

Abstract
Despite considerable research attention focused on mechanisms underlying neural spreading depression (SD), because of its association with important human CNS pathologies, such as stroke and migraine, little attention has been given to explaining its occurrence and regulation in invertebrates. In the locust metathoracic ganglion (MTG), an SD-like event occurs during heat and anoxia stress, which results in cessation of neuronal output for the duration of the applied stress. SD-like events were characterized by an abrupt rise in extracellular potassium ion concentration ([K+]o) from a baseline concentration of ∼8 to >30 mm, which returned to near baseline concentrations after removal of the applied stress. After return to baseline [K+]o, neuronal output (ventilatory motor pattern activity) from the MTG recovered. Unlike mammalian neurons, which depolarize almost completely during SD, locust neurons only partially depolarized. SD-like events in the locust CNS were suppressed by pharmacological inhibition of the nitric oxide/cyclic guanosine monophosphate/protein kinase G (NO/cGMP/PKG) pathway and were exacerbated by its activation. Also, environmental stressors such as heat and anoxia increased production of nitric oxide in the locust CNS. Finally, for the intact animal, manipulation of the pathway affected the speed of recovery from suffocation by immersion under water. We propose that SD-like events in locusts provide an adaptive mechanism for surviving extreme environmental conditions. The highly conserved nature of the NO/cGMP/PKG signaling pathway suggests that it may be involved in modulating SD in other organisms, including mammals.
Introduction
Neuronal hyperexcitability associated with elevated concentrations of extracellular potassium ions ([K+]o) is a hallmark of cortical spreading depression (CSD) in mammals (Somjen, 2001). This phenomenon is critically involved in stroke, head trauma, and migraine, and the discovery of cellular mechanisms capable of preventing or attenuating CSD has great potential health benefits. We have used a locust model of CNS function to investigate methods of manipulating SD-like events by monitoring the activity of the ventilatory central pattern generator (vCPG), which is vital for organismal health.
Under anoxic conditions, many insects enter a reversible state of coma (e.g., supplemental Video 1, available at www.jneurosci.org as supplemental material), during which electrical activity in the nervous and muscular systems is shut down. This is considered to be an adaptive strategy to conserve energy and prevent neuronal hyperexcitation (Walter and Nelson, 1975; Rodgers et al., 2007), and recovery is complete after return to normoxia, often after many hours (up to at least 6 h in locusts) (Wu et al., 2002). Anoxic conditions arise in the environment during flash floods and monsoons, and recovery occurs quickly (e.g., supplemental Video 2, available at www.jneurosci.org as supplemental material). Recently, we have shown that the arrest of CNS function during anoxia in locusts is accompanied by an abrupt increase in [K+]o in the neuropile (Rodgers et al., 2007). Similar increases in [K+]o are induced by hyperthermia and by disrupting mitochondrial function. Moreover, manipulation of ionic gradients, either by direct injection of high K+ saline into the extracellular space or by reducing Na+/K+ATPase activity with ouabain, generates waves of increased [K+]o that propagate throughout the neuropile and are associated with transient depression of motor pattern activity (Rodgers et al., 2007, 2009). These phenomena share all the major characteristics of CSD, although the extent of neuronal depolarization during the locust SD-like event is unknown.
A genetic component to hyperthermic neural depression has been demonstrated for the foraging gene in Drosophila (Dawson-Scully et al., 2007). The gene encodes for a protein kinase G (PKG), and the naturally occurring populations with different alleles of this gene (rover and sitter) differ in the levels of gene transcript and PKG (Osborne et al., 1997). Pharmacologically or genetically reducing levels of PKG, or its downstream targets, increases thermotolerance of the larval Drosophila neuromuscular junction. Similar treatments also increase the locust vCPG's tolerance to heat stress (Dawson-Scully et al., 2007), suggesting evolutionary conservation of the mechanism. Hyperthermic arrest of vCPG operation in the locust CNS is associated with SD-like increases of [K+]o that occur before irreversible cellular collapse (Rodgers et al., 2007). Hence, we reasoned that manipulation of the PKG pathway would modulate the occurrence and severity of locust SD-like events induced by direct manipulation of ion gradients, in the absence of heat. We tested this idea by measuring SD-like events induced by ouabain or by neuropile injections of K+ and by pharmacologically manipulating the NO/cGMP/PKG pathway. We monitored ventilatory motor activity in a semi-intact preparation, because of the excellent accessibility of this system during its normal operation and because the points of failure and recovery of neural function are unequivocal.
Materials and Methods
Locusts.
All experiments were performed on 3–4 week postimaginal moult male locusts (Locusta migratoria). Animals were collected from a crowded colony maintained in the Department of Biology at Queen's University. Locusts were reared under a 12 h light/dark cycle. Before experimentation, locusts were held in a ventilated 2 L plastic container. All electrophysiological experiments were performed 4–8 h after locusts were collected from the colony.
Semi-intact preparation.
Locusts were secured to a corkboard ventral side down and dissected to expose the ventral nerve cord. The metathoracic ganglion (MTG) was stabilized on a stainless steel plate, and nerves 5 on both sides of the ganglion were cut to improve access for saline and drugs. Standard locust saline (in mm: 147 NaCl, 10 KCl, 4 CaCl2, 3 NaOH, 10 HEPES buffer, pH 7.2) was used to superfuse the thoracic and abdominal cavities at 5 ml/min. Saline temperature was held at 22°C.
Water immersion.
Locusts were placed within a mesh-lidded clear plastic container which was then submerged in a larger bucket filled with water (water temperature, ∼19–21°C) for 30 min. Animals were completely submerged in water, and all were incapacitated by this treatment. After removal from the water, locusts were placed on their side on a piece of paper towel. The time to recover ventilation was calculated as the length of time between removal from the water to the start of ventilatory movements monitored by visual inspection. Motor pattern activity was assumed to have recovered at the time when repetitive movements could be observed. Time to stand was scored by measuring the time required to stand upright after removal from the water.
Motor patterns.
