

Abstract
The terminal differentiation of many developing neurons occurs after they innervate their target cells and is triggered by secreted target-derived signals that are transduced by presynaptic cognate receptors. Such retrograde signaling induces the expression of genes that are often distinctive markers of neuronal phenotype and function. However, whether long-term maintenance of neuronal phenotype requires persistent retrograde signaling remains poorly understood. Previously, we demonstrated that retrograde bone morphogenetic protein (BMP) signaling induces expression of a phenotypic marker of Drosophila Tv neurons, the neuropeptide FMRFamide (FMRFa). Here, we used a genetic technique that spatiotemporally targets transgene expression in Drosophila to test the role of persistent BMP signaling in the maintenance of Tv phenotype. We show that expression of dominant blockers of BMP signaling selectively in adult Tv neurons dramatically downregulated FMRFa expression. Moreover, adult-onset expression of mutant Glued, which blocks dynein/dynactin-mediated retrograde axonal transport, eliminated retrograde BMP signaling and dramatically downregulated FMRFa expression. Finally, we found that BMP deprivation did not affect Tv neuron survival and that FMRFa expression fully recovered to control levels after the termination of BMP blockade or Glued expression. Our results show that persistent retrograde BMP signaling is required to induce and to subsequently maintain the expression of a stably expressed phenotypic marker in a subset of mature Drosophila neurons. We postulate that retrograde maintenance of neuronal phenotype is conserved in vertebrates, and as a consequence, neuronal phenotype is likely vulnerable to neurodegenerative disease pathologies that disrupt neuronal connectivity or axonal transport.
Introduction
Maintenance of neuronal phenotype is critical to nervous system function. However, our understanding of the mechanisms that maintain the differentiated state of neurons is rudimentary. Terminal differentiation of many neurons requires retrograde signals from the target cells that they innervate, including bone morphogenetic proteins (BMPs), activins, cytokines, and neurotrophins (Ernsberger and Rohrer, 1999; Nishi, 2003; Hippenmeyer et al., 2004). Target-induced genes are often stably expressed, distinguishing phenotypic markers with critical roles in synaptic transmission, including neuropeptides (Coulombe and Kos, 1997; Patel et al., 2000; Duong et al., 2002; Allan et al., 2003), neurotransmitter biosynthetic enzymes (Ernsberger and Rohrer, 1999; Stanke et al., 2006), and ion channels (Martin-Caraballo and Dryer, 2002). However, it is currently unclear whether the maintenance of such target-induced genes requires persistent retrograde signaling in the adult nervous system.
Members of the TGFβ superfamily, including BMPs and activins, are the only known conserved mediators of retrograde neuronal differentiation from Drosophila to vertebrates (Nishi, 2003; Hippenmeyer et al., 2004; Xu and Hall, 2006). In vertebrates, target-derived activin induces the neuropeptide calcitonin gene-related peptide (CGRP) in cutaneous afferents (Ai et al., 1999). Activin retrogradely induces the neuropeptide somatostatin in choroid neurons (Darland and Nishi, 1998). BMP4 acts retrogradely to determine transcription factor expression in trigeminal neurons (Hodge et al., 2007). In Drosophila, target-derived BMP induces expression of neuropeptides, FMRFamide (FMRFa) in Tv neurons (Allan et al., 2003), and dILP7 in subsets of MP2 neurons (Miguel-Aliaga et al., 2008).
In the adult vertebrate nervous system, activin and BMP signaling modulate dendritic spine morphology and synaptic efficacy (Shoji-Kasai et al., 2007; Sun et al., 2007). Moreover, activin is upregulated in the skin after injury and acts retrogradely to increase the number of cutaneous afferents that express CGRP (Xu et al., 2005). However, only indirect evidence supports a role for retrograde TGFβ superfamily signaling in maintenance of adult neuronal phenotype. Axotomy of adult SCG (superior cervical ganglion) neurons alters their neuropeptide expression profile, an effect that is partially blocked by BMP administration (Pavelock et al., 2007). Intriguingly, BMP ligands are expressed at the target tissues of those neurons (V. May, personal communication). Similarly, systemic statins reduce CGRP expression in adult cutaneous afferents in vivo, likely by inhibition of BMP signaling (Bucelli et al., 2008). These results led the authors of both studies to propose that retrograde BMP signaling is required to maintain normal neuropeptide levels.
Here, we directly tested the hypothesis that persistent retrograde BMP signaling maintains adult neuronal phenotype. Previously, we demonstrated that induction of the neuropeptide FMRFa in Drosophila Tv neurons requires target-derived BMP signaling (Allan et al., 2003). Here, by targeting transgenic dominant blockers of BMP signaling selectively in adult Tv neurons, after their normal development, we show that maintenance of FMRFa is absolutely dependent on persistent BMP signaling. Moreover, adult-onset blockade of retrograde axonal transport inhibited nuclear BMP signaling and eliminated FMRFa expression. These data demonstrate that persistent retrograde BMP signaling is required to induce and subsequently maintain the mature phenotype of subsets of Drosophila neurons.
Materials and Methods
Fly genetics
Flies were maintained on standard cornmeal food and maintained at stable temperatures in environment rooms set at 70% humidity at 18, 25, or 29°C. The following fly strains were used: FMRFa-lacZ (WF3-T2) (Allan et al., 2003); apterousmd544 [referred to as apGAL4 here (Allan et al., 2003)]; tubP>GAL80TS [temperature-sensitive GAL80 under the control of the Drosophila tubulin 84B promoter (McGuire et al., 2003)]; UAS-nEGFP [nuclear localized enhanced green fluorescent protein (EGFP)]; UAS-wit ΔI [intracellular domain deletion (McCabe et al., 2003); referred to as UAS-witDN ]; UAS-tkv ΔGSK [GS-box and kinase domain deletion (Haerry et al., 1998); referred to here as UAS-tkvDN ]; UAS-Mad1 [Mad1 mutant that cannot bind DNA (Takaesu et al., 2005); referred to here as UAS-MadDN ]; UAS-Glued Δ84 [referred to here as UAS-GluedDN (Allen et al., 1999)]; UAS-ANF-GFP [pre-pro-atrial natruietic factor fused to emerald green fluorescent protein (emGFP) (Rao et al., 2001)]. The X chromosome insertion (Rao et al., 2001) was hopped (DWA) to chromosome III using standard methods of P-element mobilization. Flies were generously provided by Paul Taghert, Washington University School of Medicine, St. Louis, MO; Thomas Schwarz, Children's Hospital Boston, Boston, MA; Justin Kumar, Indiana University, Bloomington, IN; Stuart Newfeld, Arizona State University, Tempe, AZ; Graeme Davis, University of California San Francisco, San Francisco, CA; and the Bloomington Drosophila Stock Center (Bloomington, IN).
Spatial and temporal regulation of transgene expression using the TARGET system
Flies for TARGET-mediated transgene induction (see Figs. 3 ⇓ ⇓ ⇓–7) were generated by crossing FMRFa-lacZ,apGAL4/CyO, actin-green fluorescent protein (GFP); tub-GAL80TS,UAS-nEGFP (see Fig. 3) or apGAL4/CyO, actin-GFP; tub-GAL80TS,UAS-nEGFP (see Figs. 4 ⇓ ⇓–7) to w1118 for the control group or UAS-tkvDN;UAS-witDN (see Figs. 3 ⇓ ⇓–6), or UAS-MadDN;UAS-MadDN (see Figs. 4 ⇓–6), or UAS-GluedDN (see Fig. 7). Experiments were performed on resulting progeny bearing the appropriate genotypes (screened by loss of the Cyo, actin-GFP chromosome), as detailed in Results. All experimental and control flies were raised at 18°C to suppress GAL4/UAS-mediated transgene expression until the first day after eclosion (hatching from the pupal case). Adult flies were collected every 24 h to ensure accuracy of age for experimental conditions. Noninduced flies were subsequently maintained at 18°C for continued suppression of transgene expression for the duration of the experiment. Induced flies were switched to 29°C for the entire duration of the induction period indicated, for continuous transgene expression.
