

Abstract
The AMPA receptor subunit GluA1 (GluR-A) has been implicated to be critically involved in the expression of long-term potentiation (LTP) and memory formation. Mice lacking this subunit possess a profound spatial working memory deficit. We investigated the influence of the GluA1 subunit on the expression of LTP in pyramidal neurons of the hippocampus CA1 region and somatosensory cortex layer 2/3 for different cellular LTP protocols in adult mice. We found that the GluA1 subunit was not required for LTP in cortical pyramidal neurons. In contrast, GluA1-dependent LTP expression in CA1 pyramidal neurons was differentially dependent on the LTP induction parameters. Depolarization pairing was exclusively, theta-burst pairing was partially, and spike-timing-dependent plasticity (STDP) was independent of the GluA1 subunit. Spike-timing-dependent LTP required postsynaptic membrane fusion in CA1 pyramidal neurons. We conclude that during LTP induction at the hippocampal CA3-to-CA1 synapse the recruitment of the GluA1 subunit is controlled by particular electrical activity patterns that might reflect specific behavioral states. Furthermore, other LTP expression mechanisms exist that do not require the presence of GluA1. The previously reported spatial working memory deficits in GluA1-lacking mice (Gria1−/− mice) together with these results suggest that STDP might be a likely basis for the formation of spatial reference memory whereas it is not required for the rapid formation of spatial working memory where a fast but transient increase of synaptic efficacy might be needed.
Introduction
The AMPA receptor subunit GluA1 (GluR-A) has been implicated in the formation of hippocampus-dependent spatial working memory. Mice deficient in the GluA1 subunit (Gria1−/− mice) show a decreased performance in several behavioral tests for spatial working memory whereas in tests for spatial reference memory the Gria1−/− mice learned as efficiently as wild-type mice (Zamanillo et al., 1999; Reisel et al., 2002; Schmitt et al., 2004; Sanderson et al., 2007). The molecular basis for this difference in spatial working and reference memory formation might be a differential expression of GluA1-dependent long-term potentiation (LTP). GluA1-deficient mice lack tetanus-induced LTP at the Schaffer collateral CA3-to-CA1 synapse but show robust increases in synaptic efficacy induced by a theta-burst pairing protocol at the very same synapse (Hoffman et al., 2002). Conditional restoration of expression of the GluA1 subunit in Gria1−/− mice recovered the expression of LTP and also rescued the spatial working memory deficit (Mack et al., 2001; Schmitt et al., 2005). Thus, the formation of the hippocampal neuronal network is established by GluA1-independent forms of synaptic plasticity, but retains the potential to express GluA1-dependent LTP. Indeed, a juvenile form of LTP, which is independent of GluA1, but presumably depends on the GluA2long (GluR-Blong) subunit, is present during development (Jensen et al., 2003; Kolleker et al., 2003).
Long lasting changes in glutamatergic synaptic transmission mediated by AMPA receptors can be regulated by several biochemical mechanisms including delivery of additional novel receptors to the postsynaptic membrane (Shi et al., 1999). In this process the GluA1 subunit has been suggested to be a key element (Lüscher et al., 2000; Barry and Ziff, 2002; Malinow and Malenka, 2002; Song and Huganir, 2002). Tetanus-induced LTP has been reported to depend on SNARE proteins as part of a postsynaptic exocytotic fusion machinery (Lledo et al., 1998). Thus, rapid delivery of GluA1-containing AMPA receptors to the postsynaptic membrane by LTP-inducing stimuli is presumably mediated by exocytotic fusion events (Shi et al., 1999; Passafaro et al., 2001). Interference with the regulatory machinery of GluA1-dependent LTP also results in a behavioral impairment of spatial memory formation (Lee et al., 2003).
Since the GluA1 subunit has been implicated as a major determinant for spatial working memory formation it is important to know whether LTP in all brain regions is similarly affected by the deletion of the GluA1 subunit. We investigated the influence of the GluA1 subunit on the expression of LTP in CA1 pyramidal neurons of hippocampus and in layer 2/3 (L2/3) pyramidal neurons in somatosensory cortex of adult mice. We found that the GluA1 subunit is not required for LTP in cortical pyramidal neurons whereas in CA1 pyramidal neurons GluA1 is differentially recruited by different patterns of activity. We conclude that the requirement for GluA1-dependent LTP is brain region specific and also depends on the specific pattern of correlated activity in the presynaptic and postsynaptic cells. Therefore diverse biochemical signaling cascades, which can be selectively activated by different activity patterns, differentially regulate the subunit composition of AMPA receptors at the synapse.
Materials and Methods
Mouse lines.
For all physiological experiments, the colonies of wild-type (WT) and knock-out (Gria1−/−) mice (Zamanillo et al., 1999) were simultaneously produced by mating of heterozygous mice in the animal facility at the Interdisciplinary Research Unit (IBF) of the University of Heidelberg under the license number MPI/T-15/08. LTP experiments were performed on mice at postnatal day 39–100. Mice were genotyped by tail-PCR at postnatal day 14 (Zamanillo et al., 1999). The genotype was confirmed postmortem using another sample of tail tissue. Experiments with mice were blinded with respect to the genotype except for pharmacology. All experimental procedures were in accordance with the animal welfare guidelines of the Max Planck Society.
Slice preparation.
Mice were deeply anesthetized with halothane or isoflurane and decapitated, and the brain was quickly removed in ice-cold standard ACSF (125 mm NaCl, 25 mm NaHCO3, 2,5 mm KCl, 1.25 NaH2PO4, 1 mm MgCl2, 25 mm glucose, 2 mm CaCl2). Transversal hippocampal slices were prepared by cutting the dorsal part of the brain in parallel with its basis (Hoffman et al., 2002). Thalamocortical slices were prepared as described by Agmon and Connors (1991). The brain was glued with its cut surface onto the stage of a tissue slicer and the first 2 mm from the brain surface were discarded. Then 300-μm-thick slices were cut and stored in standard ACSF at room temperature before recording.
For whole cell recordings slices were transferred to a submerged recording chamber and perfused with standard ACSF containing 2.5 μm bicuculline and bubbled with 5% CO2 and 95% O2. 2-Amino-5-phosphonovaleric acid (APV; 100 μm) was added in some experiments. Current-clamp experiments were performed at physiological temperatures (34–37.5°C). Voltage-clamp experiments were performed at room temperature and in modified standard ACSF containing 4 mm MgCl2 and 4 mm CaCl2.
Electrophysiology.