Ventilatory motor patterns were recorded using a 0.1 mm diameter copper electromyographic (EMG) electrode placed on muscle 161. Signals were amplified using a model P15 preamplifier (Grass Instruments) and digitized using a Digidata 1322A (Molecular Devices). Data were analyzed using Clampfit 9.0 (Molecular Devices). Motor patterns were allowed to stabilize for 10 min before beginning an experiment. After dissection, motor pattern frequency is ∼1 Hz with an expiratory muscle burst duration of 250–300 ms (Armstrong et al., 2006). vCPG recovery time was calculated by examining the time difference between the last rhythmic muscle burst resulting from the KCl-evoked SD event to the first rhythmic muscle burst after recovery of the [K+]o to baseline levels. A rhythmic burst is a muscle burst having a minimum duration of 100 ms and a period of at least 300 ms for at least three consecutive cycles.
Intracellular recordings.
Intracellular recordings of neurons were made using sharp microelectrodes (1 m KAc, 30–40 MΩ) and amplified using a Model 1600 Neuroprobe amplifier (A-M Systems). To ensure that recordings were taken from regions of neurons maximally affected by the SD events, we recorded from neurites of neurons deep in the neuropile and not from neuronal somata, which are primarily located in a peripheral layer of the ganglion. Hyperpolarizing −1 or −2 nA current pulses were used to measure input resistance. Throughout the text, we refer to membrane depolarization as a decrease in membrane potential indicating a loss of the potential difference across the neuronal membrane (moving toward 0 mV) and hyperpolarization as an increase in membrane potential. Thus, a complete depolarization would result in a membrane potential of 0 mV.
Extracellular potassium-sensitive microelectrodes.
Low resistance (4–8 MΩ) microelectrodes were made from unfilamented glass pipettes with a diameter of 1 mm (World Precision Instruments) that had been washed with methanol (99.9%; Sigma-Aldrich) and dried on a hot plate before being pulled. The inner glass wall was then made hydrophobic by silanization with hot dichlorodimethylsilane (100°C; 99%; Sigma-Aldrich) vapor. After silanization, electrodes had their tips filled with Potassium Ionophore I-Cocktail B (5% Valinomycin; Sigma-Aldrich), forming an artificial membrane and then back-filled with 500 mm KCl. Reference electrodes were made from low-resistance (4–8 MΩ) filamented pipettes filled with 3 m KCl.
Extracellular potassium recordings.
Potassium-sensitive microelectrodes were connected to a DUO 773 two-channel intracellular/extracellular amplifier (World Precision Instruments) and calibrated at room temperature (22°C). KCl solutions (15 and 150 mm) were used to determine the voltage change needed to establish [K+]o (mm) using the Nernst equation (for details, see Rodgers et al., 2007).
Pharmacology.
All chemicals were obtained from Sigma-Aldrich and dissolved in standard locust saline using a minimum amount of DMSO (0.5 ml/100 ml of saline) and bath applied to the semi-intact preparations. Effective concentrations were determined in preliminary experiments and in other experimental paradigms (Dawson-Scully et al., 2007). Concentrations used to modulate activity of the NO/cGMP/PKG pathways are as follows: to augment endogenous nitric oxide (NO) levels, we bath applied 10 μm S-nitroso-N-acetylpenicillamine (SNAP) or 10 μm S-nitrosoglutathione (SNOG), a donor and a source of NO; to inhibit the endogenous production of NO, we bath applied l-NAME (10 μm), an NO synthase (NOS) inhibitor; to increase cGMP and PKG levels, we either bath applied 10 μm 8-Br-cGMP, a phosphodiesterase resistant analog of cGMP or 10 μm T-0156, a phosphodiesterase inhibitor; to inhibit PKG activity, we bath applied 10 μm KT5823, to inhibit protein phosphatase 2A (PP2A) activity (next step in the NO/cGMP/PKG pathway), we bath applied 1 μm cantharidin. To evoke repetitive SD-like events, we bath applied 100 μm ouabain, an Na+/K+ATPase inhibitor. To induce chemical anoxia, we bath applied 1 mm sodium azide, an electron transport chain inhibitor.
Pharmacology treatment for animals submerged in water was as follows: animals were injected with 10 μl of various drugs into the hemocoel (for methods, see Armstrong et al., 2006). Drug concentrations used were as follows: 100 μm 8-Br-cGMP, 100 μm T-0156, 100 μm KT5823, 100 μm Rp-8-Br-cGMP (a phosphodiesterase-resistant cGMP antagonist), 10 μm Cantharidin, 100 μm l-NAME. Control locusts were injected with standard locust saline containing 0.5% DMSO. Locusts were immersed in water 1 h after injection.
Potassium injections.
To generate K+-evoked SD-like events, we picospritzed 50 nl of saline containing an elevated concentration of KCl (150 mm; 15 times standard locust saline) directly into the anterior region of the MTG. We have previously reported that this concentration and volume is the minimum needed to generate SD-like events in the MTG (Armstrong et al., 2006; Armstrong and Robertson, 2006; Rodgers et al., 2007). Occasionally, the mechanical disturbance of the injection was sufficient to cause a disturbance in vCPG activity; however, this did not last longer than one rhythm cycle, and normal rhythm continued thereafter. Long-tipped pipettes were pulled and filled with the modified saline and attached to a picoSpritzer III (Intracell). Pressure was set to 10 psi, and pulse duration was varied to produce a bolus of 50 nl.
4,5-Diaminofluorescein-2 diacetate imaging.
Thoracic ganglia were dissected out and incubated for 1 h in 30 μm 4,5-diaminofluorescein-2 diacetate (DAF-2 DA), a selective membrane permeable marker that fluoresces in the presence of NO (Kojima et al., 1998). The ganglia were then rinsed several times over a 20 min period before being pinned to sylgard-coated chamber for examination under a fluorescence microscope (Olympus; BX50WI). Fluorescence excitation wave length was 460–490 nm, and emission wavelength was 510 nm using an Olympus FITC filter (product number U-N31001). Images were analyzed using SlideBook 4 (Intelligent Image Innovations). A perfusion system with external heater allowed saline composition and temperature to be changed while imaging. Temperature was monitored with a BAT12 thermometer and copper/constantan thermocouple placed adjacent to the ganglia.
Statistics.
SigmaStat 3.1 and JMP 7.0 were used to assess data groupings for significance. Statistical analyses used one-way and two-way ANOVA, followed by a post hoc Tukey multiple comparison test. For comparisons of percentages, we used a Z test. For nonparametric statistical tests, we used the Mann–Whitney rank sum test. For within-animal comparison of SD-like event duration and vCPG recovery time of control animals, we used a Wilcoxon signed rank test and a paired t test. Figures show significant differences using letter designations, treatments containing same letters are not significantly different from one another. Significance was assessed at p < 0.10; however, the majority of p values are <0.05. Asterisks indicate significant differences from control.