Immunohistochemistry and in situ hybridization
Antibodies.
Primary antibodies were as follows: sheep anti-digoxygenin (DIG) (1:100; Roche), mouse anti-Eya (1:100; clone 10H6; Developmental Studies Hybridoma Bank), rabbit anti-GFP (1:100; A6455; Invitrogen), rabbit anti-FMRFa (1:1000; T-4757; Peninsula Laboratories), rabbit anti-pMad (1:1000; a generous gift from C.-H. Heldin, Ludwig Institute for Cancer Research, Uppsala, Sweden), and mouse anti-β-galactosidase (1:100; 40-1a). Secondary antibodies were as follows: donkey anti-sheep Alexa 555 (1:10; Invitrogen), and donkey anti-mouse Cy5 and donkey anti-rabbit Cy2 (1:200; Jackson ImmunoResearch).
Antisense DIG-RNA probe.
DIG-uracil-tagged RNA probes were generated using T3 RNA polymerase from clone RH03963 (Drosophila Genomics Resource Center, Bloomington, IN) containing a 1584 bp FMRFa cDNA (using the Roche DIG-U-RNA Labeling kit). Probe synthesis was confirmed using gel electrophoresis.
Multiplex fluorescent in situ hybridization and immunohistochemistry.
All tissues that were compared for fluorescence intensity were processed at the same time using the same aliquots of all solutions under the same conditions. They were then mounted on the same slide. Adult ventral nerve cords were dissected in ice-cold PBS, and then incubated for 50 min in ice-cold 4% paraformaldehyde (PFA) in PBTw (PBS, 0.1% Tween 20, 0.1% DEPC-treated ddH2O). Samples were then PBTw washed and stored overnight in 100% methanol at −20°C. Samples were rehydrated and placed in 4% PFA-PBTw for 20 min. Samples were washed in PBTw and incubated in HYBE solution at 55°C for 1 h. Hybridization with DIG-tagged antisense RNA probes to FMRFa was performed at 55°C overnight on a rotating platform in a Bambino Hybridization Oven. Samples were PBTw washed and incubated for 1 h in PBTw with 5% donkey serum (PBTw-DS). Tissues were incubated overnight at 4°C in PBTw-DS containing primary antibodies. Tissues were washed (PBTw) and blocked (PBTw-DS), and then incubated in secondary antibody (in PBTw-DS) at room temperature for 3 h. Tissues were washed in PBTw, and then PBS, and slide-mounted in Vectashield (Vector Laboratories).
Immunohistochemistry.
Ventral nerve cords were dissected in ice-cold PBS and fixed in ice-cold 4% PFA in 0.1% PBT (PBS–0.1% Triton X-100) for 40 min, washed in PBT, and incubated in PBT with 5% donkey serum (PBT-DS). Primary antibodies were incubated overnight at 4°C and secondary antibodies were incubated at 3 h at room temperature. Antibodies were incubated in PBT-DS. Tissues were washed in PBT, and then PBS, and mounted in Vectashield (Vector Laboratories).
Image analysis
All images were acquired on an Olympus FV1000 confocal microscope as multiple TIFF files representing individual Z-stacks. For each data set, we quantified every identifiable Th1 and Th3 Tv neuron from each control and experimental animal. Raw files were imported into NIH ImageJ for analysis. For each Tv neuron, we compressed all Z-slices spanning whole Tv neurons using the Z-projector function, set to sum the pixel intensities from each Z-slice. Each Tv neuron was outlined and the mean of the summed pixel intensity for each neuron was measured. Background fluorescence intensity was corrected for by subtracting the average of three regions of background (of equal size to the Tv neuron) from the same summed Z-stack for each Tv neuron. The resulting value for each Tv neuron was then incorporated as a single datum point toward the mean FMRFa intensity for each experiment (shown in supplemental Tables 1–3, available at www.jneurosci.org as supplemental material). To normalize data across multiple time points and genotypes, we further expressed each datum point as a percentage of the mean of the w1118 control for that experiment (percentage intensity measurements are provided in supplemental Tables 1–3, available at www.jneurosci.org as supplemental material). For every experiment, we also collected intensity measurements for an additional pair of FMRFa-expressing neurons in Th3 in every nerve cord, using the same settings as for FMRFa in the Tv neurons. These neurons do not show expression for apGAL4;UAS-nEGFP and were therefore not subjected to transgene expression. We tabulated the expression of FMRFa in these neurons as internal controls in supplemental Tables 1–3 (available at www.jneurosci.org as supplemental material). These data show that there was no significant difference in the expression of FMRFa in these neurons between different genotypes, regardless of GAL4 induction. For images shown in Figures 1 ⇓ ⇓ ⇓ ⇓ ⇓–7, we chose representative images from Tv neurons that were stacked to show the entire ap cluster. Images that were directly compared were further processed in an identical way, simultaneously, using Adobe Photoshop CS2.
Statistical analysis
Normally distributed unpaired experimental groups data were compared using a two-tailed t test assuming equal variance, to identify significant differences between groups. For data groups that are not normally distributed, we used the nonparametric Mann–Whitney test. Normal distribution was determined using the D'Agostino and Pearson omnibus test. All statistical analysis and graph data were performed using Prism 5 software (GraphPad).
Results
The Drosophila nervous system contains six Tv neurons, one in each of the six thoracic hemisegments (Th1–3) of the ventral nerve cord (VNC) (Fig. 1 A). Before metamorphosis, Tv axons project to the ipsisegmental dorsal neurohemal organ (DNH), which protrudes dorsally from each thoracic VNC segment (Benveniste et al., 1998; Allan et al., 2003). In adults, after DNH retraction into the VNC, Tv axons form neurohaemal contacts along the dorsal VNC neural sheath (Lundquist and Nässel, 1990; Brown et al., 2006). Developing and adult Tv neurons are phenotypically characterized by expression of the neuropeptide FMRFa (O'Brien et al., 1991; Schneider et al., 1991), which they secrete into the hemolymph at neurohemal endings (Predel et al., 2004; Wegener et al., 2006). In each thoracic hemisegment, the Tv neuron is one of a cluster of four neurons that coexpress the transcription factors, apterous and eyes absent, termed the Tv cluster (Benveniste et al., 1998; Miguel-Aliaga et al., 2004). FMRFa expression in Tv neurons is determined by a specific set of transcriptional regulators and target (DNH)-derived BMP signaling (Allan et al., 2003, 2005; Miguel-Aliaga et al., 2004). Tv axons access the BMP ligand, Glass bottom boat (Gbb), at the DNH. Gbb engages the type II BMP receptor Wishful thinking (Wit) and the type I BMP receptors, Thickveins (Tkv) and Saxophone (Sax), resulting in type I receptor-mediated Mad phosphorylation (pMad) in Tv neurons (Allan et al., 2003; Marqués et al., 2003). Retrograde trafficking of the BMP signal, by an unknown mechanism, translocates pMad to the Tv neuron nucleus, where it induces FMRFa expression (Allan et al., 2003).