Patch pipettes (2–5 MΩ) for whole-cell voltage recordings were filled with intracellular solution containing (in mm) 135 K-gluconate, 10 HEPES, 10 Na-phosphocreatine, 4 KCl, 4 ATP-Mg, 0.3 GTP, and 1 mg/ml biocytin (pH 7.2 with KOH, osmolarity 290–300 mOsm) for current-clamp experiments. In some of these experiments 5 mm K-gluconate was substituted with 5 mm BAPTA. Intracellular solution for voltage-clamp experiments contained (in mm) 120 Cs-gluconate, 10 CsCl, 10 HEPES, 8 NaCl, 0.2 EGTA, 2 MgATP, 0.3 Na3GTP, and 10 phosphocreatine (pH 7.3 with CsOH, osmolarity 290–300 mOsm).
In some experiments 100 μm phalloidin in 0.1% DMSO or 1 μm botulinum neurotoxin light chain serotype B (BoNT/B) were added to the intracellular solution. For the respective control experiments, 0.1% DMSO or BoNT/B, heated to 90°C for 45 min to heat-inactivate enzymatic activity, were added to the intracellular solution. To allow for diffusional equilibration of the pharmacological agents, recording of baseline synaptic transmission started 60 min after establishing the whole-cell recording. All reagents were purchased from Sigma-Aldrich. BoNT/B was a kind gift from Dr. Thomas Binz (Medizinische Hochschule Hannover, Hannover, Germany).
Pyramidal cells in the CA1 region of the hippocampus and in layer 2/3 of barrel cortex were identified by infrared differential interference contrast video microscopy. Whole-cell voltage recordings in current-clamp mode were made with Axopatch-200A, Axopatch-200B, or Axoclamp 2B (Molecular Devices) amplifiers, voltage-clamp experiments with Axopatch-200A only. Signals were filtered at 10 kHz and digitized at 10 kHz by an analog-to-digital converter (ITC 16, Instrutech). Some slices were stained for biocytin after the experiment. To evoke synaptic potentials and currents from two independent pathways, two low-resistance (<2 MΩ) glass electrodes filled with standard ACSF were placed at a distance of ∼100 μm from the soma. Excitatory synaptic potentials were evoked by extracellular field stimulation (0.3–16 μA; 0.1 ms) at 0.125 Hz with a delay of 4 s between control and paired pathway. Series and input resistances were continuously monitored during the recording in response to a small, hyperpolarizing current or voltage step (−25 pA or −10 mV; 250 ms). In voltage-clamp experiments cells were clamped at −70 mV during EPSC recording and series resistance was compensated 50–80%.
LTP experiments.
Extracellular stimulation was adjusted until a stable baseline of EPSPs (0.8–3 mV) or EPSCs (100–200 pA) and low variability could be recorded for 10 min (75 sweeps). The peak EPSP amplitude of the synaptic response was calculated as the difference between maximum voltage in a time window of 15 ms after extracellular stimulation and the average voltage during the 10 ms preceding extracellular stimulation. The peak EPSC amplitude was calculated accordingly. In paired-pulse experiments the reference time window was reduced from 10 to 4 ms for the second EPSP. EPSP and EPSC amplitudes were normalized to the average amplitude before LTP induction.
Two pairing protocols, theta-burst pairing (TBP) and EPSP-3AP pairing, were used for LTP induction in current-clamp mode. TBP consisted of pairing five EPSPs with five postsynaptic APs (2 ms; 0.7–4 nA postsynaptic current injection) at 100 Hz with burst delay of 14 ms. Five of these paired bursts were given at theta frequency (5 Hz) to constitute a theta train. Three trains (0.1 Hz) were given in total. For EPSP-3AP pairing, 1 EPSP was paired with 3 postsynaptic APs at 50 Hz (3 ms; 0.7–3 nA postsynaptic current injection) with an EPSP-AP delay of 10 ms. This stimulation pattern was repeated 60 times at 0.125 Hz. A third pairing protocol, depolarization pairing, was used for LTP induction in voltage-clamp experiments. For depolarization pairing 180 paired pathway EPSCs were evoked at 1 Hz and were paired with continuous postsynaptic depolarization at 0 mV. After LTP induction, EPSP or EPSC sampling was continued for up to 60 min. Experiments were excluded if recording times following presynaptic and postsynaptic pairing were shorter than 30 min, if the control pathway changed by >50% or if a significant change in input resistance occurred. Cells, which depolarized constantly above −60 mV during the experiment, were excluded from data analysis.
The coefficient of variation (CV) of EPSP amplitudes was evaluated for 10 min before LTP and 20–40 min after the induction of LTP. The inverse of the square CV was calculated by 1/CV2 = m2/σ2, with m being the average EPSP amplitude and σ the standard deviation (Faber and Korn, 1991).
Values are expressed as mean ± SEM. Two-tailed Student's t tests were used for the calculation of statistical significance of differences.
Results
The activity pattern determines the expression of GluRA-dependent potentiation in hippocampus
Tetanus-induced LTP is lacking at CA3-to-CA1 synapses of hippocampal CA1 pyramidal neurons of adult Gria1−/− mice (Zamanillo et al., 1999). However, LTP can be induced in Gria1−/− mice by a theta-burst pairing protocol (Hoffman et al., 2002). We tested whether the correlated activity of a single presynaptic spike in a Schaffer collateral commissural axon paired with backpropagating action potentials (APs) in the CA1 pyramidal neuron could trigger the expression of LTP in wild-type and Gria1−/− mice. The pairing protocol, reminiscent of STDP (Markram et al., 1997; Bi and Poo, 1998), consisted of pairing an EPSP evoked by extracellular stimulation with three APs evoked by somatic current injection at 50 Hz with a time-interval between the onset of the EPSP and the onset of the first AP of Δt = 10 ms (Fig. 1A) (Nevian and Sakmann, 2006). Pairing was repeated 60 times at low repetition frequency (0.125 Hz). These patterns of activity are likely to occur during the modification of place fields where directionally selective place cells are activated in a temporal sequence (Mehta et al., 2000). Thus, STDP has been suggested to be a plausible mechanism for the modification of synaptic strength at the CA3-to-CA1 synapse in hippocampus in vivo (Dan and Poo, 2006). The burst-timing equivalent of STDP results in a more reliable expression of LTP than single presynaptic and postsynaptic spike pairings due to the increased dendritic depolarization by the additional postsynaptic spikes (Pike et al., 1999; Lisman and Spruston, 2005; Nevian and Sakmann, 2006). EPSP-3AP pairing resulted in the expression of LTP in wild-type and Gria1−/− mice (Fig. 2A). The EPSP amplitudes were significantly potentiated 20–40 min after pairing by a factor of 1.42 ± 0.11 (p < 0.001, n = 25) in wild-type and by 1.46 ± 0.11 (p < 0.01, n = 14) in Gria1−/− mice. The EPSP amplitudes gradually increased after the end of the pairing from values similar to baseline EPSP amplitudes (wild-type, 1.15 ± 0.07, p > 0.1 and Gria1−/−, 1.13 ± 0.08, p > 0.05) to the potentiated state with an identical time course in both cases (Fig. 2A). The difference plot between the normalized and averaged EPSP amplitudes of Gria1−/− and wild-type mice reflected the similarity of the time courses by showing no deviation from the zero-line.