Results
SD-like events coincide with neuronal depolarization and reduced input resistance
To determine the extent of neuronal depolarization during SD-like events, we used sharp intracellular microelectrodes to record neuronal activity in the MTG concurrently with recordings of the ventilatory motor pattern and [K+]o (Fig. 1A,B). To induce SD-like events, we either superfused the MTG with ouabain (0.1 mm) (Fig. 1C) or briefly (∼30 s) applied sodium azide (1 mm) (Fig. 1D). By inhibiting mitochondrial function, sodium azide mimics anoxic coma, whereas treatment with ouabain provides a more gradual and less extreme impairment of ionic homeostasis in the CNS. We recorded from a total of 19 neurons of which 10 were active in phase with ventilation and nine were tonically active. Both ouabain and sodium azide treatments reliably generated SD-like events characterized by surges of [K+]o (30–50 mm) associated with arrest of vCPG activity and depolarization of neurons. During SD-like events, neurons depolarized from an (mean ± SE) interburst membrane potential of −49.0 ± 2.9 mV to a maximal depolarization to −36.6 ± 2.7 mV and then repolarized to −50.9 ± 5.8 mV when [K+]o recovers to baseline values (Fig. 2A). To account for neuronal variability and for statistical comparisons, we normalized changes in membrane potential to the pre-SD-like event values (Fig. 2B). There was a statistically significant 30% decrease in membrane potential of MTG neurons during SD-like events evoked by sodium azide. Recovery of vCPG activity occurred when [K+]o returned toward normal values and neurons repolarized.

SD-like events in the MTG of the migratory locust (Locusta migratoria). A, Schematic diagram of the MTG and recording electrodes used (Int, intracellular electrode; K+ Elec., potassium ion electrode; Ref. Elec., reference electrode for measuring [K+]o; Abd. Mus. 176, abdominal muscle 176, one of the ventilatory muscles). B, Sample recording from a ventilatory interneuron (Int.) and associated motor activity (EMG). Baseline level of [K+]o is 8 mm. Lower trace (Cur.) shows −1 nA hyperpolarizing current pulses (0.2 Hz). C, Sample trace showing a preparation bath applied with the Na+/K+ATPase inhibitor, ouabain; note the two ouabain-induced SD-like events. During the surge, the neuron depolarized and then repolarized when [K+]o returned to near baseline levels. Current pulses (−1 nA) were delivered at 4.5 Hz. D, Sample trace after a brief exposure to sodium azide (30 s), which quickly induced a surge in [K+]o, coinciding with membrane depolarization and cessation of neuronal activity. After superfusion with saline, neuronal activity recovered, and membrane potential was restored.

Effect of SD-like events on membrane potential and input resistance of neurons in the MTG. A, Top, Sample intracellular recording in the neuropil from a spontaneously active neuron during an SD-like event. After a 30 s sodium azide perfusion over the MTG (NaN3), the neuron depolarized; timing coincides with the onset of SD-like event (Fig. 1). Bottom, Various time points (i–vi) were used to quantify membrane characteristics. i, Before NaN3 treatment; ii, during depolarization; iii, maximal depolarization; iv, 5 min after azide wash-off; v, before repolarization; vi, restoration of membrane potential. Note that the lower trace in each time point is showing the current pulses (cur.) (1 Hz), and the upper trace is the intracellular recording (int.). B, Changes in membrane potential normalized to initial values before sodium azide treatment show depolarization during the SD-like event with subsequent recovery. C, During SD-like events, there was a significant decrease in input resistance. During restoration of activity and resting membrane potential, input resistance was higher than pre-SD-like event values. Numbers in brackets represent sample size. Single asterisks indicate significant differences from i. Double asterisks indicate significant differences from vi (one-way ANOVA, post hoc Tukey test, p < 0.05).
To monitor changes in input resistance, we delivered hyperpolarizing −1 nA current pulses through the recording electrode and measured changes in membrane potential (Fig. 2A). Before the SD-like event, the mean input resistance from neuropil recordings was 9.3 ± 1.5 MΩ, which decreased to 4.4 ± 0.9 MΩ at the maximal depolarization of each neuron and then recovered to a value of 13.4 ± 2.1 MΩ after restoration of the K+ gradient. These values are in the same range as those reported in many previous intracellular investigations of insect neurons (e.g., ∼8 MΩ dropping to 5.6 MΩ during hypoxia) (Le Corronc et al., 1999); however, the drop in input resistance during the SD event is likely an underestimate, because it will be confounded by any loss of synaptic input (during arrest of activity) which would tend to increase input resistance. For statistical comparisons, we normalized changes in input resistance to pre-SD-like event values and found a significant decrease in input resistance in neurons during the SD-like event (Fig. 2C). Just before repolarization, input resistance increased, to the point where it was not significantly different from pre-SD-like event values. However, input resistance after restoration of K+ gradient was significantly higher than pre-SD-like event values (Fig. 2C).
These data confirm that SD-like events in the locust MTG are associated with neuronal hyperexcitation and a decrease in input resistance likely caused by opening of voltage-dependent channels.
PKG inhibition suppresses ouabain-induced SD-like events
Using a model whereby repetitive SD-like events were induced by inhibiting the Na+/K+-ATPase during a 30 min exposure to 0.1 mm ouabain, we examined if altered PKG activity would affect the occurrence of repetitive SD-like events. Coinciding with ouabain-induced cessation of vCPG activity, there was a rapid rise in [K+]o followed by restoration of [K+]o to near baseline levels corresponding with vCPG recovery (Fig. 3A).