BMP signaling and FMRFa expression persists in Tv neurons throughout larval and adult stages. A , Diagrams depicting the larval and adult ventral nerve cord, shown from thoracic segment Th1 to abdominal segment Ab8, and also in a transverse section through thoracic hemisegment Th1, to show the relative position of each Tv cluster and Tv axonal projection. These diagrams show the six thoracic Tv clusters that coexpress the transcription factors apterous (green) and eyes absent (blue) within Th1 to Th3. Each Tv cluster comprises Tv, Tvb, Tva, and Tvc neurons. In larvae, only the Tv neuron expresses FMRFa (red) and nuclear pMad (black), and innervates the DNH. In each thoracic segment, the bilateral Tv neurons innervate the ipsisegmental DNH. In adult Th1 and Th3 segments, only the Tv neuron expresses FMRFa and nuclear pMad. In adult Th2, FMRFa is expressed in Tv, Tva, and Tvc, but only the Tv neuron has nuclear pMad. There are no Tv clusters in abdominal segments, Ab1 to Ab8 in larvae or adults. B–F , The Tv neuron (arrow) is identified by coexpression of apterous (green; apGAL4;UAS-nEGFP) and FMRFa (blue; FMRFa-lacZ; anti-β-Gal) in Tv clusters. Nuclear accumulation of pMad (red; arrowhead; anti-pMad) in the Tv neuron is evident in larval stage L1 ( B ), late larval stage L3 ( C ), adult day A1 ( D ), A10 ( E ), and A20 (the oldest age examined) ( F ). Flies were maintained at 25°C.
BMP signaling persists in mature Tv neurons
To test whether BMP signaling is maintained in Tv neurons beyond the developmental period, we examined pMad immunoreactivity in the nuclei of larval and adult Tv neurons (Allan et al., 2003). Tv neurons were identified by coexpression of FMRFa (FMRFa-lacZ reporter) and apterous (apterousGAL4; UAS-nEGFP) (Allan et al., 2003). We observed strong immunoreactivity to pMad in Tv nuclei at all ages tested up to adult day 20 (Fig. 1 B–F), the last day tested. These data indicate that a BMP signal is transduced and trafficked into Tv neuronal nuclei through the life of the fly.
Transgenic blockade of BMP signaling in Tv neurons
We tested the efficacy of numerous transgenes to ablate BMP signaling in Tv neurons. In control L1 larvae (apGAL4 /+; UAS-nEGFP/+), FMRFa was expressed by 5.7 ± 0.1 Tv neurons per VNC (n = 29 VNCs), and nuclear pMad was observed in 100% of Tv neurons that expressed FMRa (n = 44 Tv neurons) (Fig. 2 A,B). Using apGAL4 to express transgenic blockers of BMP signaling in the Tv cluster, we found that coexpression of dominant-negative versions of thickveins (UAS-tkvDN ) (Haerry et al., 1998) and wishful thinking (UAS-witDN ) (McCabe et al., 2003) resulted in 100% loss of both pMad immunoreactivity (n = 34) and FMRFa expression (n = 10; p = 3.2 × 10−28) in L1 Tv neurons (Fig. 2 A,C). Expression of two copies of dominant-negative Mad (UAS-MadDN ) (Takaesu et al., 2005) resulted in 100% loss of FMRFa expression in Tv neurons (n = 10; p = 3.2 × 10−28), whereas pMad immunoreactivity was observed in 100% of Tv neurons (n = 30 Tv neurons) (Fig. 2 A,D). MadDN encodes a protein in which DNA binding is disrupted. Thus, our data show that BMP signaling upstream of pMad DNA binding had been maintained, but that DNA binding of pMad is required for FMRFa expression. FMRFa was only partially downregulated by expression of either UAS-tkvDN or UAS-witDN alone (Fig. 2 A). In summary, transgenic overexpression of MadDN or both tkvDN/witDN in the Tv cluster prevented the induction of FMRFa expression in developing Tv neurons.

Larval expression of dominant-negative Mad and dominant-negative BMP receptors blocked FMRFa expression in Tv neurons. A , Quantification of Tv neurons that express FMRFa-lacZ (anti-βGal) in L1 larvae. There are six Tv neurons per nerve cord. In w1118 control larvae (FMRFa-lacZ,apterousGAL4 /+), FMRFa expression was seen in all Tv neurons (arrows). We used apterousGAL4 to express dominant-negative BMP pathway transgenes in the Tv cluster and counted the number of Tv neurons in which FMRFa-lacZ was expressed. Expression of dominant-negative BMP type II receptor, wishful thinking (UAS-witDN ), caused a small, significant reduction in FMRFa expression (*p < 0.01). Dominant-negative BMP type I receptor thickveins (UAS-tkvDN ) caused a great reduction in FMRFa expression. FMRFa expression was 100% eliminated by coexpression of UAS-tkvDN and UAS-witDN or by expression of two copies of a dominant-negative version of the BMP-dependent transcription factor, Mad (UAS-MadDN;UAS-MadDN ). **p < 0.0001. Error bars indicate SEM. B–D , ap-expressing neurons were identified by apterousGAL4 -mediated expression of UAS-nEGFP (green). FMRFa-lacZ expression was determined using anti-βGal (blue). Immunoreactivity to pMad (red) showed its accumulation in Tv neuron nuclei (arrowhead), indicative of active BMP signaling. B , In w1118 controls, nuclear pMad accumulation was always observed in FMRFa-expressing Tv neurons. C , Coexpression of dominant-negative BMP receptors UAS-tkvDN;UAS-witDN resulted in 100% loss of FMRFa-lacZ and nuclear pMad in the Tv cluster. There was no loss of Tv cluster neurons. D , Expression of UAS-MadDN resulted in 100% loss of FMRFa-lacZ expression, but nuclear pMad was unaffected, indicating that BMP pathway activity was unaffected upstream of nuclear pMad accumulation. For details, see text.
Temporal regulation of BMP signaling in adult Tv neurons
The TARGET system (McGuire et al., 2003) enables temperature-dependent switching of GAL4/UAS-mediated transgene expression, by use of a temperature-sensitive allele of the yeast GAL4-repressor, GAL80 (GAL80TS) (for details, see Fig. 3 A,D). We generated the following genotype to spatiotemporally regulate tkvDN and witDN expression in adult Tv neurons in vivo using the TARGET system, FMRFa-lacZ, apGAL4/UAS-tkvDN; UAS-nEGFP, tub>GAL80TS/UAS-witDN . The w1118 control genotype was apGAL4/+;UAS-nEGFP, tub>GAL80TS/+.

