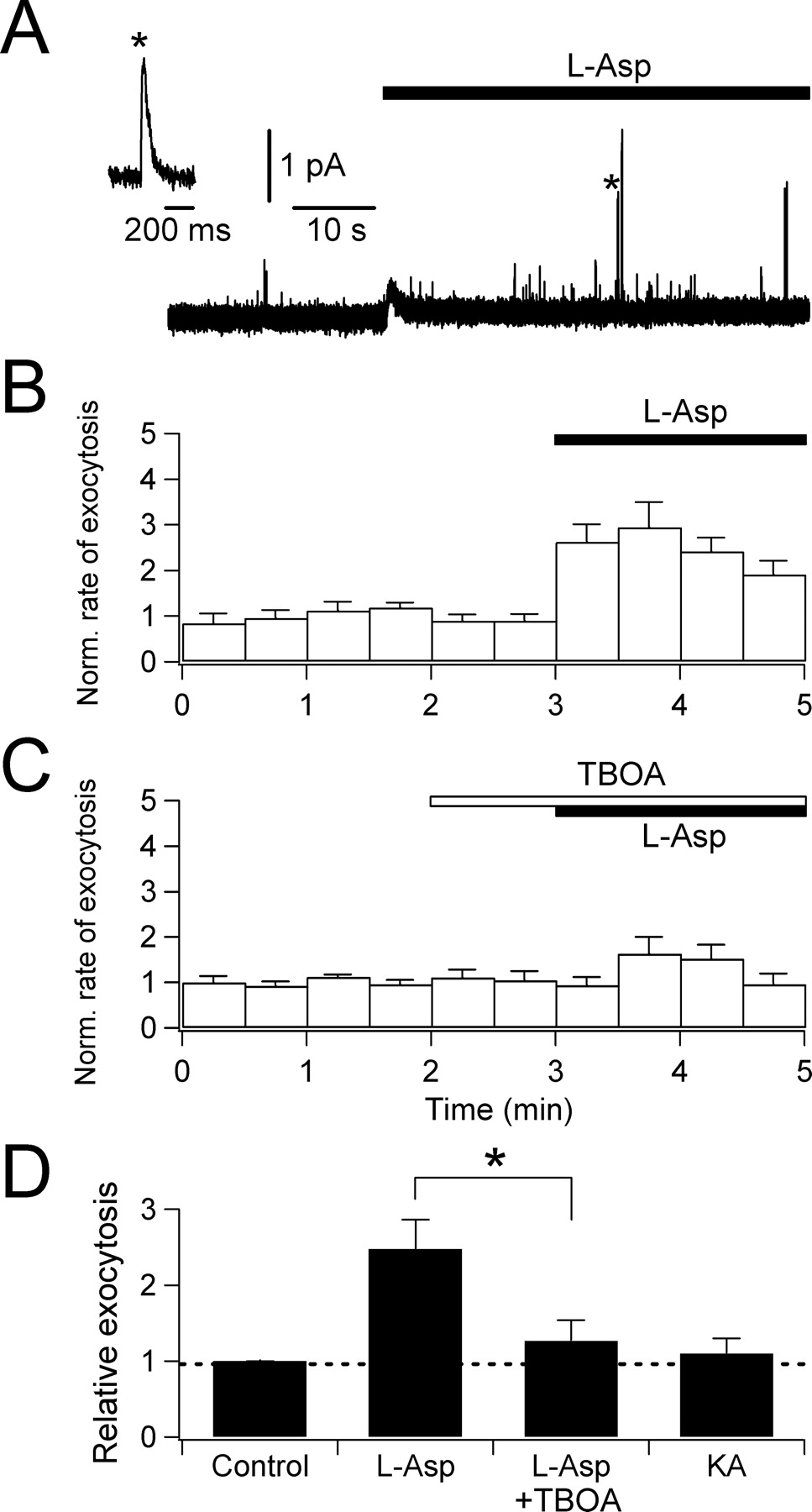
Ca2+ triggers exocytosis of SLMVs in many endocrine cells, including pinealocytes (Yamada et al., 1996a). We address whether the l-aspartate-evoked [Ca2+]i rise suffices to evoke l-glutamate secretion from SLMVs in pinealocytes. Before the measurements, SLMVs were preloaded with oxidizable dopamine as described in Materials and Methods and exocytosis was detected by carbon fiber amperometry (Fig. 6). Oxidation of dopamine released from each vesicle was recorded as an oxidative current spike (Fig. 6A); amperometry indirectly reports the release of l-glutamate from SLMVs. Addition of 1 mm l-aspartate increased the frequency of these amperometric signals. To quantify the rate of exocytosis, we counted the number of amperometric spikes per 30 s time bin and normalized this rate to the mean rate in control solution (Fig. 6B). Relative exocytosis, calculated as the mean values of normalized exocytosis, was 2.5 ± 0.4 (n = 11) during 2 min of l-aspartate application (Fig. 6B,D). This stimulation was significantly depressed by 300 μm TBOA, confirming that the exocytosis is initiated by the activation of glutamate transporters (relative exocytosis for 2 min, 1.3 ± 0.3; n = 19) (Fig. 6C,D). In contrast, 1 mm kainate failed to induce exocytosis, again indicating no activation of AMPA/kainate receptors in pinealocytes (relative exocytosis for 2 min, 1.1 ± 0.2; n = 3) (Fig. 6D).

l-Aspartate-evoked exocytosis monitored by carbon fiber amperometry. A, An amperometric trace demonstrating increase in exocytotic events after 1 mm l-aspartate application (marked by a transient deflection of base line). Inset, One spike denoted by an asterisk is expanded to show a typical quantal release. B, Rate of exocytosis in 30 s bins normalized to the control (average of 11 cells). C, Inhibition by 300 μm TBOA (average of 19 cells). D, Summary of exocytosis evoked by 1 mm l-aspartate alone (n = 11), 1 mm l-aspartate in the presence of 300 μm TBOA (n = 19; *p < 0.01 compared with l-aspartate alone), and 1 mm KA (n = 3). Error bars indicate SEM.
