

Abstract
Cellular stress caused by genetic or environmental factors are considered to be the major inducers of cell death under pathological conditions. Induction of the apoptotic function of the tumor suppressor p53 is a common cellular response to severe genotoxic and oxidative stresses. In the nervous system, accumulation of p53 and increased p53 activity are associated with neuronal loss in acute and chronic neurodegenerative disorders. Here, we show that regulation of the p53 gene (trp53) is an integral part of a synaptic activity-controlled, calcium-dependent neuroprotective transcriptional program. Action potential (AP) bursting suppresses trp53 expression and downregulates key proapoptotic p53 target genes, apaf1 and bbc3 (puma). At the same time, AP bursting activates the nuclear calcium-induced neuroprotective gene, btg2. Depletion of endogenous p53 levels using RNA interference or expression of Btg2 renders neurons more resistant against excitotoxicity-induced mitochondrial permeability transitions and promotes neuronal survival under severe cellular stresses. We propose that suppression of p53 functions together with nuclear calcium-regulated neuroprotective genes in a coordinate and synergistic manner to promote neuronal survival through the stabilization of mitochondria against cellular stresses.
Introduction
In the CNS, programmed cell death or apoptosis plays a crucial role in the clearance of inappropriately connected neurons during embryonic development (Oppenheim, 1991; Buss et al., 2006). In adult organisms, apoptosis is considered to be a major cause of the loss of neurons in several neurological disorders including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), and stroke (Mattson, 2000; Bossy-Wetzel et al., 2004). Despite differences in the genetic and environmental factors that trigger apoptosis in pathological conditions, biochemical events that execute cell death are conserved (Danial and Korsmeyer, 2004; Green, 2005). One conserved mechanism involves a shift in the mitochondrial membrane permeability, known as mitochondrial permeability transition (MPT). MPT is initiated by a sustained opening of the mitochondrial permeability transition pore, which coincides with the collapse of the mitochondria membrane potential and the release of mitochondrial apoptotic factors (e.g., cytochrome c, AIF, and caspases) into the cytoplasm (Susin et al., 1996, 1999; Bossy-Wetzel and Green, 1999; Kroemer et al., 2007). Several lines of evidence suggest a link between mitochondrial dysfunction, abnormal expression of the tumor suppressor gene trp53, and neuronal cell death (LaFerla et al., 1996; de la Monte et al., 1997; Mihara et al., 2003; Chipuk et al., 2004, 2005; Bossy-Wetzel et al., 2004; Bae et al., 2005; Culmsee and Mattson, 2005; Feng et al., 2006). For example, ectopic expression of the p53, the protein encoded by trp53, triggers apoptosis in hippocampal neurons (Jordán et al., 1997), whereas genetic deletion of trp53 renders neurons more resistant against kainate-induced neuronal cell death (Morrison et al., 1996). Moreover, increased p53 expression was observed in the several animal models of neurological disorders including AD, HD, and ALS (LaFerla et al., 1996; González de Aguilar et al., 2000; Bae et al., 2005).
In neurons, cell death can be suppressed by neurotrophic factors and the stimulation of their respective receptors as well as by neuronal activity (Levi-Montalcini, 1987; Riccio et al., 1999; Hardingham et al., 2002; Papadia et al., 2005; Lee et al., 2005; Zhang et al., 2007). Activity-dependent, sustained neuroprotection is initiated by calcium entry into the neurons through synaptic NMDA receptors and requires the propagation of calcium signals into the nucleus and the induction or repression of gene transcription (Hardingham et al., 2002; Lee et al., 2005; Papadia et al., 2005; Zhang et al., 2007). The mechanisms underlying synaptic activity- and gene transcription-dependent neuronal survival are largely unknown. In this study, we establish a link between activity-regulated genes and a process that renders mitochondria more resistant to cellular stress and toxic insults. We provide evidence for a regulatory network that involves suppression of trp53 and induction of nuclear calcium-regulated genes, ultimately promoting neuroprotection through the inhibition of mitochondrial permeability transition.
Materials and Methods
Cell culture.
Hippocampal neurons from newborn C57Black mice were prepared and maintained as described previously (Bading and Greenberg, 1991; Zhang et al., 2007), except that growth medium was supplemented with B27 (Invitrogen/BRL) and 1% rat serum. Experiments were done after a culturing period of 10–13 d in vitro (DIV), during which hippocampal neurons express functional glutamate receptors (NMDA/AMPA/kainate) and develop a rich network of synaptic contacts. The following drugs were used: PD98059, U0126, SB203580, KN62, autocamtide-2-related inhibitory peptide II (AIPII) (Calbiochem), TTX, APV (Biotrend), FK506, bicuculline (Axxora), cyclosporine A, NMDA (Sigma-Aldrich), and MK-801 (Tocris Bioscience).
Recombinant adeno-associated viruses.