Adult induction of BMP blockade in Tv neurons eliminated nuclear pMad accumulation and FMRFa reporter expression. Flies of genotype FMRFa-lacZ, apGAL4/CyO; tub-GAL80TS,UAS-nEGFP were crossed to either control w1118 flies ( B , E , G ) or UAS-tkvDN,UAS-witDN flies ( C , F , H ) to generate control and experimental progeny, which were raised at 18°C. A , D , Schematic of the TARGET system for regulated spatiotemporal expression of transgenes in the Tv cluster. GAL4 was expressed selectively in all ap neurons under control of the genomic apterous enhancer. GAL80TS was expressed ubiquitously by the tubulin promoter. A , At 18°C, GAL4 activity is not induced; GAL80TS binds GAL4 and blocks GAL4-mediated gene transactivation from UAS sites. D , At 29°C, GAL4 activity is induced; loss of GAL80TS binding to GAL4 allows GAL4 to transactivate gene expression from UAS sites, in this case in all ap neurons, including the Tv cluster. B , C , No induction: In adult day A1 animals raised at 18°C, GAL80TS blocked apGAL4 -mediated induction of UAS-nEGFP (green) and UAS-tkvDN/UAS-witDN (in C ). Note lack of nEGFP expression in both genotypes. In w1118 control flies ( B ) and UAS-tkvDN;UAS-witDN flies ( C ), there was no difference in the expression of FMRFa-lacZ (blue; anti-βGal) or nuclear pMad (red; anti-pMad; arrowhead) in Tv neurons. For details, see text. E , F , After 12 h induction at 29°C, nEGFP expression was activated (green) in both genotypes. E , FMRFa-lacZ and nuclear pMad (arrowhead) expression was maintained in Tv neurons in w1118 control flies. F , Nuclear accumulation of pMad was completely lost in UAS-tkvDN;UAS-witDN flies. FMRFa-lacZ was unaffected by 12 h induction. These data show that the BMP pathway had been blocked within 12 h of GAL4 induction. G , H , After 48 h induction at 29°C, BMP pathway blockade in UAS-tkvDN;UAS-witDN flies ( H ) resulted in a loss of pMad nuclear accumulation and a profound downregulation of FMRFa-lacZ in Tv neurons (arrow). In w1118 control flies ( G ), nuclear pMad (arrowhead) and FMRFa-lacZ expression were unaffected.
The efficacy of adult-onset BMP signaling blockade in Tv neurons was examined by pMad nuclear immunoreactivity and FMRFa-lacZ reporter expression in w1118 controls (Fig. 3 B,E,G) and tkvDN;witDN flies (Fig. 3 C,F,H). Flies were raised at 18°C up to adult day A1 (first day posteclosion). At that time, apGAL4 -mediated GAL4/UAS activity was repressed by GAL80TS, as shown by lack of nEGFP expression in the Tv cluster (Fig. 3 B,C); FMRFa-lacZ and nuclear pMad expression was observed in 100% of Tv neurons in w1118 control (n = 15 Tv neurons) and tkvDN;witDN flies (n = 18 Tv neurons) (Fig. 3 B,C).
At A1, we switched flies to 29°C to induce GAL4 activity. After both 12 and 48 h at 29°C, we observed robust nEGFP expression in both genotypes (Fig. 3 E–G). In w1118 controls, FMRFa-lacZ and nuclear pMad expression was observed in 100% of Tv neurons after 12 and 48 h at 29°C (12 h; n = 16 Tv neurons; 48 h; n = 24 Tv neurons) (Fig. 3 E,G). In contrast, in tkvDN;witDN flies, we observed 100% loss of pMad nuclear immunoreactivity at 12 h (n = 16 Tv neurons) (Fig. 3 F) and 48 h (n = 17 Tv neurons) (Fig. 3 H). In tkvDN;witDN flies, FMRFa-lacZ was maintained at 12 h (Fig. 3 F) but was profoundly reduced in 100% of Tv neurons by 48 h (Fig. 3 H).
In summary, the TARGET system provides robust spatial and temporal regulation of transgene expression in Tv neurons. We found that adult-onset blockade of BMP signaling ablated nuclear pMad immunoreactivity within 12 h in Tv neurons, and greatly reduced an FMRFa reporter in Tv neurons within 48 h.
BMP blockade in adults dramatically reduced FMRFa transcript and peptide
Using the TARGET system, we tested the effect of adult-onset BMP blockade in Tv neurons on levels of FMRFa transcript [fluorescent in situ hybridization (FISH)] (Fig. 4) and FMRFa peptide (anti-FMRFa immunofluorescence) (Fig. 5). To quantify FMRFa expression, we measured the relative pixel intensity of FISH or immunofluorescence in Tv soma in all genotypes for each experiment (tabulated in supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material). Data acquisition and quantification are described in Materials and Methods. To standardize our results across multiple time points, here we have expressed FMRFa fluorescence per Tv neuron as a percentage of the mean of the w1118 control for each experiment.
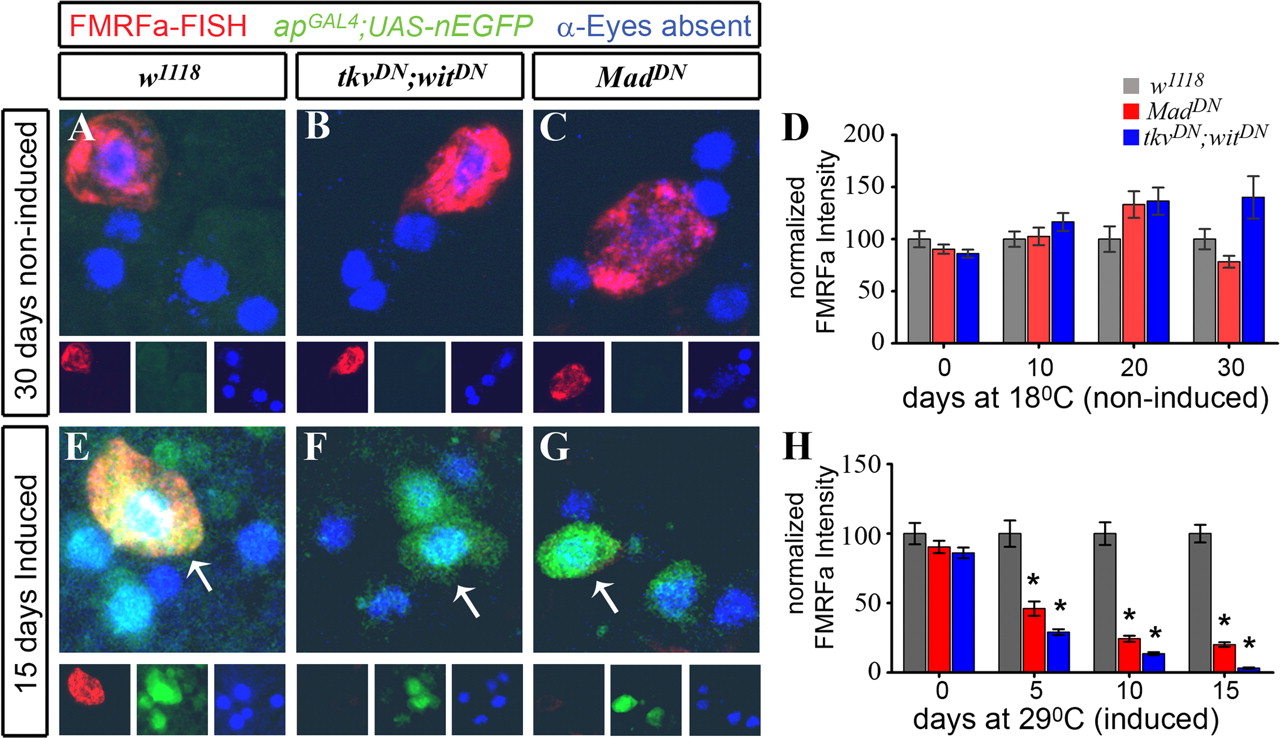
Acute blockade of the BMP pathway in adults dramatically reduced FMRFa transcript. Genotypes were as follows: w1118 control flies ( A , E ) (apGAL4 /+; tub-GAL80TS,UAS-nEGFP/+); tkvDN;witDN flies ( B , F ) (apGAL4/UAS-tkvDN; tub-GAL80TS, UAS-nEGFP/UAS-witDN ); MadDN flies ( C , G ) (apGAL4/UAS-MadDN; tub-GAL80TS, UAS-nEGFP/UAS-MadDN ). All flies were raised at 18°C until A1. On day A1, flies were subsequently kept at 18°C for up to 30 d ( A–D ), or switched to 29°C for up to 15 d ( E–H ). D , H , Experimental results comparing relative pixel intensity of FMRFa FISH at each time point, for each genotype. Each datum point was normalized to the percentage of the mean of the w1118 control for each time point. Data for each genotype, at each time point, is presented as mean ± SEM. For details, see supplemental Table 1 (available at www.jneurosci.org as supplemental material). A–C , In noninduced flies maintained at 18°C for 30 d, GAL4 activity was repressed, as shown by lack of nEGFP (green) expression in Tv cluster neurons (anti-Eyes absent; blue). High-level FMRFa FISH (red; anti-DIG) was observed in control w1118 flies ( A ), tkvDN;witDN flies ( B ), and MadDN flies ( C ). D , Quantification of FMRFa FISH intensity for flies kept at 18°C, at A1 (time 0), and at 10, 20, and 30 d later. No significant difference was observed between genotypes at any time point. E–G , In induced flies maintained at 29°C for 15 d, GAL4 activity was induced, as shown by nEGFP expression (green) in Tv cluster neurons (anti-Eyes absent; blue). FMRFa FISH (red) in Tv neurons (arrows) was observed in w1118 control flies ( E ) but was severely reduced in tkvDN;witDN ( F ) and MadDN flies ( G ). H , Quantification of FMRFa FISH intensity for flies induced at 29°C, at A1 (time 0), and at 5, 10, and 15 d later. Over the induction period, there was a progressive, dramatic reduction of FMRFa transcript levels after BMP pathway blockade until it was almost entirely absent. *p < 0.0001 compared with control at each time point.