We also measured evoked secretion of l-glutamate directly using HPLC (Fig. 7). l-Glutamate secretion was augmented 5.9-fold (n = 10 independent measurements) when cells were treated with 100 μm l-aspartate. As expected, this glutamate secretion was significantly reduced by the transporter blockers DHK and TBOA, by blocking L-type Ca2+ channels with nimodipine, and by removing extracellular Ca2+. Glutamate secretion also was sensitive to blocking vesicular proton pumps with bafilomycin A1 and to blocking vesicle fusion with botulinum neurotoxin type E (BoNT/E),as reported for the exocytosis of pineal SLMVs (Yatsushiro et al., 1997). It was not sensitive to blockers of glutamate receptors including (+)-5-methyl-10,11-dihydroxy-5H-dibenzo(a,d)cyclohepten-5,10-imine (MK-801) (a specific blocker of NMDA receptor) and CNQX. Similar results were obtained when d-aspartate was used to stimulate pinealocytes (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these results confirm that aspartate-evoked increases in intracellular [Ca2+] can trigger release of l-glutamate by vesicular exocytosis.

l-Aspartate-evoked l-glutamate secretion monitored by HPLC. l-Glutamate release evoked by 100 μm l-aspartate in the absence and presence of different blockers: blockers of glutamate transporters (500 μm DHK and 100 μm TBOA), an L-type Ca2+ channel blocker (20 μm nimodipine), removal of extracellular Ca2+ (−Ca2+), a vesicular H+-pump inhibitor (1 μm bafilomycin A1), iGluR inhibitors (10 μm MK-801 and 50 μm CNQX), or an exocytosis toxin (10 nm BoNT/E, preincubated for 24 h). Shown are means of three to four independent experiments (*p < 0.01, **p < 0.001, compared with l-aspartate alone). Error bars indicate SEM.
Activation of glutamate transporters in the pineal gland
We now test whether EAAs, l-glutamate, and l- and d-aspartate “excite” cellular responses via the glutamate transporter in freshly prepared pineal slices. Because measurement of glutamate transporters in cells positioned deep inside pineal slices suffers from slow agonist application, we studied cells on the surface of the slice. For electrophysiological experiments, we relied on a characteristic electrophysiological criterion to identify the cells: Pinealocytes but not glial cells express large transient and TEA-insensitive K+ currents activated at more than −20 mV (Aguayo and Weight, 1988; Castellano et al., 1989).