A recombinant adeno-associated virus (rAAV) vector containing a 1.3 kbp fragment of the mouse CaMKII promoter (a gift from Ali Cetin and Peter Seeburg, Max Planck Institute for Medical Research, Heidelberg, Germany) was used to express CaMKIIN, Btg2, red fluorescent protein (mRFP), and CaMBP4–mCherry. rAAV–CaMBP4–mCherry was generated by H. Eckehart Freitag (H. E. Freitag and H. Bading, unpublished observations). For expression of short hairpin RNAs (shRNAs), a rAAV vector was generated that contains the U6 promoter for shRNA expression and a CaMKII promoter driving mCherry expression. The mouse p53-targeting shRNA sequence described by Ventura et al. (2004) was extended to 21 bp (additional sequence is underlined): GTACTCTCCTCCCCTCAATAA. All rAAV vectors were generated by standard molecular biology techniques and verified by sequencing. Viral particles were produced and purified as described previously (Klugmann et al., 2005; Zhang et al., 2007). For viral infection, neurons were infected with 2–5 × 109 particles/ml on DIV 4–6 and harvested on DIV 10–13, depending on the experimental conditions.
Reporter gene assays and immunological methods.
The p53-luc (Clontech) reporter plasmid containing the p53-response element was cotransfected with a rAAV plasmid containing an expression cassette for humanized renilla luciferase (hrLuc) for normalization. Neurons were transfected with lipofectamine 2000 (Invitrogen) and harvested 48 h after transfection. Luciferase activities were measured with the Dual-luciferase Assay kit (Promega). Data represent mean value (±SEM) from at least three independent experiments, each performed in duplicates. Standard protocols for immunoblot analysis were used to detect the phosphorylation of CaMKII on threonine 286 (phospho-threonine286-CaMKII-specific antibodies; Promega), as well as the expression of p53 (1C12; Cell Signaling Technology), and tubulin (DM1A; Sigma). Immunocytochemical detection of p53 protein or Flag-tagged proteins was done by indirect immunofluorescence staining with antibodies to p53 (1C12) or anti-Flag antibodies (Sigma) and the anti-mouse Alexa 488 or Alexa 594 as secondary antibody (Invitrogen). Fluorescence images were acquired using a Leica SP2 confocal microscope (Leica).
Quantitative reverse transcriptase PCR.
cDNAs were synthesized from 2 to 3 μg of total RNA (Qiagen, RNeasy kit; Total RNA isolation kit, Roche) using High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative reverse transcriptase PCR (QRT-PCR) was done on an ABI7300 thermal cycler using universal QRT-PCR master mix with TaqMan Gene Expression Assays for the indicated genes (Applied Biosystems). The following TaqMan gene expression assays were used in this study: 18 s rRNA (4319413E), apaf1 (Mm00437530_m1), bbc3 (PUMA, Mm00519268_m1), gusb (Mm00446953_m1), and trp53 (p53, Mm01731287_m1). Expression of target genes was normalized against the expression of gusb and/or 18 s rRNA as endogenous control genes. Data represent mean value (±SEM) from at least four independent experiments.
Imaging.
Imaging of calcium signals and mitochondrial membrane potential was done as described previously (Hardingham et al., 2002), with some modifications. In brief, neurons were loaded with 3.8 μm Fluo-3 (Invitrogen) for 20 min in CO2-independent salt–glucose–glycine (SGG; 140.1 mm NaCl, 5.3 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm HEPES, 1 mm glycine, 30 mm glucose, and 0.5 mm sodium pyruvate) (Bading et al., 1993). Calcium concentrations were expressed as a function of the Fluo-3 fluorescence [(F − F min)/(F max − F)]. Ionomycin (50 μm; Sigma) and saturated MnCl2 solution was used to obtain F max and F min. For imaging of mitochondrial membrane potential, neurons were loaded with 10 μm rhodamine 123 (Rh123; Invitrogen) in SGG for 10 min followed by extensive washing step with SGG. Mitochondrial membrane potential was calculated as percentage of Rh123 fluorescence signals; maximal signals were induced with 5 μm of the mitochondrial uncoupler of oxidative phosphorylation carbonylcyanide p-(trifluoromethoxy) phenylhydrazone (FCCP; Sigma). In some experiments, 200 nm MitoTracker Red CMXRos (Invitrogen) was included into the loading solution. Tetramethylrhodamine ethylester (TMRE; Invitrogen) signals were measured as described previously (Schinder et al., 1996), with the following modifications. Neurons were loaded for 60 min with 50 nm TMRE in SGG. Baseline fluorescence (F BaSE) was measured as average normalized fluorescence emitted during the initial 200 s of the experiment.
Survival assays.
Two types of assays were used to investigate apoptotic cell death. Details of the methods have been described previously (Hardingham et al., 2002; Papadia et al., 2005; Zhang et al., 2007). Briefly, hippocampal neurons were exposed to 20 nm staurosporine (STS) (Calbiochem) for 24 h or to medium lacking growth and trophic factors for 48 h, all in the presence of TTX (1 μm). Neurons were fixed with paraformaldehyde and subjected to nuclear staining with Hoechst 33258. The percentage of neurons with a shrunken cell body and large round chromatin clumps characteristic of apoptotic death was determined. The results are given as means ± SEM from at least four independent experiments. Statistical significance was determined by ANOVA.