Acute blockade of the BMP pathway in adults dramatically reduced FMRFa peptide. Genotypes were as follows: w1118 control flies ( A , E ) (apGAL4/+; tub-GAL80TS,UAS-nEGFP/+); tkvDN;witDN flies ( B , F ) (apGAL4/UAS-tkvDN; tub-GAL80TS, UAS-nEGFP/UAS-witDN ); MadDN flies ( C , G ) (apGAL4/UAS-MadDN; tub-GAL80TS, UAS-nEGFP/UAS-MadDN ). Flies were raised at 18°C until A1. They were then kept at 18°C for up to 30 d ( A–D ), or switched to 29°C for up to 15 d ( E–H ). D , H , Experimental results comparing relative pixel intensity of FMRFa immunoreactivity at each time point, for each genotype. Each datum point was normalized to the percentage of the mean of the w1118 control at each time point. Data for each genotype at each time point are presented as mean ± SEM. For details, see supplemental Table 2 (available at www.jneurosci.org as supplemental material). A–C , In noninduced flies maintained at 18°C for 30 d, GAL4 activity was repressed, as shown by lack of nEGFP (green) expression in Tv cluster neurons (anti-Eyes absent; blue). High-level FMRFa immunoreactivity (red; anti-FMRFa) was observed in control w1118 flies ( A ), tkvDN;witDN flies ( B ), and MadDN flies ( C ). D , Quantification of FMRFa immunoreactivity intensity for flies kept at 18°C, at A1 (time 0), and at 10, 20, and 30 d later. No significant difference was observed between genotypes at any time point. E–G , In induced flies maintained at 29°C for 15 d, GAL4 activity was induced, as shown by nEGFP expression (green) in Tv cluster neurons (anti-Eyes absent; blue). FMRFa immunoreactivity (red) in Tv neurons (arrows) was observed in w1118 control flies ( E ), but was severely reduced in tkvDN;witDN ( F ) and MadDN flies ( G ). H , Quantification of FMRFa immunoreactivity intensity for flies induced at 29°C, at A1 (time 0), and at 5, 10, and 15 d later. Throughout the induction period, there was a progressive, dramatic reduction of FMRFa peptide levels after BMP pathway blockade until it was almost entirely absent. *p < 0.0001 compared with control at each time point.
We examined FMRFa expression in noninduced flies, maintained at 18°C, at 10, 20, and 30 d after A1. In these noninduced flies, GAL4 activity was blocked at all time points, as shown by lack of nEGFP expression (Figs. 4 A–C, 5 A–C). Strong FMRFa FISH (Fig. 4 A–C) and peptide immunoreactivity (Fig. 5 A–C) was observed in noninduced flies of all genotypes, at all time points. Quantification of FISH (Fig. 4 D) and peptide immunoreactivity (Fig. 5 D) showed that there was no significant difference in FMRFa levels between genotypes at each time point. Thus, genotype alone, in the absence of GAL4 induction, had no effect on FMRFa expression.
We next examined FMRFa FISH (Fig. 4 E–H) and peptide immunoreactivity (Fig. 5 E–H) in w1118 controls, tkvDN;witDN flies, and MadDN flies that had been switched to 29°C at A1, and then maintained at 29°C for 5, 10, and 15 d. These ages are comparable with noninduced flies at 10, 20, and 30 d; flies maintained at 18°C age at about one-half the rate of flies at 29°C. Flies switched to 29°C expressed nEGFP in the Tv cluster (Figs. 4 E–G, 5 E–G). After 15 d at 29°C, w1118 controls had high level FMRFa FISH (100 ± 6.4%; n = 30) (Fig. 4 E) and peptide immunofluorescence (100 ± 8.8%; n = 45) (Fig. 5 E). In contrast, expression of tkvDN;witDN for 15 d at 29°C ablated FMRFa transcript to 3.2 ± 0.5% of w1118 controls (n = 18 Tv neurons; p = 2.5 × 10−15) (Fig. 4 F) and ablated FMRFa immunofluorescence to 8.2 ± 2.7% of w1118 controls (n = 36 Tv neurons; p = 6.4 × 10−14) (Fig. 5 F). Similarly, expression of MadDN for 15 d at 29°C greatly reduced FMRFa transcript to 20.1 ± 1.6% of w1118 controls (n = 32 Tv neurons; p = 2.5 × 10−18) (Fig. 4 G) and reduced FMRFa immunofluorescence to 37.9 ± 4.6% of w1118 controls (n = 32 Tv neurons; p = 3.6 × 10−7) (Fig. 5 G).
We further quantified FMRFa transcript and peptide levels in w1118 control, tkvDN;witDN , and MadDN flies after 5 and 10 d at 29°C (Figs. 4 H, 5 H; supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material). We observed a dramatic decline of transcript levels after induction of BMP blockade at A1. In tkvDN;witDN flies, FMRFa transcript levels fell 3.5-fold by day 5, 7.4-fold by day 10, and 30-fold by day 15, when compared with w1118 controls (Fig. 4 H). Furthermore, FMRFa peptide levels fell 3-fold by day 5, 5.4-fold by day 10, and 12-fold by day 15, when compared with w1118 controls (Fig. 5 H). In MadDN flies, FMRFa transcript levels fell 2.2-fold by day 5, 4.1-fold by day 10, and 5-fold by day 15, when compared with w1118 controls (Fig. 4 H). Furthermore, FMRFa peptide levels fell 2.1-fold by day 5, 2.2-fold by day 10, and 2.6-fold by day 15, when compared with w1118 controls (Fig. 5 H).
We confirmed that loss of BMP signaling in adult Tv neurons did not cause gross retraction of Tv axons from their terminal field. We expressed a transgenic marker of dense-core vesicles, atrial natruietic factor (UAS-ANF-emGFP) (Rao et al., 2001) in Tv cluster neurons (using apGAL4 ) in w1118 control, tkvDN;witDN , and MadDN flies. After 15 d of induction at 29°C, we found that Tv axonal projections were normal in all genotypes (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), showing that Tv axons had not retracted.