We began by checking for the functional presence of glutamate transporters in the acutely prepared pineal slices. Internal KSCN solution was used to increase the anion conductance of the transporters. All pinealocytes identified in slices by the K+-current criterion exhibited transporter-mediated currents when l-aspartate was applied (n = 21) (Fig. 8A). Slowed substrate access obscured any transient peak in the response. The mean S-S current evoked by 1 mm l-aspartate was 4.4 ± 0.3 pA/pF (with KSCN internal) at −60 mV, comparable with that in cultured single cells (Fig. 3), and it was reversibly reduced by TBOA. Incubation with TBOA alone reduced the background inward current level as well as it did in cultured cells. As for isolated-cell experiments (Fig. 1B), 1 mm NMDA evoked no current, indicating no functional NMDA receptors in pineal slices. Another agonist of NMDA receptors, 1 mm d-aspartate, induced average S-S currents of 0.5 ± 0.3 pA/pF at −60 mV (with KCl internal; n = 3) apparently mediated by glutamate transporters. In the presence of 100 μm cyclothiazide, a blocker of desensitization of AMPA-type glutamate receptors, the S-S current with l-glutamate (1 mm) was 0.5 ± 0.1 pA/pF at −60 mV (n = 3), no larger than the expected glutamate transporter-mediated currents. With the same cyclothiazide conditions in dissociated cultured cells, the S-S current was 0.5 ± 0.1 pA/pF (n = 6). Hence, glutamate transporters are expressed in these more intact pineal slices, whereas there was no evidence for functional AMPA and NMDA receptors.

Glutamate transporter-mediated currents in acutely prepared pineal slices. A, Whole-cell currents evoked by bath application of 1 mm l-aspartate. An intracellular solution containing KSCN was used to enlarge transporter currents. Holding membrane potential was −60 mV. Right, At the end of each experiment, the presence of transient outward K+ currents was confirmed by voltage steps from −60 to +60 mV in 20 mV intervals in the presence of internal and external TEA. B, Whole-cell currents evoked by 50 mm KCl (−60 mV). C, Whole-cell currents activated by 135 mm KCl (−60 mV).
Next, cells in pineal slices were loaded with the Ca2+-sensitive fluorescent dye fura-2 AM and challenged with different substrates in a chamber designed to apply the molecules rapidly to the surface of the slice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Most cells in a slice showed Ca2+ rises activated by substrates of glutamate transporters, but the peak Ca2+ response to 1 mm l-aspartate or l-glutamate was quite variable, presumably because not all cells had good access to the applied molecules. The following arguments indicate that these responses are mainly attributable to glutamate transporters and not to AMPA-type iGluRs: Several cells (∼10%) in each slice showed large Ca2+ rises during application of 1 mm l-aspartate or l-glutamate, but not of AMPA or kainate (supplemental Fig. 4A,D, available at www.jneurosci.org as supplemental material). Furthermore, l-glutamate evoked responses no greater than those with l-aspartate, as was observed in dissociated cultured cells (Fig. 5A,B,E). Finally, the Ca2+ responses were significantly inhibited by TBOA and unaffected by CNQX (supplemental Fig. 4B,C,E, available at www.jneurosci.org as supplemental material).