Results
Synaptic activity suppresses p53 expression and p53 target gene expression
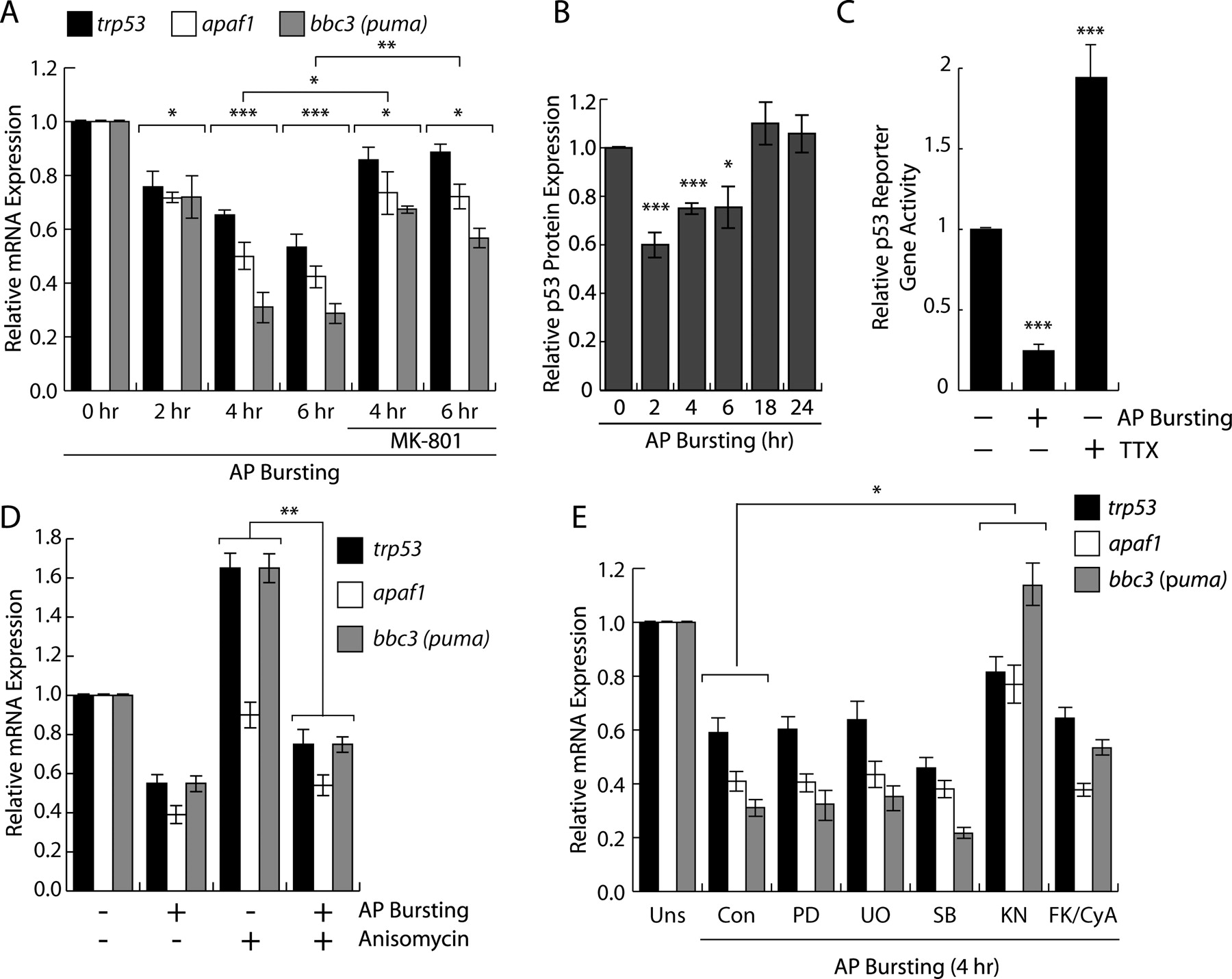
To investigate the possibility that trp53 is subject to regulation by synaptic activity, we exposed hippocampal neurons to the GABAA receptor blocker bicuculline. This treatment relieved tonic, GABAA receptor-mediated inhibition of synaptic transmission from the hippocampal network and induced periodically occurring bursts of action potential (AP) firing. Each burst is associated with a robust intracellular calcium transient that propagates to the cell nucleus, stimulates cAMP response element-binding protein (CREB)/CREB-binding protein-mediated gene expression and induces a genomic neuroprotective program (Hardingham et al., 2001, 2002; Arnold et al., 2005; Lee et al., 2005; Papadia et al., 2005; Zhang et al., 2007). QRT-PCR analysis revealed that trp53 expression is suppressed after AP bursting (Fig. 1 A). This activity-induced suppression of trp53 was dependent on activation of synaptic NMDA receptors, since pretreatment with MK-801, a selective noncompetitive NMDA receptor antagonist, significantly reduced the suppression (Fig. 1 A). Because p53 protein is a transcriptional activator known to induce expression of proapoptotic genes, we next investigated whether activity-dependent suppression of trp53 affects transcription of proapoptotic p53-target genes. We found that two prominent, putative p53 target genes, apaf1 (Fortin et al., 2001; Moroni et al., 2001) and bbc3 [also known as p53-upregulated mediator of apoptosis (puma)] (Nakano and Vousden, 2001; Yu et al., 2001) are also suppressed by AP bursting in an NMDA receptor-dependent manner (Fig. 1 A). To investigate whether activity-mediated suppression of trp53 mRNA and p53 protein expression (Fig. 1 B) leads to a decrease in p53 transcriptional activity, we performed transcription assays using a luciferase reporter gene that contains multiple copies of p53-binding sites in the promoter. We found that compared with untreated neurons, reporter gene expression was significantly reduced in neurons after AP bursting (Fig. 1 C). In contrast, neurons rendered inactive with TTX showed an ∼2-fold increase in the luciferase activity, indicating that spontaneous synaptic transmission contributes to suppression of basal p53-dependent gene transcription (Fig. 1 C). These results suggest a link between neuronal activity, synaptic NMDA receptor activation, and p53 signaling in hippocampal neurons.