Effects of PKG manipulation on ouabain-induced SD-like events in the MTG. A, SD-like events were identified as abrupt increases in [K+]o coinciding with cessation of motor output from the MTG monitored by electromyographic recordings of ventilatory motor patterns (EMG) during a 30 min exposure to 100 μm ouabain. Insets show expansions of the EMG trace, revealing the ventilatory rhythm. B, Sample recordings from a preparation pretreated with the PKG inhibitor KT5823 showing protection from ouabain-induced SD-like events. Semi-intact preparations were dissected in saline, then switched to saline containing either KT5823, 8-Br-cGMP (PKG agonist), or saline as a control for 10 min. After drug treatment, saline containing ouabain was superfused over the MTG. C, All control and 8-Br-cGMP-treated preparations cotreated with ouabain generated SD-like events, whereas only 30% of KT5823-treated preparations generated these events. D, Treatment with 8-Br-cGMP significantly increased the number of individual events occurring in a 30 min period, whereas the few animals treated with KT5823 that exhibited this phenomena (n = 3) did not differ in the number of events when compared with control preparations. Numbers in brackets represent sample size. Letters in histogram bars represent statistical groupings using a post hoc test, whereby bars with different letters are significantly different (1-way ANOVA, post hoc Tukey test, p < 0.05). E, Preparations treated with KT5823 had lower baseline [K+]o during the course of ouabain exposure than 8-Br-cGMP-treated and control preparations (n = 10 for each group). Asterisks indicate significant differences between KT5823-treated animals and control animals (2-way repeated-measures ANOVA with a post hoc Tukey multiple comparisons test, p ≤ 0.05).
All preparations treated with 0.1 mm ouabain generated SD-like events in the MTG; however, preparations pretreated with the PKG inhibitor KT5823 showed a statistically significant 70% reduction in the number of preparations generating SD-like events (z test, z = 2.813, p = 0.005) (Fig. 3B,C). All locusts pretreated with the PKG activator 8-Br-cGMP showed ouabain-induced SD-like events. Furthermore, these animals had significantly more SD-like events in the same period than locusts treated with ouabain (Fig. 3D). Moreover, treatment with the PKG activator made this model more sensitive to ouabain-induced SD-like events, observed as a reduction in the time to the first SD-like event (control, 542 ± 142 s; 8-Br-cGMP, 252 ± 26 s mean ± SEM; Mann–Whitney rank sum test, p = 0.076). We found no difference in mean amplitude or duration measured at half the maximum amplitude of each ouabain-induced SD-like event, but there was a general trend of decreased amplitude after each successive SD-like event. Preparations treated with the PKG inhibitor maintained a lower baseline level of [K+]o during the 30 min exposure to ouabain (Fig. 3E).
These data demonstrate that pharmacological manipulation of PKG has profound effects on the occurrence and severity of repetitive SD-like events induced by disturbing ion homeostasis in the locust.
PKG inhibition attenuates SD-like events evoked by extracellular K+ injection
To investigate the effect of PKG pathway manipulation on SD-like events without the confounding effects of ouabain, we tested a variety of pharmacological agents on single SD-like cycles evoked by local injections of high KCl saline directly into the MTG. All preparations, regardless of drug treatment, reliably generated SD-like events after each injection. We used a within-animal experimental design to compare various drug treatments on SD-like event duration (measured at half the maximum amplitude) and the length of time to recover vCPG function after motor pattern arrest. KCl-evoked SD-like events were compared before and after 10 min of recovery followed by 10 min of bath-applied drug treatment. Within 10–15 s of the first injection (depending on how close the ion-sensitive electrodes were to the injection site), there was a surge in [K+]o with concurrent cessation of vCPG activity (Fig. 4A). Mean SD-like event duration and vCPG recovery time of the first evoked SD-like event was 102.3 ± 9.2 s and 156.6 ± 11.1 s (n = 14; mean ± SEM), respectively. After the second injection, SD-like event duration and vCPG recovery time were longer (130.5 ± 16.0 s and 174.6 ± 10.3 s, respectively; n = 11) (Fig. 4A). The increases in mean SD-like event duration and vCPG recovery time were significant (SD-like event duration: Wilcoxon signed rank test, w = 83.00, p = 0.007; vCPG recovery time: paired t test, t = −3.308, p = 0.006).

Effects of activation and inhibition of the various steps along the NO/cGMP/PKG pathway on KCl-induced SD-like events. A, Single SD-like events were evoked by injecting saline (50 nl) containing elevated potassium chloride (150 mm vs 10 mm) directly into the MTG. During SD-like events, ventilatory motor activity monitored EMG, ceased, and recovered during restoration of the potassium ion gradient. We measured the duration of the [K+]o event at half its maximal amplitude and the time from failure of the ventilatory motor pattern to the recovery of the rhythm. After recovery (20 min) of ventilatory motor pattern activity, a second injection was delivered into the MTG, evoking a second SD-like event, permitting within-animal comparisons. Before the second injection (10 min), various pharmacological antagonists or agonists were bath applied to manipulate steps along the NO/cGMP/PKG pathway or protein phosphatase 2A activity. B, In control preparations, the second event was ∼30% longer than the first. Event duration was increased by T-0156 (phosphodiesterase inhibitor), although not by 8-Br-cGMP, and reduced by KT5823 and cantharidin (PP2A antagonist). The effect of T-0156 was offset by cantharidin but not significantly reduced. C, In control preparations, motor pattern recovery time was ∼10% longer for the second event compared with the first. The recovery time was increased by 8-Br-cGMP and T-0156 and reduced by KT5823 and cantharidin. The effect of T-0156 was significantly offset by cotreatment with cantharidin. These data indicate that PKG inhibition (KT5823) reduces the duration of SD-like events and hastened the time to recover neural function after KCl-induced SD-like events, whereas activation of the pathway (8-Br-cGMP, T-0156) had the opposite effect. D, Animals were treated with either the NOS inhibitor l-NAME, the NO donors SNAP or SNOG, or a combination of l-NAME and SNOG 10 min before the second injection. Animals treated with l-NAME had significantly shorter SD-like event durations than animals treated with SNOG. E, After l-NAME treatment, the length of time taken to recover CPG function after KCl-evoked SD-like events was shorter than for all other drug treatments. This effect was abolished by concurrent exposure to the NO donor SNOG (l-NAME plus SNOG). Letters represent statistical groupings using a post hoc test, whereby bars with different letters are significantly different and bars that share a letter are not (1-way ANOVA, Tukey, p < 0.10).