Synaptic potentiation induced by different presynaptic and postsynaptic activity patterns. A, Bright-field image of a biocytin-stained CA1 pyramidal neuron in adult mouse hippocampus. The stimulation electrode in stratum radiatum indicates the recording configuration for hippocampal LTP experiments. EPSP-3AP pairing is depicted schematically to the right and the bottom. This spike-timing-dependent plasticity (STDP) protocol consisted of pairing a synaptically evoked EPSP (synaptic stim) followed with a time delay of Δt = 10 ms by 3 APs (50 Hz) evoked by somatic current injection (somatic I-inj). Pairing was repeated 60 times at 0.125 Hz. B, EPSP amplitudes over time for the induction of LTP with the EPSP-3AP pairing protocol depicted in A. EPSPs (traces on bottom) were averaged during 10 min baseline (gray trace) and 0–2 min (light gray) and 20–40 min (dark gray) postpairing recording as indicated by the corresponding bars. EPSP amplitudes showed a slow increase after STDP pairing to a potentiated state. C, The theta-burst pairing protocol consisted of five synaptically evoked EPSPs at 100 Hz paired with five APs at 100 Hz evoked by somatic current injection. This pattern was repeated five times at theta frequency. This sequence was delivered three times at 0.1 Hz. D, EPSP amplitudes over time for the induction of LTP with the theta-burst pairing protocol depicted in C. EPSPs (traces on bottom) were averaged during 10 min baseline (gray trace) and 0–2 min (light gray) and 20–40 min (dark gray) postpairing recording as indicated by the corresponding bars. EPSP amplitudes showed a rapid increase after theta-burst pairing, which decayed to a potentiated state during the remainder of the experiment.

Spike-timing-dependent plasticity in hippocampal CA1 pyramidal neurons is independent of GluA1. A, Normalized and pooled EPSP amplitudes for EPSP-3AP pairing in wild-type (open circles, n = 25) and Gria1−/− (filled circles, n = 14) mice. The difference between wild-type and Gria1−/− mice (solid line) showed no difference in the expression of LTP. B, Normalized and pooled EPSP amplitudes for theta-burst pairing in wild-type mice (open squares, n = 70) and Gria1−/− mice (filled squares, n = 43). The difference between wild-type and Gria1−/− mice (solid line) revealed a GluA1-dependent component of potentiation for the first 10 min after pairing. C, Change in EPSP amplitude normalized to baseline for a 2 min average directly after and a 20 min average 20 min following theta-burst (squares) or EPSP-3AP (circles) pairing protocols in wild-type (open symbols) or Gria1−/− (filled symbols) mice. Only the theta-burst stimulation showed a significant difference between wild-type and Gria1−/− mice, whereas the STDP protocol resulted in identical development of LTP in both mouse lines. All protocols resulted in similar potentiation 20–40 min after pairing.
In contrast, induction of LTP by theta-burst pairing revealed a difference in the time course of expression of LTP between wild-type and Gria1−/− mice (Fig. 2B), as shown before (Hoffman et al., 2002). Theta-burst pairing consisted of pairing a burst of five synaptically evoked EPSPs at 100 Hz with a coincident burst of five APs at 100 Hz repetitively at theta frequency (five times at 5 Hz) (Fig. 1C). Theta-band activity is prevalent in hippocampus during exploratory investigation of a novel environment (Csicsvari et al., 1999) and implicated in the formation of long-term memory traces (Jensen and Lisman, 2005). The initial phase after theta-burst pairing depended on the availability of AMPA receptors containing the GluA1 subunit. In wild-type mice the EPSP amplitude was potentiated during the first 2 min after theta-burst pairing by a factor of 1.80 ± 0.09 (p < 0.0001, n = 70), whereas potentiation in Gria1−/− mice was only 1.39 ± 0.06 (p < 0.0001, n = 43). At 20–40 min after pairing, wild-type and Gria1−/− mice showed the same amount of LTP (wild-type, 1.34 ± 0.06, p < 0.0001 and Gria1−/−, 1.33 ± 0.08, p < 0.001). Whereas the initial increases in EPSP amplitude were significantly different between wild-type and Gria1−/− mice (p < 0.001, unpaired t test), the final potentiated EPSP amplitudes were indistinguishable (p = 0.9, unpaired t test). The difference plot between the normalized and averaged EPSP amplitudes of Gria1−/− and wild-type mice after theta-burst pairing showed that the GluA1 subunit was transiently required for increases in EPSP amplitudes for the first 10 min after pairing, consistent with previous results (Hoffman et al., 2002).
Theta-burst and EPSP-3AP pairing required Ca2+ influx through NMDA receptors for the induction of LTP. Bath application of the NMDA receptor antagonist APV (100 μm) blocked the expression of LTP in CA1 pyramidal neurons (theta-burst, 0.92 ± 0.11, p > 0.6, n = 12; EPSP-3APs, 0.89 ± 0.07, p > 0.05, n = 15). Postsynaptic loading of the cells with the Ca2+ chelator BAPTA (5 mm) also abolished the increase in EPSP amplitude (theta-burst, 0.97 ± 0.13, p > 0.8, n = 12; EPSP-3APs, 0.96 ± 0.06, p > 0.4, n = 9).
We conclude that the GluA1-dependent form of increases in EPSP amplitude are transient and depend on the presynaptic and postsynaptic activity patterns during the induction phase. The final magnitude of potentiation is independent of the pairing protocol and independent of the presence of GluA1 subunit-containing AMPA receptors. Our results show that an STDP pairing protocol (EPSP-3AP) results in significant LTP in hippocampal CA1 pyramidal neurons in Gria1−/− mice.
Neocortical LTP is independent of GluA1
We tested the same pairing protocols as used in hippocampus in layer 2/3 pyramidal neurons of primary somatosensory cortex in adult wild-type and Gria1−/− mice (Fig. 3). Pairing an EPSP with 3 APs (50 Hz) at Δt = 10 ms resulted in significant potentiation of the EPSP amplitude 20–40 min after pairing in wild-type (1.39 ± 0.13, p < 0.01, n = 22) and Gria1−/− (1.39 ± 0.14, p < 0.05, n = 15) mice. The time course of the increase in EPSP amplitude in both cases was similar as reflected in the difference plot between the normalized and averaged EPSP amplitudes of Gria1−/− and wild-type mice (Fig. 3A).