Synaptic activity suppresses trp53 and p53 target gene expression. A , QRT-PCR analysis of trp53, apaf1, and bbc3 expression in hippocampal neurons after the indicated times after AP bursting induced by bicuculline (50 μm) in the presence or absence of the NMDA receptor antagonist, MK-801 (10 μm). Statistical analysis was determined by ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001. A transcriptome analysis revealed that in hippocampal neurons the expression levels of several thousand genes did not change at 2 and 4 h after AP bursting induced by bicuculline (Zhang et al., 2007). B , Immunoblot analysis of p53 expression in hippocampal neurons after the indicated times after AP bursting induced by bicuculline (50 μm). The quantifications of the immunoblots are shown (n = 4). C , Analysis of p53-dependent transcription from a p53-luc reporter gene in hippocampal neurons 4 h after AP bursting induced by bicuculline (50 μm) or after 16 h of treatment with TTX (1 μm). Mean values ± SEM from three independent experiments are shown. Statistical analysis was determined by ANOVA; ***p < 0.001. D , QRT-PCR analysis of trp53, apaf1, and bbc3 expression in hippocampal neurons 4 h after AP bursting induced by bicuculline (50 μm) in the presence or absence of the protein synthesis inhibitor, anisomycin (10 μg/ml). E , QRT-PCR analysis of trp53, apaf1, and bbc3 expression in hippocampal neurons 4 h after AP bursting induced by bicuculline (50 μm) in the presence or absence of the indicated drugs. PD98059 (PD; 50 μm), U0126 (UO; 10 μm), SB203580 (SB; 10 μm), KN62 (KN; 5 μm), FK506 plus cyclosporine A (FK/CyA; 1 μm each). Drugs were applied 60 min before induction of AP bursting.
To determine whether activity-dependent suppression of trp53, apaf1, and bbc3 is mediated by a pre-existing machinery, we treated hippocampal neurons before the induction of AP bursting with the protein synthesis inhibitor, anisomycin. We found that anisomycin led to elevated basal levels of trp53 and bbc3 (not of apaf1) but did not inhibit AP bursting-induced suppression of trp53, apaf1, and bbc3 (Fig. 1 D). These results indicate that the protein machinery that mediates the suppression of trp53, apaf1, and bbc3 is pre-existing and targeted by synaptic activity controlled signaling pathways.
Role of calcium-activated signaling pathways in activity-induced suppression of trp53
Neuronal activity and calcium entry into neurons can control gene expression through several distinct intracellular signaling cascades (Cruzalegui and Bading, 2000; Flavell and Greenberg, 2008). To delineate activity-regulated signaling pathways involved in transcriptional suppression of trp53 and p53 target genes, we used pharmacological and recombinant tools. The following drugs were used and their efficiency in inhibiting the appropriate pathway was confirmed: PD98059 and UO126 [extracellular signal-regulated protein kinase–mitogen-activated protein (MAP) kinase pathway], SB203580 (p38 MAP kinases/stress-activated protein kinase), FK506 plus cyclosporine A (calcineurin), and KN-62 [calcium/calmodulin-dependent (CaM) protein kinases]. With the exception of KN-62, none of the inhibitors had a significant effect on AP bursting-induced suppression of trp53 and its transcriptional targets apaf1 and bbc3 (Fig. 1 E). The effect of KN62 on trp53 and p53 target genes suggests an involvement of CaM kinases in the gene repression process.
Previous studies have established that the generation of nuclear calcium transients after synaptic activity and the stimulation of nuclear CaM kinase IV are key events in synapse-to-nucleus communication and the control of activity-induced, neuroprotective genomic responses (Bading, 2000; Hardingham et al., 1997, 1999, 2001; Chawla et al., 1998; Papadia et al., 2005; Zhang et al., 2007). Indeed, several nuclear calcium-regulated genes with roles in neuronal survival have been described, which includes btg2 and bcl6 (Zhang et al., 2007). However, also CaMKII, which is predominantly a non-nuclear CaM kinase (Griffith et al., 2003), and CaMKI (Picciotto et al., 1995), have been implicated in neuronal gene expression (Soderling, 1999; Cruzalegui and Bading, 2000; Hook and Means, 2001; Schmitt et al., 2004; Wayman et al., 2006; Zhou et al., 2006; Flavell and Greenberg, 2008). To investigate whether calcium signaling in the cell nucleus and thus the activating of nuclear CaM kinases are necessary for AP bursting-induced suppression of trp53, apaf1, and bbc3, we expressed CaMBP4 in hippocampal neurons. CaMBP4 is a nuclear protein that consists of four tandem repeats of the calmodulin-binding peptide, M13, from myosin light chain kinase. CaMBP4 binds to the nuclear calcium/calmodulin complex, thereby preventing nuclear calcium/calmodulin-regulated events from being activated (Wang et al., 1995; Papadia et al., 2005; Zhang et al., 2007). We constructed an rAAV containing an expression cassette for a fusion protein of CaMBP4 and the red fluorescent protein, mCherry. In hippocampal neurons infected with rAAV–CaMBP4–mCherry, expression of CaMBP4–mCherry was readily detectable (Fig. 2 A). Expression of CaMBP4–mCherry efficiently blocked AP bursting-induced expression of btg2, a known nuclear calcium-regulated gene (Zhang et al., 2007) (Fig. 2 A). However, CaMBP4–mCherry, also in combination with the MEK1 inhibitor UO126, did not significantly affect the AP bursting-induced suppression of trp53, apaf1, or bbc3 (Fig. 2 B). An rAAV-expressing mRFP was used as control; rAAV–mRFP did not affect any of the transcriptional responses analyzed (Fig. 2 A,B,D). These results indicate that activity-induced suppression of trp53, apaf1, and bbc3 does not require nuclear calcium signaling.