Consistent with the ouabain experiments, single KCl-evoked SD-like events were significantly altered by PKG inhibition. To quantify the change in SD-like event duration and vCPG recovery time after drug treatments, we normalized these measures to the values obtained as a result of the first injection [e.g., normalized SD-like event duration equals (2nd injection SD-like event duration minus 1st injection SD-like event duration) divided by first SD-like event duration time]. A normalized change in SD-like event duration of zero would indicate that there was no change between the first and second SD-like event duration. Treatment with the PKG inhibitor KT5823 shortened SD-like event durations more than all other treatments (Fig. 4B). Using cantharidin to inhibit PP2A, a downstream target of PKG that acts on K+ channels (Zhou et al., 1996), we found a similar decrease in SD-like event duration (Fig. 4B). Neither PKG activation (via 8-Br-cGMP) or phosphodiesterase inhibition (via T-0156) (Mochida et al., 2002) significantly affected SD-like event duration beyond that of control experiments, although there was a general trend of increased SD-like event duration.
Perhaps a more relevant measure of neuronal activity recovery after KCl-evoked SD-like events is the time taken for motor pattern activity to recover operation (see Materials and Methods for assessment of vCPG recovery time). Preparations treated with either KT5823 or cantharidin after their first KCl-evoked SD-like event recovered vCPG activity quicker (Fig. 4C). This was in contrast to control preparations and those treated with 8-Br-cGMP or T-0156 which were statistically longer (Fig. 4C). In addition, preparations cotreated with cantharidin and T-0156 had shorter vCPG recovery times, confirming that inhibition of downstream steps can prevent the exacerbating effects of upstream activation of the PKG pathway by T-0156 (Fig. 4C).
Thus, the severity of the depression of neural activity during individual cycles of SD-like events can be modulated by altering levels of PKG and one of its downstream targets.
Inhibition of NOS attenuates SD-like events evoked by extracellular K+ injection
In the CNS of invertebrates and vertebrates, NO-activated soluble guanylate cyclase is one of the enzymes responsible for the production of cGMP and subsequent activation of PKG (Moncada et al., 1991; Elphick et al., 1993), although other activators of PKG do exist (Morton and Giunta, 1992; Müller, 1997). Histochemical techniques and immunocytochemistry have been used to identify cells in the locust thoracic nerve cord that contain NOS, the key enzyme that produces NO as it converts l-arginine to l-citrulline (Moncada et al., 1991; Müller, 1997; Bullerjahn and Pflüger, 2003). To examine the role of NO in modulation of KCl-evoked SD-like events, we used the same double injection experimental paradigm used previously. We either treated preparations with l-NAME (NOS inhibitor) or we augmented endogenous NO using the NO donors SNAP or SNOG. Although treatment with l-NAME or the NO donors did not significantly alter SD-like event duration compared with control preparations, preparations treated with l-NAME generated significantly shorter SD-like event durations than preparations treated with SNOG (Fig. 4D). There was a general trend of increased duration after bath application of both NO donors (Fig. 4D). There was a strong effect of l-NAME on the time to recover circuit function which was significantly different from all other treatments (Fig. 4E). The presence of NO donors did not significantly increase the length of time needed for vCPG activity to recover but did offset the effect of l-NAME, suggesting that NO levels may be at saturation under control conditions.
These data demonstrate that inhibition of NOS mimics the effect of reduced PKG activity on reducing SD-like event duration in the MTG.
Abiotic stress increases NO production in the MTG
A plausible interpretation of anoxic and hyperthermic comas is that they are adaptive, conserving energy during periods of abiotic stress (Walter and Nelson, 1975; Rodgers et al., 2007). Thus, we looked for evidence that abiotic stressors could affect the NO/PKG pathway by measuring the production of NO in the MTG. We dissected out the MTG and mesothoracic ganglion and incubated both for 1 h in 30 μm DAF-2 DA, a selective membrane permeable marker, which is converted by cellular esterases to DAF-2 that fluoresces in the presence of NO (Kojima et al., 1998; Brown et al., 2000). Changes in NO levels were estimated as the difference in fluorescence between the DAF-2 loaded neurons and surrounding tissue arising during a 500 ms exposure. Fluorescence readings were collected every 5 min. Labeled cell body patterns were similar to previously reported cells that contain NOS in the desert locust, Schistocerca gregaria (Müller and Bicker, 1994; Ott and Burrows, 1998). Cell sizes ranged from 9.4 to 39.0 μm with an average size of 18.9 ± 2.9 μm (mean ± SEM; n = 11).
In untreated ganglia, DAF-2 fluorescence (corrected for background) remained nearly constant for 65 min. The modest (not significant) decrease with time in these ganglia is likely attributable to a combination of low NO production with photobleaching and leakage. When ganglia were treated with sodium azide, there was no significant change in DAF-2 fluorescence during the 15 min treatment, but after azide removal, DAF-2 fluorescence increased linearly for the next 25 min. After this point, DAF-2 fluorescence remained greater than untreated cells but did not further increase, suggesting either a reduction in NO production or saturation of the available DAF-2 (Fig. 5A,B). When azide-treated cells were compared with untreated cells, DAF-2 fluorescence was significantly increased [2-way repeated-measures (rm) ANOVA, p < 0.001; n = 6] (Fig. 5C). To assess the role of NOS in this phenomenon, we pretreated cells with the NOS inhibitor l-NAME by including it as a pretreatment during DAF-2 loading (n = 3). Under these conditions, l-NAME completely abolished the azide-induced increase in DAF-2 fluorescence (2-way rm ANOVA, p = 0.449) (Fig. 5C).