Cortical LTP does not depend on GluA1. A, Normalized and pooled EPSP amplitudes for EPSP-3AP pairing in wild-type (open circles, n = 22) and Gria1−/− (filled circles, n = 15) mice. The difference between wild-type and Gria1−/− mice (solid line) showed no difference in the expression of LTP. B, Normalized and pooled EPSP amplitudes for theta-burst pairing in wild-type (open squares, n = 21) and Gria1−/− (filled squares, n = 6) mice. No difference (solid black line) in the expression of LTP between wild-type and Gria1−/− mice was revealed. The difference plot for the expression of LTP after theta-burst pairing in wild-type cortical L2/3 pyramidal neurons and hippocampal CA1 pyramidal neurons of Gria1−/− mice showed no deviation from zero (solid gray line). C, Change in EPSP amplitude normalized to baseline for a 2 min average directly after and a 20 min average 20 min following theta-burst (squares) or EPSP-3AP (circles) pairing protocols in wild-type (open) or Gria1−/− (filled symbols) mice. No differences between wild-type and Gria1−/− mice were found for the different pairing protocols.
Theta-burst pairing resulted in a rapid potentiation of EPSP amplitudes in wild-type (1.36 ± 0.08, p < 0.01, n = 21) and Gria1−/− (1.42 ± 0.09, p < 0.01, n = 6) mice directly after pairing. This initial increase in EPSP amplitude relaxed to a potentiated state 20–40 min after pairing in both cases (wild-type, 1.24 ± 0.08, p < 0.05; Gria1−/−, 1.22 ± 0.08, p < 0.05). The time course of the changes in EPSP amplitude in wild-type and Gria1−/− mice were again similar (Fig. 3B). Strikingly, the averaged time course of changes in EPSP amplitude after theta-burst pairing in cortical L2/3 pyramidal neurons in wild-type mice and theta-burst pairing in hippocampal CA1 pyramidal neurons in Gria1−/− mice were identical (Fig. 3B). These results suggest that cortical L2/3 pyramidal neurons are similar to CA1 pyramidal neurons in Gria1−/− mice in terms of expression of LTP.
As in hippocampus, theta-burst and EPSP-3AP pairing required Ca2+ influx through NMDA receptors for the induction of LTP in cortical L2/3 pyramidal neurons. Bath application of the NMDA receptor antagonist APV (100 μm) blocked the expression of LTP for theta-burst pairing (1.08 ± 0.08, p > 0.1, n = 15) and even resulted in a reduction in EPSP amplitude for EPSP-3AP pairing (0.76 ± 0.08, p < 0.05, n = 8). Postsynaptic loading of the cells with the Ca2+ chelator BAPTA (5 mm) abolished the increase in EPSP amplitude (theta-burst, 0.93 ± 0.07, p > 0.3, n = 16; EPSP-3APs, 1.04 ± 0.17, p > 0.9, n = 9).
These experiments suggest that L2/3 pyramidal neurons in primary somatosensory cortex do not require the presence of the GluA1 subunit for the expression of LTP.
A transient decrease in paired-pulse ratio is accompanied by theta-burst pairing in the hippocampus independent of GluA1
To investigate the initial GluA1-dependent phase of synaptic potentiation further we first determined the paired-pulse ratio (PPR) of EPSPs after induction of LTP with theta-burst pairing in hippocampal CA1 pyramidal neurons in wild-type and Gria1−/− mice. A change in PPR is thought to indicate a presynaptic contribution to the expression of LTP. The PPR for two EPSPs evoked at 20 Hz was measured before and after the induction of LTP with theta-burst pairing in wild-type and Gria1−/− mice (Fig. 4). PPRs before pairing were similar (p > 0.2, unpaired t test) in wild-type (PPR = 1.38 ± 0.03, n = 26) and Gria1−/− (PPR = 1.35 ± 0.04, n = 21) mice. For the first 2 min after theta-burst pairing the first and second EPSP amplitudes were significantly increased in both cases (wild-type, ΔEPSP1 = 1.71 ± 0.14, p < 0.001, ΔEPSP2 = 1.51 ± 0.10, p < 0.001; Gria1−/−, ΔEPSP1 = 1.33 ± 0.07, p < 0.001, ΔEPSP2 = 1.22 ± 0.05, p < 0.001). Additionally, the PPR was significantly reduced in wild-type (PPR = 1.24 ± 0.04, p < 0.001) and Gria1−/− (PPR = 1.26 ± 0.04, p < 0.05) mice. PPRs recovered to the initial prepairing values (wild-type, 1.41 ± 0.04, p > 0.5; Gria1−/−, 1.33 ± 0.03, p > 0.6). Thus, PPRs for wild-type and Gria1−/− mice were similar for the time course of the experiment (Fig. 4D). The control pathway in wild-type mice had initially a similar PPR to the paired pathway (PPR = 1.37 ± 0.04, p > 0.6), which was constant throughout the recording (20–40 min, PPR = 1.44 ± 0.06, p > 0.1) and did not show the decrease in PPR directly after pairing (PPR = 1.37 ± 0.08), indicating that the transient decrease in PPR was pathway specific (Fig. 4C). The Gria1−/− mice lacked the large initial increase in EPSP amplitude after theta-burst pairing, as described above for the first and second EPSP when compared with wild-type mice. Nevertheless PPRs decreased similarly in both cases, suggesting that these changes in PPR were not related to the expression of LTP. We confirmed that the decrease in PPR after theta-burst stimulation was unrelated to the expression of LTP by application of a dissociated theta-burst protocol, in which the evoked EPSPs were not coincident with the postsynaptic APs. This protocol resulted in a slight, but not significant increase in EPSP amplitude directly after dissociated pairing (ΔEPSP1 = 1.22 ± 0.09, p > 0.05, ΔEPSP2 = 1.17 ± 0.07, p > 0.05, n = 5). Nevertheless this protocol resulted in a significant decrease in the PPR directly after dissociated pairing (baseline PPR = 1.31 ± 0.12, 0–2 min PPR = 1.20 ± 0.10, p < 0.05), which increased to baseline values 20–40 min after pairing (PPR = 1.37 ± 0.12, p > 0.1), similar to the time course in the paired pathways in wild-type and Gria1−/− mice (Fig. 3C).