In summary, we show that maintenance of FMRFa expression in adult Tv neurons requires persistent BMP signaling. Interestingly, the decline of FMRFa peptide closely parallels that of FMRFa transcript. The simplest explanation is that FMRFa peptide is rapidly translated and trafficked to Tv axon termini for secretion into the hemolymph, and that a high level of FMRFa transcription is required to maintain that.
Restoration of FMRFa transcript after recovery from acute BMP blockade in adults
We tested the capacity of Tv neurons to restore FMRFa transcript after a period of BMP blockade. We raised w1118 control, tkvDN;witDN , and MadDN flies at 18°C until A1. They were switched to 29°C for 10 d to induce GAL4 activity, and reduce FMRFa transcript (∼13 and ∼24% of w1118 control levels in tkvDN;witDN and MadDN flies, respectively) (Figs. 4 H, 6 A–D; supplemental Table 1, available at www.jneurosci.org as supplemental material). Flies were then returned to 18°C for an additional 10 d of “recovery,” in which GAL4 no longer drove transgene expression and transgene levels presumably decline. Remarkably, after recovery, FMRFa FISH levels in tkvDN;witDN and MadDN flies was fully restored to control levels (Fig. 6 E–H; supplemental Table 1, available at www.jneurosci.org as supplemental material). In w1118 controls, FMRFa FISH intensity was 100 ± 7.3% of the mean of w1118 controls (n = 53 Tv neurons). In tkvDN;witDN flies, FMRFa FISH was 109.8 ± 10.3 [n = 53 Tv neurons; no significant difference to control (NSD)] (Fig. 6 F). In MadDN flies, FMRFa intensity was 111.2 ± 7.4% (n = 53 Tv neurons; NSD) (Fig. 6 G). We noted a weak persistent EGFP expression in the Tv cluster in “recovered” flies (Fig. 6 E–G), confirming that transgene expression had been induced previously.

FMRFa transcript was fully restored after recovery from acute BMP blockade in adults. Genotypes were as follows: w1118 control flies ( A , E ) (apGAL4/+; tub-GAL80TS,UAS-nEGFP/+); tkvDN;witDN flies ( B , F ) (apGAL4/UAS-tkvDN; tub-GAL80TS, UAS-nEGFP/UAS-witDN ); MadDN flies ( C , G ) (apGAL4/UAS-MadDN; tub-GAL80TS, UAS-nEGFP/UAS-MadDN ). Flies were raised at 18°C until A1. They were then switched to 29°C to induce GAL4 activity for 10 d ( A–D ), or switched to 29°C for 10 d to induce GAL4 activity ( E–H ), and then returned to 18°C for 10 d to allow neurons to recover from BMP blockade. D , H , Scatter plots show relative pixel intensity of FMRFa FISH in individual Tv neurons of w1118 control (w), tkvDN;witDN flies (TW), or MadDN flies (M). FMRFa FISH intensity was expressed as a percentage of the mean of the control for each experiment. Mean ± SEM is shown. For details, see supplemental Table 1 (available at www.jneurosci.org as supplemental material). A–C , Induction of BMP blockade in adult Tv cluster neurons (blue; anti-Eyes absent) for 10 d reduced FMRFa transcript (red) in Tv neurons (arrow). High nEGFP (green) expression showed GAL4 activity. D , Scatter plot shows that BMP blockade in tkvDN;witDN flies (TW) and MadDN flies (M) significantly reduced FMRFa expression in all Tv neurons, when compared with w1118 control flies (w). Note that these data are included in Figure 4 H for the 10 d time point. *p < 0.0001 compared with w1118 control. E–G , After 10 d of recovery from BMP blockade, FMRFa FISH (red) was fully restored to control levels. Note low-level nEGFP expression (green), indicative of nEGFP expression during the induction period. H , Scatter plot shows that recovery from BMP blockade resulted in a complete restoration of FMRFa FISH levels, with no significant difference seen between w1118 control flies (w), tkvDN;witDN flies (TW), or MadDN flies (M).
In summary, these data show that adult Tv neurons retain the capacity to reestablish normal FMRFa expression after an acute 10 d block of BMP signaling that had dramatically reduced FMRFa transcript levels.
Late-onset expression of mutant Glued in adult Tv neurons blocked retrograde BMP signaling and downregulated FMRFa, in a reversible manner
The above data led us to the hypothesis that neurodegenerative disease pathologies may disrupt gene expression in adult neurons by disrupting retrograde transport of essential target-derived signals. We tested this hypothesis using mutant Glued (GluedDN ) (Allen et al., 1999). Glued mutation has been linked to familial and sporadic amyotrophic lateral sclerosis (ALS) (Puls et al., 2003; Münch et al., 2004). Moreover, mutant Glued disrupts dynein motor function in Drosophila (Martin et al., 1999) and vertebrates (Levy et al., 2006) and recapitulates key features of ALS in mouse models (Chevalier-Larsen et al., 2008; Laird et al., 2008).
We generated flies to spatiotemporally target GluedDN expression (UAS-GluedDN ) in adult Tv neurons using the TARGET system [genotype (FMRFa-lacZ),apGAL4/UAS-GluedDN; UAS-nEGFP, tub>GAL80TS/+]. We tested the effect of late-onset GluedDN expression on maintenance of FMRFa expression. We raised w1118 control and GluedDN flies at 18°C until A1. First, we subsequently maintained flies at 18°C for 10 d to control for genotype in the absence of GAL4 activity, which was verified by lack of nEGFP expression (Fig. 7 A,B). After 10 d at 18°C, the intensity of FISH for FMRFa transcript, normalized to the mean of w1118 control, was not significantly different between w1118 control (100 ± 12.4; n = 15 Tv neurons) and GluedDN flies (106.3 ± 14.2%; n = 20; NSD) (Fig. 7 C; supplemental Table 3, available at www.jneurosci.org as supplemental material). Second, we induced GluedDN for 5 d at 29°C; FMRFa expression in GluedDN flies fell to 28.8 ± 4.9% (n = 20; p = 1.7 × 10−13), compared with that of w1118 control flies (100 ± 4.7%; n = 34) (Fig. 7 D–F; supplemental Table 3, available at www.jneurosci.org as supplemental material). Third, we tested whether Tv neurons were able to recover from that acute period of GluedDN induction that downregulated FMRFa expression. We raised w1118 control and GluedDN flies at 18°C until A1. They were then switched to 29°C for 5 d to induce GAL4 activity. Flies were then returned to 18°C for an additional 10 d of recovery. Remarkably, after the 10 d recovery period, FMRFa levels were restored to 194.6 ± 24.9% (n = 15; p = 0.00011) of w1118 control flies (100 ± 4.8%; n = 21) (Fig. 7 G–I; supplemental Table 3, available at www.jneurosci.org as supplemental material). In summary, overexpression of GluedDN in adult Tv neurons led to a profound downregulation of FMRFa expression.