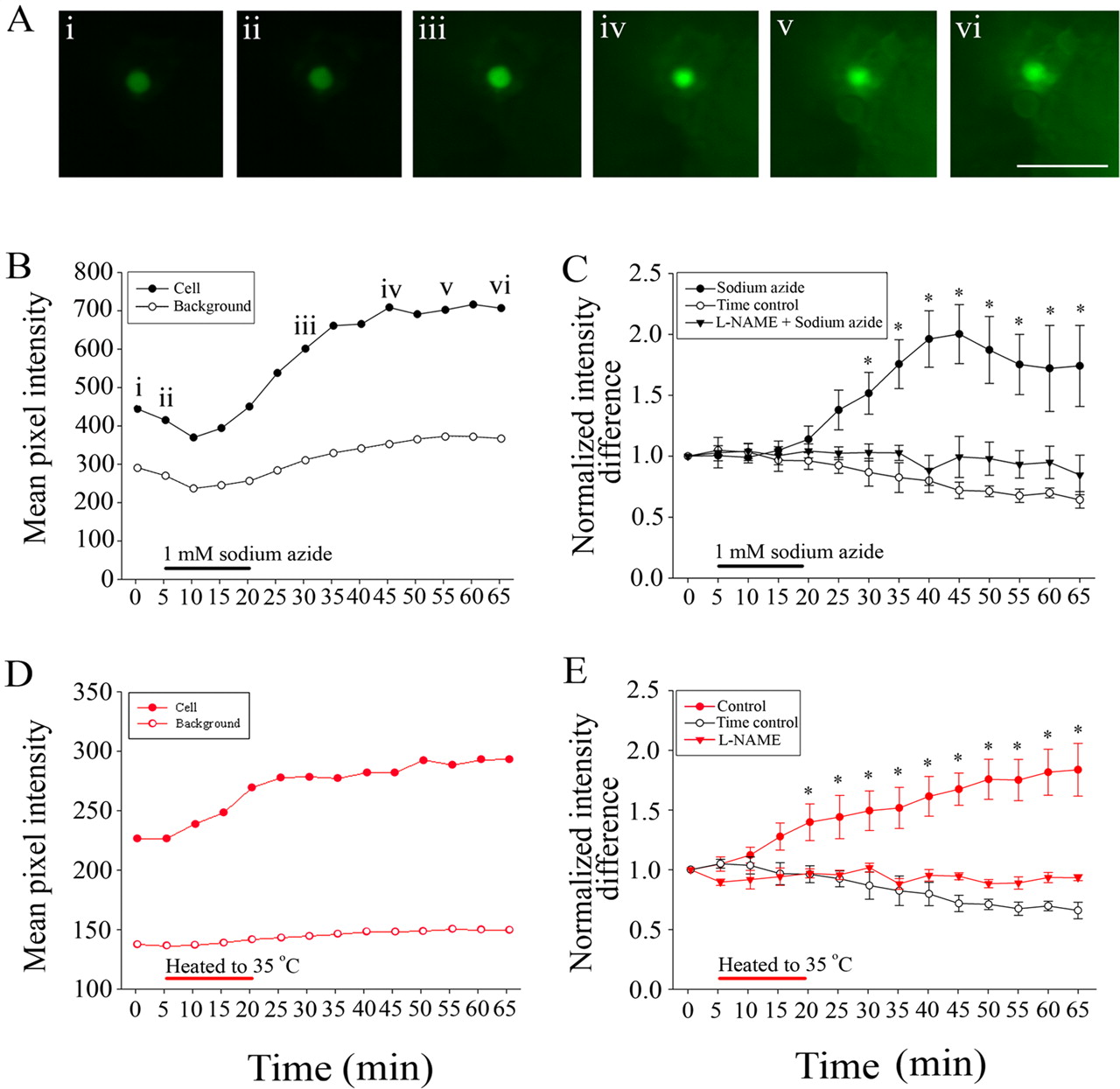
Chemical anoxia and heat stress increase NO production in thoracic ganglia. A, Example images of a DAF-2 DA-loaded cell body at various time points during exposure to 1 mm sodium azide. Scale bar, 50 μm. B, Cell body and background fluorescence changes after chemical anoxia. C, Normalized change in intensity difference (cell body to background) fluorescence over a 65 min time period in tissue treated with sodium azide (n = 6), tissue treated with sodium azide and the NOS inhibitor l-NAME (n = 3), and non-treated tissues (time control, n = 3). Exposure to sodium azide significantly increased fluorescence, but this rise in fluorescence was not observed in tissue first treated with l-NAME. D, Cell body and background fluorescence changes after exposure to 35°C. E, Control cells in ganglia exposed to heat (control, n = 4) showed a significantly larger increase in intensity difference than those in non-heated time controls (n = 3). l-NAME-treated tissues (n = 4) did not significantly produce NO when exposed to heat. Asterisk indicates significant differences from time controls (2-way ANOVA with a Tukey test, p < 0.05).
We also investigated if temperature stress would alter NO production in these cells. We briefly raised the temperature of saline flowing over these ganglia to 35°C and then allowed the saline to return to room temperature levels. We found a significant increase in intensity difference between control preparations that were heated and unheated preparations (2-way rm ANOVA, p < 0.001) (Fig. 5D,E). In alignment with what we found after exposure to sodium azide, in l-NAME and DAF-2 DA cotreated tissues, exposure to heat did not significantly increase the fluorescence intensity difference (2-way rm ANOVA, p = 0.156) (Fig. 5E).
These experiments demonstrate that the stressors that induce coma associated with SD-like events in the MTG also increase the production of NO in this ganglion. This suggests that NO may be the upstream effector of the PKG pathway that modulates SD-like event duration.
Recovery from coma induced by suffocation under water is faster after inhibition of the NO/cGMP/PKG pathway
In a final data set, we examined if the length of time required to recover from anoxic coma in intact behaving animals could be altered by modulating the NO/cGMP/PKG signaling pathway. To accomplish this, we used a similar before and after experimental paradigm as used in the previous sections. This consisted of immersing intact animals in water for 30 min. All animals were incapacitated (no movement) within 10 min after immersion in water and recovered ventilatory motor pattern activity and stood upright after removal from the water. After recovery (all animals standing) from this initial immersion in water, an injection of 10 μl into the hemocoel was administered to each animal. These injections contained some of the drugs previously used. After the injections (1 h), locusts were again immersed in water for 30 min, removed, and the times to start ventilation and stand were recorded. All animals recovered and survived to reproduce normally. To quantify the change in the time to start of ventilation and the change in time to stand, we normalized these measures to the values obtained after the first water immersion. In alignment with our previous data, locusts injected with l-NAME and KT5823 recovered ventilatory activity significantly faster, and animals treated with l-NAME and Rp-8-Br-cGMP took less time to stand upright than control preparations which received sham injections (Fig. 6A,B). There was no difference between the results for KT5823 and Rp-8-Br-cGMP, indicating that effects of cGMP not mediated by PKG are unlikely to be involved. Animals treated with 8-Br-cGMP and T-0156 took significantly longer to start ventilating again than control animals, but no differences were found in the length of time required to stand upright (Fig. 6B).