Paired-pulse facilitation is transiently reduced in CA1 pyramidal neurons after theta-burst pairing. A, Normalized and pooled EPSP amplitudes for the first EPSP (upper graph) and second EPSP (middle graph) in paired-pulse stimulation experiments (inter-EPSP interval 50 ms) for theta-burst pairing in wild-type mice and the resulting paired-pulse ratio (PPR, lower graph, n = 26). Example voltage recordings of paired-pulse EPSPs (20 Hz) before (bottom gray trace) and 20–40 min after (bottom black trace) LTP induction show the increase in EPSP amplitudes. The bottom right traces show the same traces normalized to the first EPSP illustrating that the PPR has not changed. B, Normalized and pooled EPSP amplitudes for the first EPSP (upper graph) and second EPSP (middle graph) in paired-pulse stimulation experiments for theta-burst pairing in Gria1−/− mice and the resulting PPR (lower graph, n = 21). Example voltage recordings of paired-pulse EPSPs (20 Hz) before (bottom gray trace) and 20–40 min after (bottom black trace) LTP induction show the increase in EPSP amplitudes. Normalization to the first EPSP illustrates that the PPR has not changed. C, PPR during baseline recording, a 2 min average directly after theta-burst pairing, and a 20 min average 20 min after theta-burst pairing. Wild-type (open squares) and Gria1−/− (filled squares) mice and a dissociated theta-burst pairing protocol in wild-type mice (black squares, n = 5) showed similar changes in PPR. No change was observed in the unpaired control pathway of wild-type mice (open diamonds). D, No difference in the PPR between wild-type and Gria1−/− mice was found.
We conclude that the transient decrease in PPR, which corresponds to an increase in release probability after pairing, is a short-term effect that can be related to the high-frequency stimulation of the presynaptic axons, regardless of associative pairing.
Locus of LTP expression is not different in wild-type and Gria1−/− mice
The previous experiments suggested that the theta-burst-induced expression of LTP 20–40 min after the induction is postsynaptic. This is consistent with some studies (Margrie et al., 1998; Poncer and Malinow, 2001) while others indicate a mixed presynaptic and postsynaptic (Sjöström et al., 2007) or purely presynaptic locus of LTP expression (Stanton et al., 2005). In Gria1−/− mice it was suggested that the expression of LTP is presynaptic in contrast to a mixed presynaptic and postsynaptic expression in wild-type mice (Hardingham and Fox, 2006). Analysis of the coefficient of variation (CV) of EPSP amplitudes before and after induction of LTP showed linear increases of the normalized CV−2 with the normalized changes in EPSP amplitude along the line of unity for all experimental conditions (Fig. 5). These results are consistent with a mixed locus of LTP expression. Nevertheless, given the nonminimal stimulation conditions in our experiments, the same results could also be obtained by purely postsynaptic modifications (Faber and Korn, 1991). No significant differences in CV−2 were found between Gria1−/− and wild-type mice for all induction protocols tested (p > 0.05) suggesting that the locus of synaptic modification after LTP induction in both CA1 and L2/3 pyramidal neurons was the same, independent of the pairing protocol and independent of the presence of the GluA1 subunit.

CV analysis of LTP. To estimate the locus for the change in synaptic efficacy in wild-type and Gria1−/− mice, the normalized 1/CV2 is plotted against the normalized change in EPSP amplitude. The dashed line indicates unity and might suggest a mixed presynaptic and postsynaptic locus for LTP expression. A, CV analysis for CA1 pyramidal neurons for LTP induction by EPSP-3AP pairing (left) and theta-burst pairing (right). B, CV analysis for layer 2/3 pyramidal neurons for LTP induction by EPSP-3AP pairing (left) and theta-burst pairing (right). Open symbols indicate wild-type and solid symbols indicate Gria1−/− mice. No differences between the two groups were found in any case.
GluA1-dependent LTP requires postsynaptic membrane fusion mediated by SNARE proteins
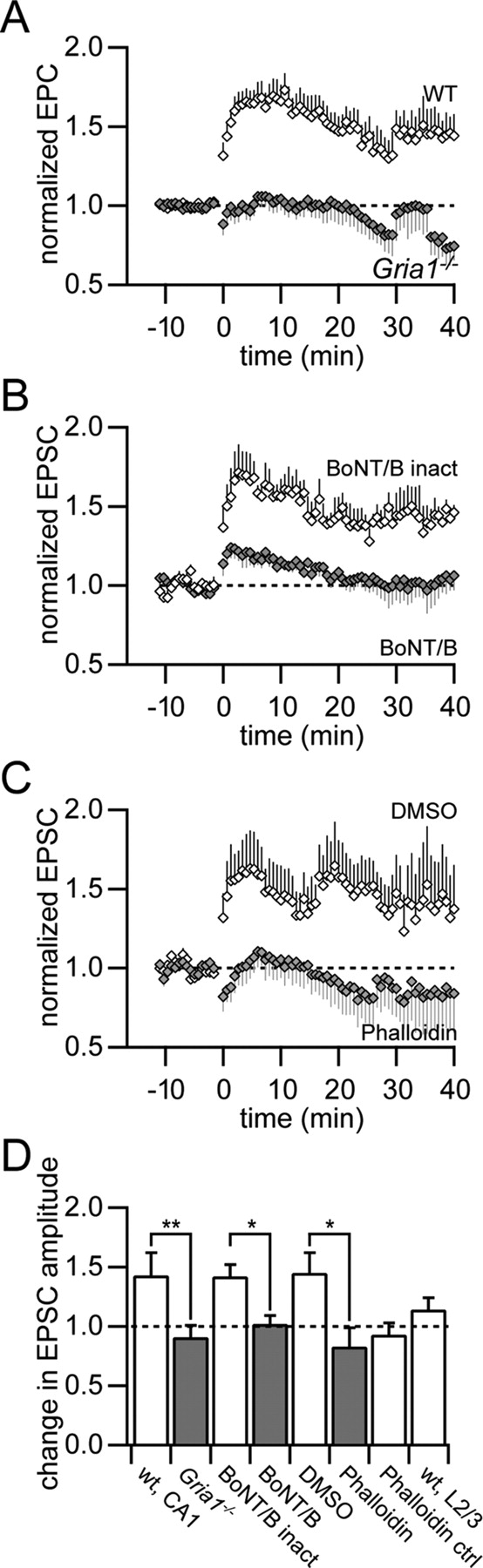
The postsynaptic component of the expression of LTP might reflect the insertion of new AMPA receptors into the postsynaptic membrane (Shi et al., 1999). To elucidate the mechanism of the GluA1-dependent increase in synaptic efficacy in CA1 pyramidal neurons we induced LTP in the voltage-clamp configuration by pairing postsynaptic depolarization to 0 mV with low-frequency synaptic stimulation (1 Hz) for 3 min (Fig. 6). This pairing protocol results in LTP, which is solely mediated by GluA1-containing AMPA receptors (Jensen et al., 2003). As a control we reconfirmed the GluA1 dependence of the depolarization pairing protocol. Wild-type mice showed a strong potentiation in EPSC amplitude 20–40 min after pairing (1.38 ± 0.11, p < 0.01, n = 9). In contrast, Gria1−/− mice showed no increase in EPSC amplitude (0.91 ± 0.10, p > 0.2, n = 9), which was significantly different from wild-type mice (p < 0.01, unpaired t test) (Fig. 6D).