Role of calcium-activated signaling pathways in activity-induced suppression of trp53. A , B , QRT-PCR analysis of expression of btg2 ( A ; right) and trp53, apaf1, and bbc3 ( B ) 4 h after AP bursting induced by bicuculline (50 μm) in the presence or absence and U0126 (10 μm) in uninfected hippocampal neurons and in hippocampal neurons infected with rAAV–mRFP or rAAV–CaMBP4–mCherry. Data represent mean ± SEM (n ≥ 4). Statistical analysis was determined by ANOVA; *p < 0.05, **p < 0.01. A , Left, Photomicrographs (confocal images) of hippocampal neurons infected with rAAV–mRFP or rAAV–CaMBP4–mCherry illustrating the subcellular localization of mCherry and CaMBP4–mCherry. Hoechst staining identified nuclei. Scale bar, 20 μm. C , Left, Immunoblot blot analysis of CaMKII phosphorylation at threonine residue 286 4 h after AP bursting induced by bicuculline (50 μm) in uninfected neurons and in neurons infected with rAAV–mRFP or rAAV–CaMKIIN–Flag. Antibodies to the Flag tag identify CaMKIIN–Flag; tubulin was used as loading control. Right, Immunostaining analysis (using antibodies to the Flag tag) of hippocampal neurons infected with rAAV–CaMKIIN–Flag. Hoechst staining identified nuclei. Representative examples are shown. Scale bar, 25 μm. D , QRT-PCR analysis of expression of trp53, apaf1, and bbc3 4 h after AP bursting induced by bicuculline (50 μm) in uninfected hippocampal neurons and in hippocampal neurons infected with rAAV–mRFP or rAAV–CaMKIIN–Flag. Data represent mean ± SEM (n ≥ 4). Statistical analysis was determined by ANOVA; **p < 0.01; NS, not significant.
To investigate a possible role of CaMKII in the activity-induced suppression of trp53, we expressed the CaMKII inhibitor, CaMKIIN, in hippocampal neurons. CaMKIIN acts by interfering with CaMKII autophosphorylation at the threonine residue 286 (Chang et al., 1998). Western blot analysis revealed that rAAV-mediated expression of CaMKIIN inhibited both basal as well as AP bursting-induced autophosphorylation of CaMKII at threonine 286 by ∼50% (Fig. 2 C). However, neither expression of CaMKIIN nor inhibition of CaMKII using a synthetic, cell-permeable AIPII (Ishida et al., 1995) affected activity-induced suppression of trp53, apaf1, and bbc3 (Fig. 2 D) (data not shown). These results argue against a role of CaMKII in activity-induced suppression of trp53, apaf1, and bbc3, although we cannot rule out the possibility that the residual CaMKII activity in CaMKIIN-expressing or AIPII-treated hippocampal neurons is sufficient to mediate these genomic events.
Suppression of p53 activity promotes neuronal survival
We next investigated a possible causal link between the activity-induced suppression of trp53 expression and neuroprotection afforded by synaptic activity. We used RNA interference (RNAi) to decrease p53 expression in hippocampal neurons, which were subsequently subjected to cell death assays. A shRNA designed to target the mouse trp53 mRNA was inserted downstream of the U6 promoter of a rAAV vector. The resulting rAAV, termed rAAV–trp53–RNAi, also harbors an expression cassette for mCherry (Fig. 3 A). To control for nonspecific effects of infections with rAAVs carrying an expression cassette for shRNAs, a rAAV was used that contains a control shRNA (rAAV–control–RNAi), which has no significant sequence similarity to the mouse, rat, or human genome. Analysis of mCherry expression allowed us to assess infection rates, which ranged from 80 to 95% of the neuron population (Fig. 3 A). QRT-PCR and immunoblot analysis revealed that in neurons infected with rAAV–trp53–RNAi, but not in neurons infected with rAAV–control–RNAi, p53 expression was reduced (Fig. 3 A,B). Analysis of p53 target gene expression revealed a reduction of apaf1 but not of bbc3 mRNA levels in hippocampal neurons infected with rAAV–trp53–RNAi (Fig. 3 B). This indicates that basal levels of p53 in hippocampal neurons are critical for apaf1 expression, whereas reduced levels of p53 or proteins other than p53 are sufficient to uphold expression of bbc3.