Late-onset expression of mutant Glued blocked retrograde BMP signaling and downregulated FMRFa, in a reversible manner. Genotypes were as follows: w1118 control flies ( A , D , G ) (apGAL4/+; tub-GAL80TS,UAS-nEGFP/+); GluedDN flies ( B , E , H ) (apGAL4/UAS-Glued Δ84 ; tub-GAL80TS, UAS-nEGFP/+). Flies were raised at 18°C until A1. They were then kept at 18°C for 10 d to block GAL4 induction ( A–C ), switched to 29°C for 5 d to induce GAL4 activity ( D–F ), or switched to 29°C for 5 d ( G–I ), and then returned to 18°C for 10 d to allow neurons to recover from GluedDN expression. C , F , I , Scatter plots show relative pixel intensity of FMRFa FISH in individual Tv neurons of w1118 control (w), and GluedDN flies (G). FMRFa FISH intensity was expressed as a percentage of the mean of the control for each experiment. Mean ± SEM is shown. For details, see supplemental Table 1 (available at www.jneurosci.org as supplemental material). A , B , Flies were kept at 18°C for 10 d. Note the lack of nEGFP (green) in Tv cluster neurons (blue; anti-Eyes absent), showing lack of GAL4 activity. FMRFa FISH (red) was high in noninduced w1118 control flies ( A ) and GluedDN flies ( B ). C , Scatter plot shows that there was no significant difference in FMRFa transcript in individual Tv neurons between noninduced w1118 control (w), and GluedDN flies (G). D , E , Five days of induced GluedDN expression in adult Tv cluster neurons (blue; anti-Eyes absent) significantly reduced FMRFa FISH (red) in Tv neurons (arrow). High nEGFP (green) expression showed induction of GAL4 activity. F , Scatter plot shows that FMRFa transcript was downregulated in all Tv neurons in GluedDN flies (G), compared with w1118 control flies (w). **p < 0.0001 compared with control. G , H , After 10 d of recovery from GluedDN expression, FMRFa FISH (red) was restored to above control levels (194.6 ± 24.9%). Note low nEGFP expression (green), indicative of nEGFP expression during the induction period. I , Scatter plot shows that, after recovery from GluedDN expression, FMRFa transcript was fully restored to above control levels (G), when compared with w1118 control flies (w). *p < 0.001 compared with control. J–M , Flies of genotype FMRFa-lacZ, apGAL4/CyO; tub-GAL80TS,UAS-nEGFP were crossed to either control w1118 flies ( J , L ) or UAS-GluedDN flies ( K , M ) to generate progeny that were raised at 18°C. J , K , No induction, A1 flies maintained at 18°C. Note lack of nEGFP expression in both genotypes. In w1118 control flies ( J ) and UAS-GluedDN flies ( K ), there was no difference in the expression of FMRFa-lacZ (blue; anti-βGal) or nuclear pMad (red; anti-pMad) in Tv neurons. L , M , After 12 h induction at 29°C, nEGFP expression was activated (green) in both genotypes. L , In w1118 control flies, FMRFa-lacZ and nuclear pMad (arrowhead) expression was maintained in Tv neurons. M , In UAS-GluedDN flies, FMRFa-lacZ expression was maintained, but nuclear accumulation of pMad was entirely absent in Tv neurons.
We next examined whether BMP retrograde signaling is blocked by GluedDN, by testing nuclear pMad accumulation in Tv neurons in w1118 control and GluedDN flies (Fig. 7 J–M). We raised w1118 control and GluedDN flies at 18°C until A1. First, we examined pMad immunoreactivity in Tv nuclei at A1, before induction at 29°C. In both w1118 control and GluedDN flies, 100% of Tv neurons expressed FMRFa-lacZ and pMad immunoreactivity (n = 6; n = 12 Tv neurons, respectively). Next, we induced apGAL4 activity by switching animals to 29°C. After just 12 h of induction, we found that pMad nuclear accumulation was observed in 0% of Tv neurons in GluedDN flies (n = 29 Tv neurons), as opposed to 100% of Tv neurons in w1118 controls (n = 15). This shows that acute GluedDN expression in adult Tv neurons rapidly blocked persistent retrograde transport of the BMP signal in adult Tv neurons.
In summary, these data support the hypothesis that maintenance of FMRFa expression in adult Tv neurons requires persistent retrograde BMP signaling. Moreover, we provide the first direct evidence that neurodegenerative disease-related disruption of axonal transport leads to the downregulation of phenotypic markers, such as the neuropeptide FMRFa, that depend on persistent target-dependent signaling. These findings highlight a novel mechanism by which neurodegenerative diseases may disrupt normal neuronal function. Importantly, our data further demonstrate that the effect of GluedDN is fully reversible, in that neurons can restore normal target-dependent FMRFa expression after recovery from mutant Glued expression.
Discussion
Previously, we demonstrated that target-dependent neuronal differentiation is conserved from invertebrates to vertebrates by showing that expression of FMRFa in developing Drosophila Tv neurons is induced by retrograde BMP signaling (Allan et al., 2003). FMRFa is a stably expressed phenotypic marker of Tv neurons expressed throughout Drosophila life (Schneider et al., 1993). Despite the growing number of identified target-induced genes in vertebrates and Drosophila, it is unclear whether maintenance of the expression of those genes requires persistent retrograde signaling, or switches to cell-autonomous maintenance. Here, we address this question for FMRFa in Tv neurons. By cell-autonomous blockade of either BMP signaling or retrograde axon transport in Tv neurons, selectively in adult flies, we show that persistent retrograde BMP signaling is absolutely required to maintain FMRFa expression. To our knowledge, this is the first explicit demonstration that a specific retrograde signaling pathway is required to induce and then to maintain the expression of a stably expressed phenotypic marker in neurons.
TGFβ superfamily ligands are conserved regulators of circulating peptide hormone levels
Tv neurons secrete FMRFa into the fly circulatory system, the hemolymph, which is akin to the secretion of peptide hormones from the mammalian pituitary and pancreas into the circulation. Intriguingly, persistent BMP signaling is required to maintain the expression of insulin in pancreatic β-islet cells (Goulley et al., 2007), and persistent activin signaling is required to maintain the expression of follicle-stimulating hormone subunit β (FSHβ) in pituitary gonadotrophs (DePaolo et al., 1992a,b; Guo et al., 1998; Kumar et al., 2003). Together with our results showing that BMP signaling maintains neuropeptide expression in Drosophila neuroendocrine neurons, these findings demonstrate that TGFβ superfamily signaling is a conserved mechanism for maintenance of circulatory peptide hormone levels in adults.
Circulating peptide hormones in vertebrates are typically under homeostatic regulation. Does BMP signaling participate in the homeostatic regulation of FMRFa in Drosophila? A precedent for homeostatic peptide hormone regulation by TGFβ-type signaling comes from the well characterized regulation of FSHβ during the estrous cycle by activin. The level of FSHβ transcription depends on the level of activin receptor signaling in pituitary gonadotrophs, which in turn is modulated by the balance of locally produced activin and circulating levels of inhibin, an activin antagonist secreted from the gonads (Gregory and Kaiser, 2004; Bilezikjian et al., 2006). In Drosophila, the level of BMP ligand or antagonist accessible to Tv neurons may be responsive to an unknown cue that reads through to physiologically instructive levels of FMRFa. We have not detected any periodicity in FMRFa expression, which would be suggestive of homeostatic modulation, but neither have we ruled out such a possibility. BMP signaling has a well documented role in neuromuscular efficacy in Drosophila larvae (McCabe et al., 2003; Keshishian and Kim, 2004), and pharmacological administration of FMRFa enhances neuron-evoked contractility of larval Drosophila body wall muscles (Hewes et al., 1998; Clark et al., 2008). These findings have led to the untested proposal that activity-dependent BMP signaling feeds back to FMRFa transcription to influence neuromuscular efficacy (Keshishian and Kim, 2004). However, there is no evidence to rule out a simpler alternative hypothesis that retrograde BMP signaling may simply function to maintain high level FMRFa transcription independent of any homeostatic regulatory role. Future studies will need to discriminate between these models and define the role(s) of FMRFa in adult flies.