Glutamatergic paracrine interactions in pineal slices
Glutamate transporters, when activated by exogenous EAAs, supported significant Ca2+ rise and glutamate release from pinealocytes. We reasoned that transporters might be activated by the EAAs released from neighboring cells (Fig. 8B, inset). Release of EAAs from the surrounding cells was evoked by an external solution containing 50 mm KCl to depolarize the membrane (Fig. 4B,D), increase [Ca2+]i (supplemental Fig. 6A, available at www.jneurosci.org as supplemental material), and evoke EAA release (Yamada et al., 2002). Activity of the transporters was monitored electrically using the KSCN internal solution (Fig. 8B). In this condition, two types of inward current were expected during the KCl application: augmented transporter current and K+ inward current. KCl applied to the slice induced an inward current of 2.6 ± 0.4 pA/pF (n = 7), which could be reduced by 54 ± 6% with 300 μm TBOA. The remaining current after TBOA was apparently TEA-insensitive K+ current. The current activated by 135 mm KCl was larger and often quite fluctuating (variance, 66.2 ± 24.0 pA2; n = 4; compared with control, 5.2 ± 1.6 pA2) (Fig. 8C). We interpret the noisy current during 135 mm KCl stimulation as caused by overlapping transient transporter currents activated by quantal release of EAAs from the neighboring pinealocytes. In summary, the EAAs released by KCl stimulation from pinealocytes can activate glutamate transporters in neighboring cells in the compact pineal gland.
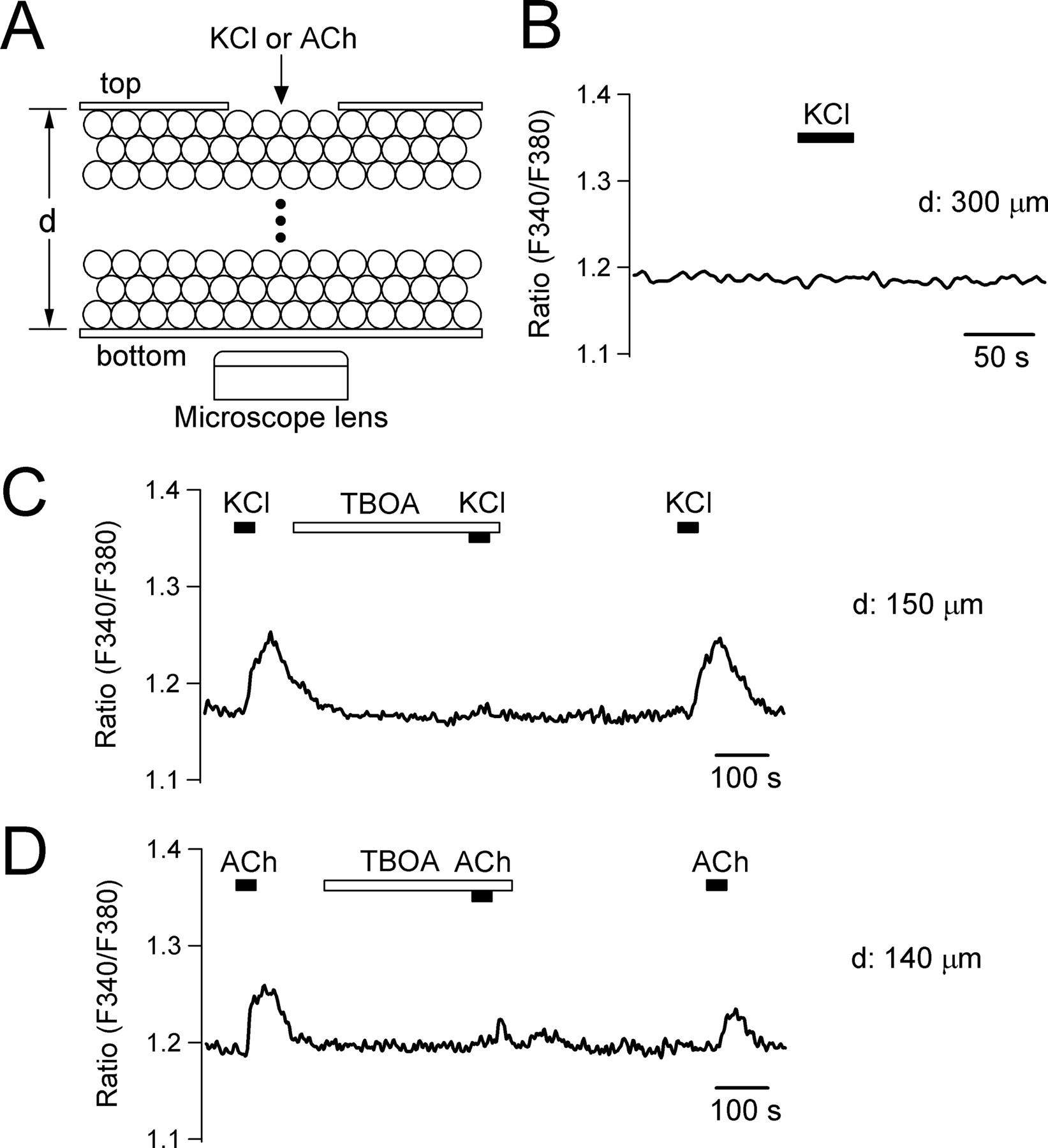
Because we had observed “glutamate-induced glutamate release” in single pinealocytes, we asked whether the endogenous EAAs released by KCl stimulation from a pinealocyte could evoke release of EAAs from other neighboring cells. Such a chain reaction would result in propagation of EAA release inside the pineal gland. To test the possibility, we designed a chamber (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) to allow us to apply 50 mm KCl to one side of a pineal slice and to monitor Ca2+ signals on the opposite side (Fig. 9A). Pineal slices were loaded with fura-2 dye and installed in the chamber. Insertion of the inner chamber covered the top of the slice except for a hole of ∼400 μm. With thin slices (150–200 μm), application of 50 mm KCl to the slice through the hole frequently increased Ca2+ in pinealocytes on the bottom (8 of 13 tested slices) (Fig. 9C). With thicker slices (250–300 μm), KCl did not evoke any Ca2+ signals in cells on the bottom (n = 5) (Fig. 9B). The Ca2+ signals were abolished by TBOA (relative Ca2+ rise compared with the control response; 0.05 ± 0.03; n = 5), suggesting that EAA signals propagated through several cell layers via glutamate