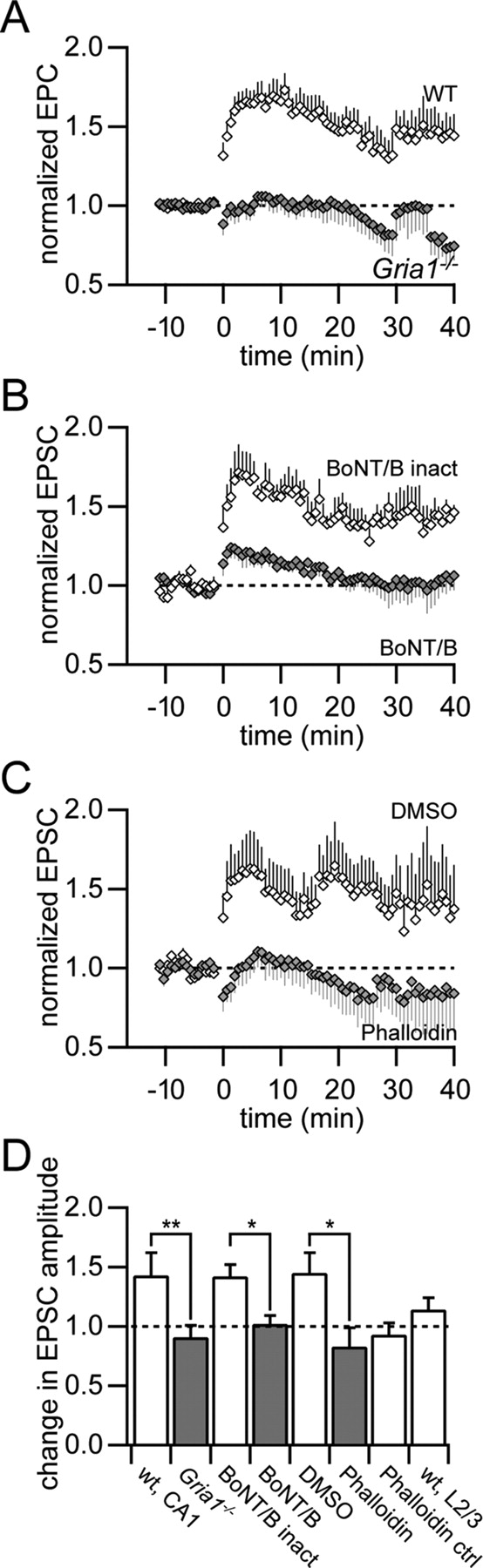
Depolarization pairing-induced LTP depends on the presence of the GluA1 subunit, exocytosis, and spine stabilization. A, Normalized EPSC amplitudes for a low-frequency synaptic stimulation coincident with postsynaptic depolarization pairing protocol in wild-type (open diamonds, n = 9) and Gria1−/− (filled diamonds, n = 9) mice in voltage clamp. B, Intracellular loading of the light chain of botulinum toxin serotype B (BoNT/B), a blocker of exocytosis, via the patch pipette abolished the expression of depolarization pairing-induced LTP (filled diamonds, n = 6) compared with the heat-inactivated form of BoNT/B as control (open diamonds, n = 4). C, Application of the actin filament stabilizer phalloidin (filled diamonds, n = 5) blocked the expression of LTP compared with the DMSO control (open diamonds, n = 5). D, Summary of changes in EPSC size 20–40 min after pairing synaptic stimulation with postsynaptic depolarization. The expression of LTP was significantly reduced in Gria1−/− mice (**p < 0.01), in the presence of BoNT/B and phalloidin (*p < 0.05). The unpaired pathway in the presence of phalloidin (Phalloidin ctrl) showed a similar reduction in EPSC amplitude as the paired pathway. Depolarization pairing did not result in significant LTP in cortical L2/3 pyramidal neurons (wt, L2/3).
Intracellular application of the exocytosis blocker botulinum toxin serotype B light chain (BoNT/B) blocked the expression of LTP by depolarization pairing (1.02 ± 0.07, p > 0.7, n = 6), whereas the heat-inactivated form of BoNT/B had no effect on LTP (1.42 ± 0.10, p < 0.05, n = 4). These experiments suggested that GluA1-dependent LTP required membrane fusion mediated by SNARE protein complexes.
The dynamic insertion of GluA1-containing AMPA receptors might involve the remodeling of the structure of the postsynaptic spine apparatus (Kim and Lisman, 1999). Indeed, stabilization of actin filaments by intracellular perfusion of phalloidin (dissolved in DMSO) resulted in a significant block of LTP by depolarization pairing compared with control (DMSO-only, p < 0.05, n = 5, unpaired t test) (Fig. 6C). The unpaired control pathway showed a slight decrease in EPSC amplitude in the presence of phalloidin similar to the paired pathway in this case (Fig. 6D).
Only slight potentiation of EPSC amplitude was found in cortical L2/3 pyramidal neurons with the depolarization pairing protocol (1.14 ± 0.10, p > 0.1, n = 5), in contrast to hippocampal CA1 pyramidal neurons (Fig. 6D). This is consistent with our finding that the expression of LTP in cortical pyramidal neurons is independent of the GluA1 subunit.