Suppression of endogenous p53 confers robust neuroprotection in hippocampal neurons. A , Top left, Schematic drawing of rAAV vector for shRNA expression. Bottom left, Immunoblot analysis of p53 expression in hippocampal neurons infected with rAAV–control--RNAi or rAAV–trp53–RNAi. Tubulin was used as loading control. Right, Immunostaining analysis (using antibodies to p53) in hippocampal neurons infected with rAAV–control--RNAi or rAAV–trp53--RNAi. Photomicrographs of p53 immunoreactivity, mCherry fluorescence, and Hoechst staining are shown. Scale bar, 20 μm. B , QRT-PCR analysis of trp53, apaf1, and bbc3 expression in uninfected hippocampal neurons and in hippocampal neurons infected with rAAV–control–RNAi or rAAV–trp53–RNAi. Data represent means ± SEM (n = 4). Statistically significant differences were analyzed by ANOVA; ***p < 0.001; NS, not significant. C , Analysis of apoptosis after STS treatment or growth factor withdrawal (−GF) in uninfected hippocampal neurons and in hippocampal neurons infected with rAAV–control–RNAi or rAAV–trp53–RNAi with or without a 16 h period of AP bursting induced by bicuculline (50 μm) before STS treatment or GF withdrawal. Bars represent means ± SEM (n = 4). Statistical analysis was determined by ANOVA; ***p < 0.001; NS, not significant.
We next investigated the ability of rAAV–trp53–RNAi to confer neuroprotection. Two types of assays were used: growth factor (GF) withdrawal and treatment with a low concentration of STS, a classical inducer of apoptotic cell death. We found that compared with uninfected neurons or neurons infection with rAAV–control–RNAi, cell death induced by either GF withdrawal or STS treatment was inhibited in neurons infected with rAAV–trp53–RNAi (Fig. 3 C). The inhibition was comparable with the known inhibition of cell death afforded by AP bursting (Hardingham et al., 2002; Lee et al., 2005; Papadia et al., 2005; Zhang et al., 2007) (Fig. 3 C) or after infection of hippocampal neurons with rAAV–btg2, which gives rise to expression of Btg2, a nuclear calcium-regulated, survival-promoting gene (Zhang et al., 2007). The combination of both neuroprotective treatments (i.e., AP bursting and infection with rAAV–trp53–RNAi) did not yield an additive effect on neuronal survival (Fig. 3 C). These results indicate that the suppression of trp53 is an integral part of an activity-controlled neuroprotective transcriptional program.
Synaptic activity, suppression of trp53, and expression of Btg2 render neurons resistant against mitochondrial permeability transition
Mitochondrial dysfunction may be involved in several (perhaps all) neurodegenerative condition including AD, HD, and stroke (Wallace, 2005; Lin and Beal, 2006; Kroemer et al., 2007). We, therefore, reasoned that stabilization or strengthening of mitochondria may be a common end point of many neuroprotective processes. To test this hypothesis, we investigated the mitochondrial membrane potential and the stimulus-induced breakdown of it in hippocampal neurons that have been rendered more resistant to toxic stimuli either by AP bursting, suppression of trp53, or expression of Btg2. The breakdown of mitochondrial membrane potential is considered the first step toward MPT, which can lead to mitochondrial dysfunction, loss of energy supply/ATP production, and ultimately to cell death (Zamzami et al., 1995; Bossy-Wetzel and Green, 1999; Kroemer et al., 2007). MPT was initiated by inducing NMDA receptor-mediated excitotoxicity, and changes in mitochondrial membrane potential were assesses using Rh123 imaging (Keelan et al., 1999; Hardingham et al., 2002). Since NMDA receptor-mediated excitotoxicity is a calcium-dependent process, we first investigated whether rAAV infections used in this study affect the NMDA-induced increases in intracellular calcium concentration. We found that infection of hippocampal neurons with rAAV–mRFP, rAAV–control–RNAi, rAAV–empty–RNAi, rAAV–trp53–RNAi, rAAV–btg2 had no significant effect on NMDA-induced calcium transients (Fig. 4).

Depletion of p53 does not affect NMDA-induced calcium transients. A , Fluo-3 calcium imaging of uninfected hippocampal neurons and hippocampal neurons infected with the indicated rAAVs after exposure to NMDA (10 μm). Traces represent mean values obtained from at least 150 neurons from more than five independent experiments. B , C , Quantitative analysis of NMDA-induced calcium responses. B , Peak values were calculated from the average Fluo-3 signals within a phase of 16.35 s after NMDA application above baseline. C , The area under the curve represents the integral of the Fluo-3 signals above baseline for the duration of NMDA treatment. Mean values from at least five independent experiments were used for the analysis. Statistical analysis was determined by ANOVA; NS, not significant.