Inhibition of NO production and PKG activity reduced the time to recover from coma as a result of suffocation by immersion in water. We measured the length of time it took for animals to begin ventilatory behavior (A) and the length of time it took for locusts to stand upright after a 30 min period of water immersion (B). Immediately after standing, locusts received an injection into their hemocoel of various pharmacological antagonists or agonists to manipulate steps along the NO/cGMP/PKG pathway or protein phosphatase 2A activity. This permitted within-animal comparisons. Locusts treated with the NOS inhibitor (l-NAME) or a PKG inhibitor (KT5823 or Rp-8-Br-cGMP) recovered ventilatory motor pattern activity significantly faster, and animals treated with l-NAME took less time to stand upright than control animals. Conversely, locusts treated with the PKG activator (8-Br-cGMP) and the phosphodiesterase inhibitor (T-0156) took a significantly longer time to recover ventilatory motor pattern activity and to stand than control animals. Letters represent statistical groupings, whereby bars with different letters are significantly different (1-way ANOVA, Tukey, p < 0.05).
This data set confirms that activity of the NO/cGMP/PKG signaling pathway in intact animals can alter the time required to recover from incapacitating environmental conditions. Inhibition of this signaling pathway reduced the time to recover ventilatory activity and reduced the time before the animal is able to stand upright.
Discussion
Genetic and pharmacological downregulation of the cGMP/PKG pathway in Drosophila attenuates hyperthermic neural failure, and similar pharmacological treatments modulate the temperature sensitivity of the vCPG in L. migratoria (Dawson-Scully et al., 2007). Given that hyperthermic ventilatory arrest in the locust is an SD-like event (Rodgers et al., 2007), our primary goal in this investigation was to test the hypothesis that inhibition of PKG would reduce the severity of SD-like events monitored in the locust MTG. We found a striking effect of PKG manipulation on ouabain-induced SD-like events, such that PKG inhibition completely suppressed SD-like events evoked by 10−4 m ouabain in 70% of preparations. Conversely, PKG activation reduced the time to SD-like event onset and exacerbated it by increasing the frequency of individual SD-like events. These results were confirmed in a model of SD-like events induced by KCl injection and were extended to show that inhibition of a downstream target of PKG (PP2A) reduced the duration of SD-like events. This effect could offset the exacerbating effect of phosphodiesterase inhibition to increase PKG levels. We have also shown that manipulation of NO levels also affects SD-like events induced by KCl injection. In addition, we demonstrated that two cellular stressors known to generate SD (chemical anoxia and hyperthermia) increase the production of NO in the thoracic ganglia of this system and that this can be prevented by incubation with an inhibitor of NO synthase. Thus, we conclude that the NO/cGMP/PKG pathway acting through PP2A has an important role in modulating the incidence and severity of SD-like events in this system. Finally, we show that differing activity levels of this pathway can alter the time taken to recover from incapacitating environmental conditions such as water immersion.
To consider the potential mechanisms underlying the attenuation of SD-like events by inhibition of the NO/cGMP/PKG pathway, we propose the following model for SD-like events in the locust MTG (Fig. 7). This model is based on models proposed for CSD (Kager et al., 2002; Somjen, 2002; Cestèle et al., 2008) and the described characteristics of locust SD-like events, whereby [K+]o shows accelerating transitions between two stable states (Rodgers et al., 2007). The core feature is the balance between K+ accumulation and K+ clearance in a relatively restricted extracellular volume. Normally, extracellular volume buffering and mechanisms of K+ clearance are more than sufficient to accommodate the increased flux of K+ associated with increased neural activity. However, when processes leading to K+ accumulation predominate over processes of clearance sufficiently to increase [K+]o and reduce the K+ equilibrium potential, the system becomes unstable and enters a positive feedback cycle (Fig. 7A). This requires either movement of large quantities of ions or a small extracellular volume. Reduction of the K+ equilibrium potential depolarizes neurons, which in turn leads to more K+ accumulation and further depolarization until neuronal activity ceases when voltage-dependent Na+ channels remain inactivated: SD-like event. The positive feedback accounts for the acceleration and rapidity of the increase of [K+]o to a new equilibrium. Different manipulations that predispose toward the generation of an SD-like event act at different points in the model. Thus, hyperthermia and other forms of hyperexcitation increase K+ accumulation, whereas metabolic inhibition and inhibition of the Na+/K+ ATPase decrease K+ clearance, and K+ injection is a direct disturbance of K+ homeostasis, but all eventually result in the same end state of an SD-like event.

Model of spreading depression-like events in the locust metathoracic ganglion. A, Onset. SD-like event is evoked when extracellular K+ accumulation exceeds clearance under conditions when [K+]o can appreciably affect the K+ equilibrium potential and cause neuronal depolarization, thus entering a positive feedback cycle (+). This can be induced by increasing accumulation, decreasing clearance, or directly manipulating [K+]o. B, Recovery. If cessation of neural activity is sufficient to enable K+ clearance to predominate then the system enters a positive feedback cycle that restores [K+]o and membrane potentials to pre-SD-like event levels. Further details in Discussion.
Some treatments are extreme enough that the SD-like event is maintained until normal conditions are restored (e.g., anoxic coma). Nevertheless, the repetitive nature of ouabain-induced SD-like events, seen here, and occasionally in hippocampal slices (Balestrino et al., 1999), is striking and can be accommodated in the model (Fig. 7B). When the reduction of K+ clearance is modest (e.g., using 10−4 m ouabain), then the onset of an SD-like event reduces K+ accumulation by arresting neuronal activity and allows processes of clearance to predominate again. At this point, the system enters the restorative positive feedback cycle, whereby decreasing [K+]o increases the K+ equilibrium potential, hyperpolarizes neurons, and inactivates voltage-dependent K+ channels, which decreases [K+]o. At some threshold, neuronal activity can be restored, but this causes processes of accumulation to predominate, and the cycle repeats (Fig. 3A).