transporter signals. Pineal slices are compactly packed with pinealocytes and have a high density of binding sites for TBOA (i.e., glutamate transporters). Therefore, we took several precautions to ensure that TBOA was able to reach the glutamate transporters in the slice: (1) The TBOA concentration was increased to 600 μm, 100-fold higher than needed for half-maximal inhibition of EAAT2 (Ki = 5.7 μm) (Shimamoto et al., 1998). (2) Slices were preincubated in TBOA for 6 min. Note also that TBOA has a small molecular weight (239.2), comparable with acetylcholine (ACh) (181.7). The possibility that TBOA blocks KCl-induced Ca2+ responses of the top cells was excluded in Ca2+ imaging experiments using cultured single pinealocytes (relative Ca2+ rise in the presence of 600 μm TBOA compared with the control KCl response; 0.86 ± 0.03; n = 8) (supplemental Fig. 6A, available at www.jneurosci.org as supplemental material).

Propagation of [Ca2+]i signals via glutamate transporters in pineal slices. A, Schematic drawing of experimental setup. A solution containing 50 mm KCl was applied to cells on the top side of a pineal slice. [Ca2+]i response was monitored on the cells at bottom of the slice. See supplemental Figure 5 (available at www.jneurosci.org as supplemental material) for the detailed design of the chamber. “d” denotes the thickness of pineal slices. B, Ca2+ signal measured in the bottom cells of a thick slice (300 μm) on application of 50 mm KCl to the upper surface of the slice. C, The KCl stimulation evoked Ca2+ response at bottom of a thinner slice (150 μm). Preincubation of the slice with 600 μm TBOA reversibly abolished the Ca2+ signal. Shown is a representative record from eight similar experiments. D, Same experiment performed with a thin slice (140 μm) and 200 μm ACh. Shown is a single record from 11 similar experiments.
The pineal gland is innervated with cholinergic nerve terminals (Phansuwan-Pujito et al., 1999). Pinealocytes also express nicotinic acetylcholine receptors and their activation results in membrane depolarization, Ca2+ rise, and glutamate secretion (Letz et al., 1997; Yamada et al., 1998b). Therefore, we tested whether EAAs released by ACh also evoked a propagating signal. In single cells, 200 μm ACh increased [Ca2+]i (1.3 ± 0.3 μm; n = 22) as efficiently as 50 mm KCl. In pineal slices, the ACh stimulation was able to initiate the propagating EAA signals (11 of 16 tested slices; 140–180 μm) (Fig. 9D). Again, the Ca2+ signals were significantly reduced by 600 μm TBOA (relative Ca2+ rise compared with control; 0.33 ± 0.07; n = 10). There was a time delay (∼10 s) between the KCl/ACh stimulations and Ca2+ rise at the bottom of the slices, presumably reflecting a slow cell-to-cell propagation rate.