Rh123 imaging revealed that in uninfected hippocampal neurons and in hippocampal neurons infected with rAAV–mRFP or rAAV–empty–RNAi, bath application of NMDA (30 μm) caused a rapid breakdown of mitochondrial membrane potential, which is evident as a robust increase in Rh123 fluorescence (Fig. 5). Even after washout of NMDA, Rh123 fluorescence continued to increase (Fig. 5). In contrast, in hippocampal neuron that had either been infected with rAAV–trp53–RNAi or rAAV–btg2 or had undergone a period of AP bursting to boost survival activity (Hardingham et al., 2002; Lee et al., 2005; Papadia et al., 2005; Zhang et al., 2007), the NMDA-induced loss of mitochondrial membrane potential was delayed, occurred with slower kinetics, and reached significantly lower magnitudes (Fig. 5). Consistent with the role of synaptic NMDA receptor activation in the build-up of neuroprotection by AP bursting, we found that the protection of mitochondria from membrane potential breakdown failed to develop when the period of AP bursting took place in the presence of the NMDA receptor antagonist, APV (Fig. 5). We also assessed mitochondrial function using TMRE. Unlike the green-shifted fluorescence of Rh123, TMRE exhibits a red-shifted fluorescence, and is, therefore, compatible with the expression of GFP. Because TMRE looses its fluorescence during mitochondrial membrane potential breakdown, we used the half-decay time of TMRE (τTMRE) after application of NMDA as an indicator for the degree of protection. Similar to the result obtained with Rh123, we observed a significant increase in τTMRE in hippocampal neurons after AP bursting and in neurons infected with rAAV–trp53–RNAi or rAAV–btg2 compared with uninfected, unstimulated hippocampal neurons, neurons that had undergone a period of AP bursting in the presence of APV, and neurons infected with rAAV–gfp or rAAV–empty–RNAi (Fig. 6). Thus, suppression of p53 and induction of nuclear calcium-regulated neuroprotective genes, both triggered by synaptic activity and NMDA receptor activation, render mitochondria more resistant to death-inducing conditions. These findings support a concept in which the strengthening of mitochondrial functions serve as one common end point of a diverse range of neuronal survival promoting signaling pathways and genes.

Rh123 imaging of NMDA-induced breakdown of mitochondrial membrane potential. A , Rh123 imaging of uninfected hippocampal neurons and hippocampal neurons infected with the indicated rAAVs. Rh123 imaging of uninfected hippocampal neurons 16 h after induction of AP bursting in the presence or absence of APV (250 μm) is shown for comparison. Neurons were stimulated with NMDA (30 μm) for 4 min followed by washout of NMDA and treatment at the indicated time with the mitochondrial uncoupler, FCCP (5 μm) to obtain the maximal Rh123 signals. Representative traces are shown; the thick line represents the mean value. B , C , Quantitative analysis of Rh123 measurements. The NMDA-induced percentage increase in Rh123 fluorescence after NMDA application is shown in B . The area under the curve represents the integral of the Rh123 signals above baseline beginning at the time of NMDA application until the application of FCCP ( C ). Data represent mean ± SEM (n ≥ 4 independent experiments, with at least 150 single cells). Statistical analysis was determined by ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001.

TMRE imaging of NMDA-induced breakdown of mitochondrial membrane potential. A , TMRE imaging of uninfected hippocampal neurons and hippocampal neurons infected with the indicated rAAVs. TMRE imaging of uninfected hippocampal neurons 16 h after induction of AP bursting in the presence or absence of APV (250 μm) is shown for comparison. Neurons were stimulated with NMDA (50 μm) for 400 s followed by washout of NMDA. Baseline fluorescence (F Base) was defined as average normalized TMRE fluorescence emitted during the initial 200 s of the experiment. Representative traces are shown; the thick line represents the mean value. Dashed lines indicate τTMRE. B , Quantitative analysis of TMRE measurements. The half-decay time of TMRE fluorescence (τTMRE) after NMDA application is shown. Data represent mean ± SEM (n ≥ 4 independent experiments, with at least 110 cells analyzed each). Statistical analysis was determined by ANOVA; **p < 0.01, ***p < 0.001.
Discussion
In this study, we show that the tumor suppressor gene, trp53, and two of its targets are subject to regulation by synaptic activity and NMDA receptor signaling. We provide evidence that suppression of trp53 acts in concert with neuronal activity and nuclear calcium-induced neuroprotective genes to promote neuronal survival through a mechanism that results in inhibition of mitochondrial permeability transition. Until now, anti-apoptotic Bcl-2 family members (some of which can be induced by CREB) have been considered primarily responsible for the inhibition of mitochondria-dependent apoptosis in neurons (Riccio et al., 1999; Rong and Distelhorst, 2008). For example, Bcl-2 inhibits the mitochondrial release of proapoptotic factors, prevents excessive mobilization of calcium from the endoplasmic reticulum, and the activation of calcium-dependent proteases of the calpain family (Kluck et al., 1997; Bossy-Wetzel and Green, 1999; Pinton and Rizzuto, 2006; Rong et al., 2008; Rong and Distelhorst, 2008). The results presented here provide a complementary mechanism through which neuronal activity controls and limits mitochondrial permeability transition and prevents apoptosis.