Our findings with respect to the model described above suggest that activation of the NO/cGMP/PKG pathway predisposes toward K+ accumulation (by increasing accumulation and/or by decreasing clearance), whereas inhibition of this pathway predisposes toward K+ clearance (Fig. 7A). Although the details of the mechanisms in this system remain to be determined, it is probable that numerous and diverse targets contributing to a coordinated tissue response to stress are involved. Nevertheless, there is evidence from other systems to suggest direct effects on K+ conductances and the Na+/K+ ATPase. Notably, reduced PKG activity in the sitter variants of the Drosophila foraging gene is associated with reduced peak and steady-state voltage-activated K+ currents, and this can be mimicked by pharmacological inhibition of PKG (Renger et al., 1999). Moreover, activation of K+ conductances by the NO/cGMP/PKG pathway has been demonstrated in mammalian systems (Kubokawa et al., 1998; Cuong et al., 2006). Elevated NO both activates leak K+ current through a PKG-dependent mechanism in forebrain neurons (Kang et al., 2007) and can indirectly decrease Na+/K+ ATPase activity when ATP production is impaired in cortical neurons (Tavalin et al., 1997). NO responses that are normally stabilizing and protective (Andoh et al., 2002), such as stress-induced activation of K+ conductances to hyperpolarize neurons and prevent excitotoxicity, could promote SD-like events under conditions when there are insufficient resources to maintain K+ homeostasis, particularly with a restricted extracellular volume.
NO modulation of neural function (Calabrese et al., 2007) has been implicated in several important processes, including synaptic plasticity (Antonov et al., 2007), tuning of locomotor circuits in the spinal cord (McLean and Sillar, 2002), in response to increased CNS activity in insects (Zayas et al., 2002) and as a response to environmental stressors including hypoxia (Wingrove and O'Farrell, 1999). Indeed, NO has been shown to modulate the threshold for spreading depolarization in human and rodent cortex (Petzold et al., 2008), although in this study the threshold was reduced by decreasing levels of NO and was primarily dependent on endothelial NOS. In insects, the neuroanatomical substrate has been well described (Bicker, 1998), especially in locusts (Ott and Burrows, 1998; Bullerjahn and Pflüger, 2003), and several studies have investigated its effect on motor patterning, sensory processing, and neuronal excitability (Müller and Hildebrandt, 2002; Newland and Yates, 2007). In contrast, the conditions under which NO production increases are less well known, and thus the behavioral relevance of the motor pattern modulation is not always clear. We found that treatments that mimic environmental stress (chemical anoxia and hyperthermia) increased DAF-2 fluorescence of particular neurons in the thoracic ganglia. Some caution in interpretation is warranted, because ascorbic acid and dehydroascorbic acid react with DAF-2 to produce fluorescent compounds (Zhang et al., 2002). However, treatment with l-NAME did produce less fluorescence, suggesting that a significant portion of measured fluorescence was derived from NO. Nevertheless, taking all the observations together, we believe that in this system stress activates the NO pathway, and the subsequent adaptive responses make the system more likely to generate SD-like events. Several possibilities exist, such as increasing K+ conductance to prevent neuronal hyperexcitation (as mentioned above) or reducing energy consumption by the ion pumps. Downregulation of this pathway would have immediate consequences of reducing the likelihood of generating SD-like events but could have unforeseen, longer-term consequences.
We have argued previously that the phenomena we describe here as SD-like in the MTG of the locust and CSD in vertebrates are essentially the same. We provide further support for this by demonstrating that metathoracic neurons depolarize during SD-like events, and this is associated with a large decrease in input resistance suggesting the opening of voltage-dependent membrane channels. The levels of depolarization we recorded (∼13 mV) are consistent with previous reports in insects [(10–25 mV during hypoxia in cockroaches) (Le Corronc et al., 1999); 10–18 mV during hyperthermic failure in flight motoneurons (Wu et al., 2001)] but are markedly smaller than the depolarizations to zero, evident in mammalian neurons (Collewijn and Harreveld, 1966). It is unclear why insect neurons do not depolarize to zero during SD-like events, but it may account for the ability of insects to survive long periods of anoxic coma without neuronal damage, and thus it would be of considerable interest to find out.
In mammals, CSD and anoxic depolarization are considered to be related phenomena which differ primarily in severity and mechanism of induction (Somjen, 2001; Anderson and Andrew, 2002). Although CSD is closely associated with migraine pathology, it is generally thought to be benign, whereas anoxic depolarization lasting longer than a few minutes is damaging, mainly attributable to cell swelling (Jarvis et al., 2001). In our preparation, SD-like events are similarly related to the depolarization and neural failure associated with anoxic coma. A major difference from mammals is that insects recover from several hours of anoxic coma, apparently none the worse for the experience (see supplemental Videos, available at www.jneurosci.org as supplemental material). It is hard to avoid the interpretation that anoxic coma in insects (and thus the SD-like event that generates it) is adaptive in nature, allowing insects to cope temporarily with extreme environmental conditions by entering a state of neural arrest that conserves energy and prevents complete neuronal depolarization and associated cell swelling and damage. The idea still requires conclusive substantiation, but the discovery of a genetically encoded pathway that can modulate SD-like events, particularly its initiation and duration, lends support to this interpretation. Conversely, in humans, it is hard to accept the interpretation that SD and migraine might have adaptive roles, although arguments along these lines have been made (Loder, 2002); indeed, it has been shown that CSD can provide protection against ischemia (Yanamoto et al., 2004). Despite these fundamentally different perspectives, and given the strong functional similarity at the cellular level that allows invertebrates to be used as genetic models for human diseases, we propose that much can be learned about general mechanisms of CSD by investigations of SD-like events in the locust MTG. Moreover, it is a reasonable assumption that anoxic coma in Drosophila, which can be manipulated genetically (Haddad et al., 1997), is generated by the same mechanisms as SD-like events in the locust. This would permit powerful, high throughput, molecular genetic tools to be brought to bear on the problem (Farahani and Haddad, 2003).
In conclusion, we have shown in the locust that the generation of SD-like events can be manipulated in the long term by pretreatments requiring protein synthesis like heat shock (Rodgers et al., 2007) and in the short term by targeting well defined cellular signaling pathways like the NO/cGMP/PKG pathway. The fact that these pathways are evolutionarily conserved suggests that insect models of SD-like events will be fruitful avenues for future investigation.
Footnotes
-
This research was funded by the Natural Sciences and Engineering Research Council of Canada to R.M.R. and G.A.B.A. We thank Donald H. Maurice, David R. Andrew, Chris D. Moyes, Ken D. Dawson-Scully, and Barry A. Trimmer for helpful discussion and comments on this manuscript.
- Correspondence should be addressed to R. Meldrum Robertson, Department of Biology, Queen's University, Biosciences Complex, Kingston, ON K7L 3N6, Canada. robertrm{at}queensu.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}