In Discussion, we consider the stimulation of melatonin synthesis by norepinephrine (NE) and a negative feedback on that stimulation by l-glutamate. Our working hypothesis is that a propagating glutamate signal inhibits melatonin synthesis via inhibitory metabotropic glutamate receptor (Kus et al., 1994; Yamada et al., 1997a, 1998a; Ishio et al., 1998). A preliminary experiment (Fig. 10) indicates that the inhibition of melatonin secretion requires glutamate transporters. Melatonin secretion evoked by 10 μm NE from intact pineal glands was inhibited by 500 μm l-aspartate. This inhibition by l-aspartate was blocked by 300 μm TBOA (p < 0.01, compared with NE plus l-aspartate).

Effect of glutamate transporters on NE-evoked melatonin secretion. Pineal glands were treated with a combination of 10 μm NE, 500 μm l-aspartate, and 300 μm TBOA for 6 h at physiological 37°C, and released melatonin was measured (four independent experiments for each condition). Error bars indicate SEM. *p < 0.01.
Discussion
Properties and functions of pineal glutamate transporters
In excitatory synapses of the CNS, glutamate activates iGluRs, transiently depolarizing and increasing the excitability of the postsynaptic membrane (McBain and Mayer, 1994; Dingledine et al., 1999). Apparently, in pinealocytes, a similar function is performed by glutamate transporters while they clear excitatory neurotransmitters released from other cells. In this study, we conclude that EAA-evoked responses of pinealocytes are mainly mediated by the glutamate transporter not by iGluR for the following reasons: (1) They are almost equally activated by l-glutamate and l- or d-aspartate; (2) they are insensitive to iGluR agonists and antagonists; (3) they are sensitive to transporter blockers; (4) there are no outward currents; (5) S-S currents are dependent on the internal K+; and (6) the conductance increases when Cl− is replaced by SCN−. These properties were shown in isolated pinealocytes, and most were also shown with pineal slices.
Which subtype of glutamate transporters is expressed in pinealocytes? The pineal transporters exhibit many of the known properties of glutamate transporters (Grewer et al., 2000; Otis and Kavanaugh, 2000; Bergles et al., 2002; Tzingounis and Wadiche, 2007). We previously demonstrated with reverse transcription-PCR, Northern and Western blots, and immunocytochemistry that pinealocytes express glutamate transporters of the GLT-1-type and not GLAST or EAAC1 (Yamada et al., 1997b). In agreement, we find here that pineal glutamate transporters are readily inhibited by DHK, which blocks GLT-1 at concentrations 130-fold lower than for GLAST or EAAC1 (Arriza et al., 1994). Thus, we favor the hypothesis that our observations here concern GLT-1.
The relatively small (10–15 pA) inward current mediated by the glutamate transporters depolarizes the membrane potential enough to activate Ca2+ channels in pinealocytes (Figs. 4, 5). The specific electrophysiological properties of pinealocytes are well suited for this scenario: The resting membrane potential of dissociated pinealocytes is not very negative and variously estimated at −43 mV (Letz et al., 1997), −31 mV (Aguayo and Weight, 1988), and −45.4 mV (our measurement). The input resistance of pinealocytes around the resting membrane potential is high [>1 GΩ (Aguayo and Weight, 1988); 1–2 GΩ (n = 3) (J. Myung and D.-S. Koh, unpublished observation)]. The L-type Ca2+ channels expressed in pinealocytes begin to open at −30 mV when measured with 20 mm Ba2+ (Aguayo and Weight, 1988; Chik et al., 1995). Considering that screening effect by high Ba2+ induces a right shift of the activation curve of voltage-gated channels (∼15 mV with 20 mm Ba2+) (Hille et al., 1975), the channels are expected to be active even at −40 mV at the normal 2 mm Ca2+, just 10 mV above the resting potential. Therefore, transporter-mediated peak and S-S currents in the picoampere range can depolarize the membrane and open Ca2+ channels. Positive to −20 mV, voltage-gated delayed rectifier K+ channels activate, preventing additional depolarization (Aguayo and Weight, 1988). The modulation of membrane potential by pineal glutamate transporters appears to operate in this narrow range to support glutamate secretion.