Transcriptional suppression of p53: new therapeutic opportunities for cancer treatments and neurological diseases
The regulation of p53 at the level of gene transcription is rather unexpected, given the wealth of literature on post-translational control of p53 function (Oren, 1999; Vogelstein et al., 2000; Harris and Levine, 2005; Vousden and Lane, 2007) and sparse information on trp53 gene regulation (Wu and Lozano, 1994; Ray et al., 1997; Raman et al., 2000). Although the precise mechanism through which neuronal activity regulates trp53 remains to be investigated, the new findings presented here may have an impact on current therapeutic strategies to prevent cell death associated with elevated p53 levels. One application for a selective inhibition of trp53 expression is the protection of normal cells from the detrimental side effects of chemotherapy or radiotherapy, which aim at activating a p53-dependent apoptotic program in tumor cells (Komarov et al., 1999; Gudkov and Komarova, 2005). Exploiting the ability of neurons to suppress trp53 expression may be beneficial in the treatment of aggressive brain tumors, where the application of high therapeutic doses of gamma irradiation is complicated by the detrimental effects of radiation-induced elevated p53 levels and cell death in the surrounding healthy brain tissues. A selective and transient suppression of trp53 expression using, for example, pharmacological activators of neuronal activity may help prevent therapy-induced collateral damage in healthy tissue. Another potential application is in the therapy of neurodegenerative diseases. Inhibition of p53 function either using pharmacological blockers (Culmsee et al., 2001; Duan et al., 2002) or RNAi, or through stimulating the activity-induced suppression of trp53 transcription, may provide efficient neuroprotection and attenuate cell death associated with stroke or chronic neurodegenerative conditions including PD and AD.
Role of the p53-nuclear calcium-signaling mitochondria nexus in neurodegenerative diseases
The observed link between activity-induced suppression of trp53, induction of nuclear calcium-regulated neuroprotective genes, and mitochondrial function raise the question whether persistent lack of neuronal activity resulting from disease-specific factors might render affected neurons more vulnerable to cellular stresses, such as respiratory oxidative stress. Neurons deprived of synaptic activity may be prone to apoptosis because of the gradual accumulation of p53 and lack of expression of nuclear calcium-regulated survival genes, both leading to destabilization of mitochondria. Neuronal death is, therefore, not a direct consequence of disease-specific factors but rather reflects the failure of the cellular and mitochondrial defense systems to neutralize cellular stresses that in healthy conditions would not harm the neurons. In this model, selective vulnerability of particular brain regions that is characteristic of several neurodegenerative diseases (Coyle et al., 1983; Dawson and Dawson, 2003; Boilllée et al., 2006; Orr and Zoghbi, 2007) may result from the disease-specific factors causing an impairment of synaptic transmission only within a local neuronal circuit (Lotharius and Brundin, 2002; Selkoe 2002; Li et al., 2003; Smith et al., 2005; Palop et al., 2006). The lack of neuronal activity renders those neurons more susceptible to apoptosis through the described p53-nuclear calcium-signaling mitochondria nexus. Extended periods of synaptic dysfunction, perhaps combined with an aging-related decline of intrinsic cellular defense systems [such as antioxidant systems (Papadia et al., 2008)], may be required to reach a dysfunctional, pathological state, which explains the late onset of many neurodegenerative disease.
Mitochondria: a common end point of neuroprotective processes?
As mitochondrial failure is the gate to death, survival-promoting signaling pathways or genes may ultimately provide neuroprotection through a common process that preserves and strengthens mitochondria and makes them more resistant to cellular stress and toxic insults. Thus, mitochondria may be the converging point of a variety of neuroprotective cellular paths. In this study, we provide examples of two distinct activity-regulated genes that can attenuate mitochondrial dysfunction. Several other calcium signal-regulated genes with putative roles in survival have been identified (Zhang et al., 2007) that may act in a similar manner. We predict that endogenous, activity-induced neuroprotection is mediated by a calcium signal-regulated gene network controlling mitochondrial function.
Footnotes
-
This work was supported by the Alexander von Humboldt Foundation (Wolfgang-Paul-Prize to H.B.), the Deutsche Forschungsgemeinschaft (DFG) Graduate College 791 (D.L.), SFB488 and SFB636 from the DFG, Graduate Academy (Excellence Initiative) of Heidelberg University, the European Union (EU) Network of Excellence NeuroNE, and the EU Project Glutamate Receptor Interacting Proteins as Novel Neuroprotective Targets. H.B. is a member of the Excellence Cluster CellNetworks at Heidelberg University. We thank Peter Seeburg and Ali Cetin (Max Planck Institute for Medical Research, Heidelberg, Germany) for providing the AAV expression vector containing the 1.3 kb mouse CaMKIIα promoter, Roger Tsien (University of California, San Diego, San Diego, CA) for providing pRSetB–mRFP, Peter Schultz (Scripps, San Diego, CA) for the p53-luc reporter, Thomas Soderling (Vollum Institute, Portland, OR) for CaMKIIN expression vector, H. Eckehard Freitag for the construction of rAAV-expressing CaMBP4–mCherry, Iris Bünzli-Ehret for hippocampal neurons, Anja Eder and Peter Bengtson for advice on live-cell imaging, and Ruth Jelinek for virus purification.
- Correspondence should be addressed to Hilmar Bading, Department of Neurobiology, Interdisciplinary Center for Neurosciences, University of Heidelberg, Im Neuenheimer Feld 364, 69120 Heidelberg, Germany. Hilmar.Bading{at}uni-hd.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}