Spike-timing-dependent LTP requires postsynaptic exocytotic membrane fusion
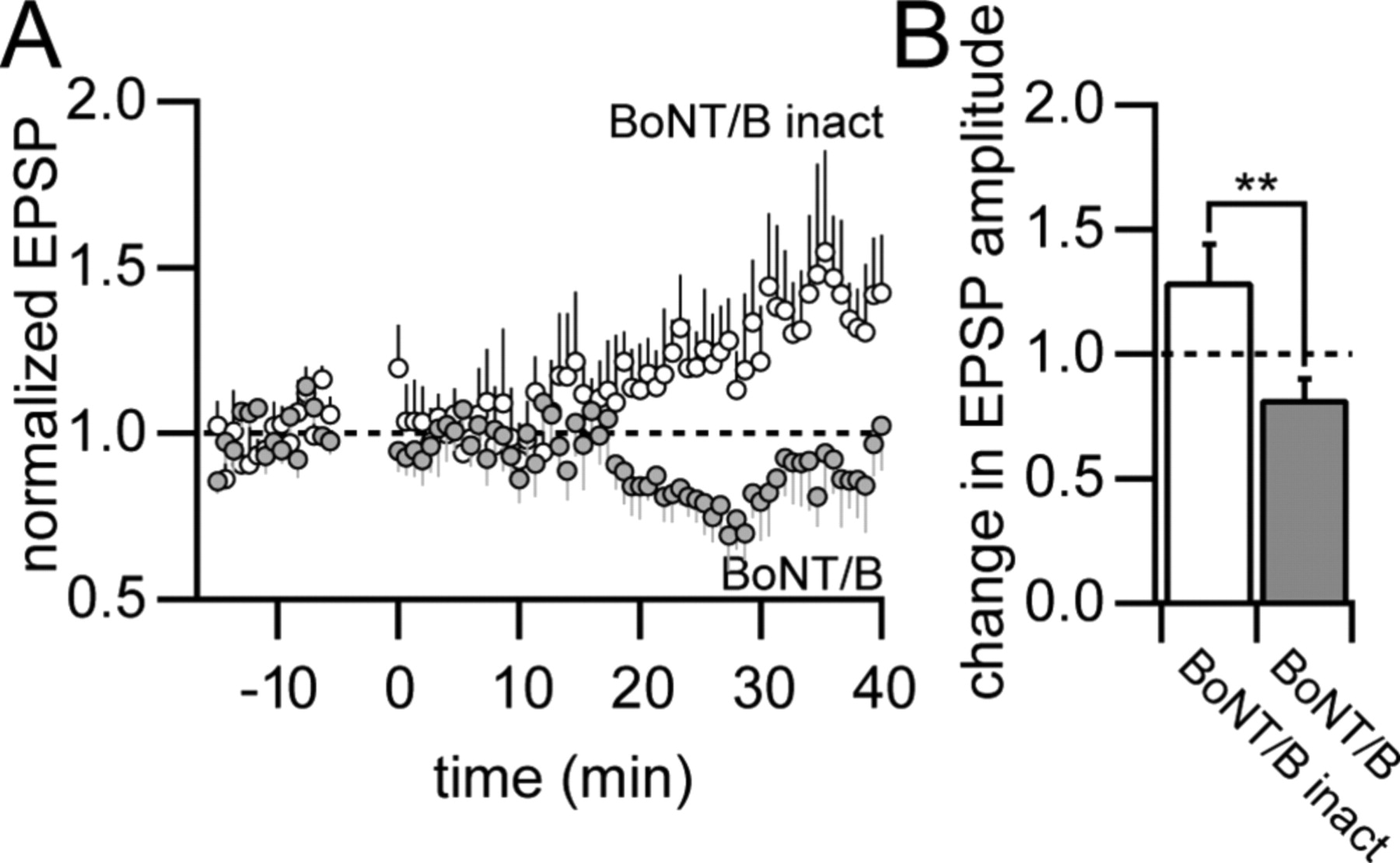
The activity-dependent insertion of AMPA receptors into the postsynaptic membrane that results in potentiation is thought to depend critically on the presence of the GluA1 subunit (Shi et al., 2001). Our results, however, suggest that the GluA1 subunit is not required for the expression of spike-timing-dependent LTP. What could be the mechanism for the expression of GluA1-independent LTP in this case? We tested whether this form of LTP required exocytotic membrane fusion (Fig. 7). Pairing an EPSP with 3 APs at Δt = +10 ms in hippocampal CA1 pyramidal neurons in the presence of intracellularly applied BoNT/B resulted in a slight decrease in EPSP amplitude (0.82 ± 0.08, p < 0.05, n = 9). This block of LTP was significant compared with the results with the heat-inactivated form of BoNT/B (p < 0.01, unpaired t test). Thus, these results suggest that spike-timing-dependent LTP, which is independent of the GluA1 subunit, nevertheless requires exocytosis presumably of vesicles containing AMPA receptors.

Potentiation by STDP requires membrane fusion. A, Normalized EPSP amplitudes for EPSP-3AP pairing in CA1 pyramidal neurons during intracellular application of BoNT/B (filled circles, n = 9) and its heat-inactivated form (open circles, n = 6). B, Summary data of average EPSP amplitudes 20–40 min after the induction of LTP. Potentiation was significantly reduced by BoNT/B compared with the heat-inactivated form of BoNT/B (**p < 0.01).
Discussion
We showed that the GluA1 subunit is not necessarily required for the expression of LTP in hippocampus. Furthermore, LTP is independent of GluA1 in the somatosensory cortex of adult mice. Pairing EPSPs with postsynaptic APs, a protocol reminiscent of STDP, resulted in pronounced LTP in hippocampus and cortex even in the Gria1−/− mice. STDP has been implicated in the refinement and formation of memory traces in the hippocampus (Bi and Poo, 1998; Bi and Poo, 2001; Dan and Poo, 2004). The requirement of repeated pairing for this form of plasticity might be a correlate to the acquisition of spatial reference memory. A mouse trained in the water-maze spatial reference memory task can integrate sensory stimuli over an extended period of time (Morris et al., 1990; Morris, 2006). Thus the occurrence of repeated coincident activity during this time period can strengthen synapses via an STDP mechanism (Lisman and Spruston, 2005). Performance in the water maze depends on the activation of NMDA receptors in the hippocampus (Davis et al., 1992). Consistently, blocking NMDA receptors by APV abolished the expression of STDP in hippocampus. Therefore we suggest that STDP is a potential synaptic mechanism that can explain the acquisition of spatial reference memory even in the Gria1−/− mouse (Reisel et al., 2002, 2005).
In contrast, theta-burst pairing requires increased synchronous and cooperative activity of high-frequency presynaptic and postsynaptic bursts for a brief pairing epoch. This pattern of activity is frequently observed in hippocampus during exploration of the environment and has been indicated to code for the position in a place field (Csicsvari et al., 1999; Buzsáki, 2002; Buzsáki et al., 2002). This presumably “stronger” pairing protocol compared with STDP is most effective if local dendritic spikes are generated (Hoffman et al., 2002). Theta-burst pairing results in a strong and rapid potentiation of synaptic transmission at the end of the pairing period, which is dependent on the GluA1 subunit. Therefore it was suggested that this initial increase in synaptic strength correlates with the observed spatial working memory deficit in the Gria1−/− mouse (Reisel et al., 2002; Bannerman et al., 2003; Schmitt et al., 2005).