The effect of glutamate transporters on the membrane potential depends on the subtype of the transporter. For example in the EAAT4- and EAAT5-type transporters, the nonstoichiometric Cl− current can be high enough to counteract the inward stoichiometric transporter current. With this kind of transporter, the outward Cl− currents can hyperpolarize the cell, possibly facilitating EAA transport (Eliasof and Jahr, 1996; Dudel and Schramm, 2003). However, in the GLT-1-, GLAST- and EAAC1-type glutamate transporters the Cl− conductance is lower and the glutamate transport rates higher (Grewer and Rauen, 2005). Expressing GLT-1 allows pinealocytes to be depolarized on EAA stimulation (Fig. 4).
Depolarization of the membrane by glutamate transporters leads to [Ca2+]i rises (Fig. 5). A significant rise occurs with 10 μm l-aspartate, and larger rises occur at higher substrate concentrations. Perhaps both the S-S transporter currents (well activated by 10 μm substrate) and the peak currents contribute to the underlying depolarization. Because there is no evidence in the pineal gland for release of EAAs into very confined spaces, their concentration probably never rises to the millimolar range that might be seen in some synapses. Nevertheless, the sensitivity to EAAs is sufficient for Ca2+-dependent exocytotic responses.
Glutamate transporter-mediated EAA release
With amperometry and HPLC, we found that the glutamate transporter evoked Ca2+ influx, exocytosis, and glutamate secretion in the pineal gland (Figs. 6, 7). A few previous studies have raised the possibility of secretion induced by glutamate transporters including reports of the following: (1) increase in intracellular Ca2+ via a high-affinity glutamate transporter in GH3 pituitary cells (secretion of thyroid stimulating hormone was not tested) (Villalobos and Garcia-Sancho, 1995); (2) a depolarization mediated by electrogenic transporters in hippocampal slices (Frenguelli et al., 1991); and (3) transporter-evoked depolarization and Ca2+-dependent glutamate release from cerebral cortical synaptosomes (McMahon et al., 1989).
The glutamate transporter of the pineal gland triggers exocytosis of its own substrates, EAAs stored in SLMVs. Such stimulus–secretion coupling suggests that transporters might not clear extracellular EAAs efficiently. What would be the physiological significance of glutamate-evoked glutamate release? Pineal glands are compactly packed with pinealocytes (∼90% in rat) together with some minor cell types such as interstitial glial cells (Pévet, 1983). With this structural organization, perhaps EAAs released into the interstitial space from a pinealocyte could recruit the release of EAAs from neighboring cells. Whether this regenerative paracrine mechanism exists and, if so, how far the glutamate signal propagates should depend on several factors. They include the amount of EAA secreted from a stimulated cell (efficiency of the stimulus–secretion coupling), the volume of the interstitial space (determining the dilution of secreted EAAs), and the type of EAAT expressed (determining net inward vs net outward transporter current). Our results using pineal slices suggest that spreading or propagation of the EAA signals is possible (Figs. 8, 9).
Potential role of EAA signals
The pineal gland secretes melatonin with a well defined daily rhythm (Ganguly et al., 2002). The synthesis of melatonin is under a variety of neural and hormonal controls (Simonneaux and Ribelayga, 2003). The best understood is the sympathetic input from the superior cervical ganglion, which, in turn, is driven by suprachiasmatic nucleus, the master circadian clock (Ganguly et al., 2002). At night, norepinephrine released from the sympathetic terminals activates serotonin-N-acetyltransferase (NAT), the rate-limiting enzyme in melatonin production, mainly by acting through β1-adrenergic receptors to increase cAMP.