Retrograde maintenance of neuronal phenotype in adults
After neurons differentiate, they must maintain their differentiated state for the lifetime of the animal. Blau and Baltimore (1991) postulated that the differentiated state of a cell requires persistent active regulation, rather than lapsing into a passive “locked-in” state. Evidence has emerged from work in Caenorhabditis elegans to show that core “terminal selector” transcription factors, which differentiate neuronal phenotype during development, are subsequently required to maintain that identity after development. Notably, the expression of these transcription factors is maintained in an autoregulatory manner, providing an intrinsic, cell-autonomous mechanism for maintenance of neuronal phenotype (Hobert, 2008; Etchberger et al., 2009). Maintenance of extrinsically induced gene expression poses a distinct problem for maintenance. In neuronal progenitor cells, genes that are induced by inductive extrinsic signals subsequently switch to an intrinsic mechanism for maintenance (Edlund and Jessell, 1999). In contrast, our data now show that maintenance of extrinsically activated genes in postmitotic Drosophila neurons requires persistent extrinsic signaling, rather than switching to intrinsic, cell-autonomous regulation. This is the first demonstration that persistent TGFβ-type signaling is required to maintain neuronal phenotype, and highlights the possibility that many of the identified target-induced genes in the vertebrate nervous system may likewise require persistent retrograde signaling for their maintenance.
Work in vertebrates has provided compelling evidence that neurotrophins act retrogradely to maintain adult neuronal phenotype. Axotomy of adult sympathetic and sensory neurons alters their expression of specific neurotransmitters and neuropeptides (Zigmond et al., 1998). As nerve growth factor (NGF) is expressed at the targets of those neurons, it is notable that NGF administration partially blocks axotomy-induced phenotypic changes (Verge et al., 1995) and that NGF function-blocking antibodies partially recapitulate axotomy-induced changes in intact neurons (Shadiack et al., 2001). Similarly, cholinergic markers are downregulated in murine CBF (cholinergic basal forebrain) neurons in transgenic mice that express an anti-NGF antibody or after target tissue ablation (Sofroniew et al., 1993; Capsoni et al., 2000).
Such vertebrate studies indicate that retrograde signaling maintains neuronal phenotype, but in most cases it has been difficult to discriminate between (1) loss of gene expression as a direct consequence of loss of a specific retrograde signal and (2) loss of gene expression as an indirect consequence of neuronal injury, degeneration, or regeneration. The use of conditional mouse knock-outs and transgenics will circumvent many of these issues and promises a wealth of insight. For example, conditional BDNF knock-outs have uncovered a role for anterograde BDNF signaling in the maintenance of serotonin receptor expression in the adult prefrontal cortex (Rios et al., 2006). In our study, we have circumvented these issues using the TARGET spatiotemporal transgene targeting system (McGuire et al., 2004), which provides several advantages: (1) Using reversible temperature-dependent transgene expression, we were able to block retrograde BMP signaling cell-autonomously at any time point in intact Tv neurons in freely moving flies. (2) We were able to examine FMRFa expression in the same individually identifiable neurons between experimental groups. (3) The use of cell-specific markers that are not BMP-dependent, apterous and eyes absent, allowed us to identify Tv neurons in the absence of FMRFa expression, confirming that Tv neurons do not require retrograde BMP signaling for survival. Using this methodology, our results provide direct evidence to strengthen the conclusions from vertebrate work indicating a role for retrograde signaling in the maintenance of neuronal phenotype.
Inhibition of persistent retrograde signaling: a mechanism for disrupting neuronal phenotype in neurodegenerative disease
This study has demonstrated a requirement for persistent retrograde signaling in the maintenance of a gene critical to subset-specific neuronal phenotype and function. These data are of particular significance in light of the many neurodegenerative disorders, such as ALS, Alzheimer's disease, and Huntington's disease, that have been increasingly attributed to defects in axon transport (Gunawardena and Goldstein, 2004; Chevalier-Larsen and Holzbaur, 2006; De Vos et al., 2008). To block retrograde transport, we overexpressed a dominantly acting Glued mutant in Tv axons. Importantly, dominantly acting Glued mutations have been linked to ALS (Puls et al., 2003; Münch et al., 2004) and mouse models recapitulate many cellular and functional deficits of ALS (Levy et al., 2006; Chevalier-Larsen et al., 2008; Laird et al., 2008). Glued is a critical component of the dynactin complex necessary for dynein motor function in retrograde axon transport (Chevalier-Larsen and Holzbaur, 2006; De Vos et al., 2008), and together with data showing similar phenotypes have been described in mouse dynein mutants or on dynamitin overexpression (LaMonte et al., 2002; Hafezparast et al., 2003; Laird et al., 2008), numerous lines of evidence converge to indicate that axonal transport defects may be causative for ALS.
It is still unclear how reduced axon transport leads to neuronal dysfunction and degeneration. A widely held hypothesis proposes that disrupted axon transport diminishes target-derived neurotrophin signaling (Salehi et al., 2003; Blesch, 2006), depriving neurons of necessary trophic support and leading to degeneration. Evidence for this link has come from elegant work using mice trisomic for amyloid precursor protein (APP) to model early-onset Alzheimer's disease in Down's syndrome. This showed that APP overexpression inhibited retrograde NGF transport, leading to presynaptic neuron atrophy and reduced expression of the p75 low-affinity NGF receptor (Cooper et al., 2001; Salehi et al., 2006).
Our data show that blockade of dynein-mediated axonal transport eliminates retrograde BMP signaling in adult neurons. We further show that this directly impacts neuronal phenotype by eliminating the expression of a stably expressed, target-dependent gene that defines the identity and function of the neuron. Importantly, this occurs in the absence of overt neuronal degeneration. Thus, we postulate that the maintenance of neuronal phenotype may be particularly vulnerable to pathologies that diminish retrograde signaling, even in the absence of overt neuronal degeneration or death. Importantly, both our work and that of others suggest that the loss of neuronal phenotype resulting from disrupted axonal transport may be reversible. Cooper et al. (2001) found that reduced retrograde NGF transport in cholinergic basal forebrain neurons led to neuronal atrophy and reduced p75 expression, but that both could subsequently be fully reversed by NGF infusion. Similarly, our recovery data show that FMRFa expression was fully restored after retrograde BMP signaling was reestablished. Both studies strongly suggest that loss of neuronal phenotype in neurodegenerative disease may be treatable.
Footnotes
-
This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) and the EJLB Foundation. All imaging was performed on a confocal microscope that was supported from infrastructure awards from the Canadian Foundation for Innovation and The British Columbia Knowledge Development Fund. D.W.A. is a CIHR New Investigator, a Michael Smith Foundation for Health Research Career Scholar, an EJLB Foundation Investigator, and a Tula Scholar. We gratefully acknowledge Drs. Vanessa Auld, Shernaz Bamji, Timothy O'Connor, Jane Roskams (University of British Columbia, Vancouver, British Columbia, Canada), and Victor May (University of Vermont, Burlington, VT) for their valuable comments, and members of the Allan Laboratory for technical support and intellectual input. We also thank Justin Kumar, Carl-Henrick Heldin, Thomas Schwarz, Stuart Newfeld, Graeme Davis, Paul Taghert, and the Bloomington Drosophila Stock Centre for fly stocks and reagents.
- Correspondence should be addressed to Douglas W. Allan, Department of Cellular and Physiological Sciences, Room 2420 Life Sciences Centre, 2350 Health Sciences Mall, University of British Columbia, Vancouver, British Columbia, Canada V6T 1Z3. dwallan{at}interchange.ubc.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}