Our results support this view and they additionally suggest that synaptic modifications by STDP are not required for the formation of spatial working memory since this form of plasticity was not impaired in the Gria1−/− mouse. In hippocampus, both theta-burst and spike-timing protocols resulted in the same magnitude of LTP after 40 min, suggesting that the final change in synaptic strength is mediated by the same molecular mechanisms. The initial phase of potentiation is only activated by the appropriate stimulation pattern; that is strong stimulation by theta-burst activity. Thus, our results might be able to explain the conundrum why the working memory deficit in the Gria1−/− mouse has no consequence for the acquisition of spatial reference memory (Sanderson et al., 2008). Working memory might require the presence of burst activity to code for the immediate task that triggers independent signaling cascades for the rapid modification of synaptic strength, whereas a reference memory is formed by repeated coincident activity that employs a different biochemical signaling machinery. Eventually both pathways converge so that theta-burst pairing might also have a contribution to the formation of spatial reference memory. These results are consistent with the apparently normal development of neuronal circuitry in Gria1−/− mice (Mack et al., 2001; Schmitt et al., 2005). STDP is a plausible model for experience-driven refinement of neural circuits and could therefore serve as one mechanism for the establishment of appropriate synaptic weight distributions during development. Furthermore, a juvenile form of LTP, which is independent of the GluA1 subunit is presumably also involved in strengthening of synapses during development (Jensen et al., 2003). The here-presented view that certain patterns of correlated activity that result in LTP in vitro have a functional link to memory systems in vivo, namely spatial working or spatial reference memory, might be an overinterpretation. The stereotyped in vitro pairing protocols might not occur in isolation in vivo, where more complex patterns of activity are presumably prevalent. Therefore, paired in vivo recordings of the cellular electrical activity patterns in hippocampus during spatial working memory tasks will be required to corroborate these suggestions.
Our results indicate that the strength and pattern of stimulation differentially recruits GluA1-containing AMPA receptors for the expression of LTP in hippocampal CA1 pyramidal neurons. First, depolarization pairing results in LTP which is completely dependent on the GluA1 subunit. Second, theta-burst pairing results in an initial strong, GluA1-dependent but transient increase in synaptic efficacy which is accompanied by overlapping, additional GluA1-independent potentiation. Thirdly, STDP is GluA1 subunit independent. What could be the potential mechanisms for this different recruitment? Ca2+ influx through the NMDA receptor is required for the induction of LTP by all of the pairing protocols (Hoffman et al., 2002; Jensen et al., 2003; Nevian and Sakmann, 2006). Nevertheless, the magnitude, duration, and spatial extent of the Ca2+ transients will be different, which can activate diverse downstream signaling cascades. Pairing an EPSP with backpropagating APs results in a supralinear increase in Ca2+ that is localized to the activated spine heads and lasts only on the order of 100 ms (Nevian and Sakmann, 2004, 2006). Our results show that this STDP protocol requires a postsynaptic exocytotic membrane fusion event for the expression of LTP. Recently, it was suggested that exocytotic vesicles containing AMPA receptors can be targeted directly into the postsynaptic density (Gerges et al., 2006). The local rise in spine Ca2+ could be the trigger for this event. Theta burst pairing results in a large Ca2+ influx in the activated spines and the adjacent dendritic shaft (Golding et al., 2002; Nevian and Helmchen, 2007). The intracellular Ca2+ presumably stays elevated for 1 s in each pairing epoch. Therefore Ca2+ sensors with different affinities might get activated, resulting in the rapid delivery of GluA1-containing AMPA receptors to the membrane of the postsynaptic cell (Shi et al., 1999; Malinow and Malenka, 2002). Expression of LTP by theta-burst pairing can be completely blocked by BoNT/B, suggesting membrane fusion as a critical step (Yang et al., 2008b, 2008a). The location for membrane fusion is thought to be extrasynaptic and might even be dendritic with subsequent diffusion and capture of the AMPA receptors at the postsynaptic density (Passafaro et al., 2001; Borgdorff and Choquet, 2002; Andrásfalvy et al., 2003; Lisman and Raghavachari, 2006; Oh et al., 2006). In addition, the previously described mechanism for GluA1-independent potentiation is also activated, explaining the potentiation by theta-burst pairing in the Gria1−/− mice. The expression of the GluA1-dependent component of potentiation is only transient, suggesting that GluA1-containing AMPA receptors are lost from the postsynaptic density. Finally, depolarization pairing results in a prolonged elevation of Ca2+ in the activated spines and in the adjacent dendritic shaft for 3 min (Conti and Lisman, 2002). This form of plasticity triggers exocytosis-dependent insertion of GluA1-containing AMPA receptors and actin-dependent remodeling of the postsynaptic membrane as demonstrated here and in previous studies (Kim and Lisman, 1999; Shi et al., 1999, 2001; Boehm et al., 2006; Kopec et al., 2007). These receptors are stabilized in the postsynaptic density for a much longer time than in theta-burst-induced potentiation. Furthermore, the exclusive dependence of this form of plasticity on the presence of GluA1-containing AMPA receptors suggests that the previously described mechanism triggering STDP at this synapse is either not activated or overridden by the prolonged elevated levels of Ca2+.
Overexpression of the GluA1 subunit by viral transfection in the barrel cortex of young rats has suggested that it is critical for the experience-dependent refinement of the somatosensory cortical circuitry (Takahashi et al., 2003). Our results show that LTP in somatosensory cortex of adult rats does not require the GluA1 subunit. Furthermore, depolarization pairing, which results in potentiation solely dependent on the GluA1 subunit fails to induce LTP in adult somatosensory cortex, but effectively triggers LTP in more juvenile animals (Feldman, 2000; Yoshimura et al., 2003). Together, these findings suggest that there is a developmental shift in the recruitment of the GluA1 subunit in synaptic plasticity in cortex. This is consistent with low levels of GluA1 expression in the adult cortex compared with hippocampus (Pellegrini-Giampietro et al., 1991; Martin et al., 1993, 1998; Kim et al., 2006) and reports that the rules for synaptic plasticity are age dependent (Feldman et al., 1998). Notably, the developmental switch in the utilization of the GluA1 subunit in cortical L2/3 pyramidal neurons seems to be opposite to that of CA1 pyramidal neurons in hippocampus (Jensen et al., 2003). Nevertheless, if GluA1 subunits are overexpressed in cortical neurons by viral transfection they get recruited and inserted into synapses (Takahashi et al., 2003).
We conclude that the presynaptic and postsynaptic activity patterns during the induction of LTP differentially recruit GluA1-containing AMPA receptors. This recruitment is development and brain region specific suggesting diverse and specific biochemical machineries for different kinds of spatial memory.
Footnotes
-
This study was supported by the Max Planck Society, the German Research Foundation SFB636-A4 (R.S.), and the Swiss National Science Foundation, Grant 3100A0-118395 (T.N.). We thank Bert Sakmann and Peter Seeburg for their advice and continuous support, Marlies Kaiser for excellent technical assistance, and Verena Bosch for providing the Gria1−/− and wild-type mice.
- Correspondence should be addressed to Thomas Nevian, Department of Physiology, University of Berne, Bühlplatz 5, CH-3012 Bern, Switzerland. nevian{at}pyl.unibe.ch





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}