The pineal is also innervated by parasympathetic cholinergic inputs from peripheral ganglia and the CNS (Phansuwan-Pujito et al., 1999). Acetylcholine evokes release of l-glutamate and l-aspartate by activating nicotinic acetylcholine receptors present on pinealocytes (Letz et al., 1997; Yamada et al., 1998b). The nicotinic receptor induces membrane depolarization, Ca2+ rise through L-type voltage-gated Ca2+ channel, and Ca2+-evoked exocytosis of SLMV, much as glutamate transporters do. From these considerations, it is tempting to suggest that EAAs are initially released from pinealocytes innervated by cholinergic nerve terminals (Yamada et al., 1998b), and the EAAs in turn activate neighboring cells via glutamate transporters and evoke additional release of EAA (Yamada et al., 1996b). Then regenerative spread of EAA signals in the pineal gland can reduce melatonin synthesis by activating class II metabotropic glutamate receptors (mGluRs), in turn reducing the activity of the serotonin-N-acetyltransferase (Kus et al., 1994; Yamada et al., 1998a). Indeed, we have shown previously that exogenous acetylcholine inhibits melatonin synthesis in a manner involving glutamate release and mGluR activation (Yamada et al., 1998b). In summary, cholinergic input, when coupled to this putative paracrine mechanism using EAAs, could effectively inhibit melatonin synthesis and counteract the stimulatory norepinephrine system as originally suggested by Phansuwan-Pujito et al. (1999). At night, when it is desirable to maintain low levels of glutamate in the pineal, the transporters probably take on their usual role of clearing background glutamate and generate insufficient depolarization to stimulate regenerative release. This would avoid unnecessary activation of inhibitory mGluRs that has a high affinity to glutamate (Pin and Duvoisin, 1995).
In the pineal, d-aspartate is highly accumulated (Lee et al., 1997; Schell et al., 1997) and has been implicated in control of melatonin secretion (Ishio et al., 1998; Takigawa et al., 1998). d-Aspartate, partly synthesized in other tissues, is taken up into pinealocytes by the glutamate transporter and distributed in the cytoplasm but not into SLMVs. The cytosolic d-aspartate is known to be released significantly by norepinephrine. Previously, we and others have shown that d-aspartate inhibited the norepinephrine-evoked serotonin-N-acetyltransferase activity and melatonin synthesis by a novel mechanism (Ishio et al., 1998; Takigawa et al., 1998): A hypothetical glutamate receptor responding to d-aspartate was suggested to explain the result. However, based on our present results, the released d-aspartate evokes l-glutamate secretion mediated by the glutamate transporters, and this l-glutamate inhibits serotonin-N-acetyltransferase via an inhibitory mGluR. In this case, the glutamate transporter-mediated glutamate secretion forms a central part of the negative feedback for sympathetic input. In line with this idea, all substrates of pineal glutamate transporters decreased melatonin synthesis induced by NE, whereas d-glutamate had no effect (Kus et al., 1994; Yamada et al., 1997a, 1998a; Ishio et al., 1998). Our preliminary experiment (Fig. 10) does suggest that the inhibition of melatonin secretion was mediated by the activation of glutamate transporters.
Together, our work presents the first clear evidence that glutamate transporters can trigger vesicular exocytosis of neurotransmitters and suggests a novel function of glutamate transporter-mediated EAA release in the pineal gland.
Footnotes
-
This work was supported by grants from the Korean Ministry of Commerce, Industry and Energy (D.-S.K.), the Japanese Ministry of Education, Science, Sports and Culture (Y.M.), and National Institutes of Health Grant GM083913 (B.H.). M.-H.K. was supported by Korea Research Foundation Grant KRF-2006-612-C00011 and Brain Korea 21 to S. H. Kim (POSTECH). We thank J.-H. Cho, J. Myung, S. Han, H. Yamada, and M. Kinoshita for help with experiments and J. G. Duman for the comments on this manuscript.
- Correspondence should be addressed to Dr. Duk-Su Koh, Department of Physiology and Biophysics, University of Washington, Box 357290, Health Sciences Building, Seattle, WA 98195-7290. koh{at}u.washington.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}