

Abstract
To date, long-term consequences of septic encephalopathy on cerebral metabolism, cognition, learning, and memory capabilities and factors involved are poorly understood. In this study, we used a murine sepsis model to demonstrate that bacterial lipopolysaccharide (LPS) causes long-term cognitive deficits in mice. Two months after LPS treatment, wild-type mice committed more working and reference memory errors than controls. The behavioral impairment was independent of the cerebral glucose uptake as evidenced by 18F-Fluordeoxyglucose small animal positron emission tomography. In contrast, mice deficient for the inducible nitric oxide synthase gene (NOS2−/−) did not show any cognitive changes when challenged with LPS. Immunohistochemical analysis demonstrated that LPS did not lead to neuronal cell death but caused sustained microglial activation in wild-type as compared to NOS2−/− mice. Expression analysis showed that LPS-treated NOS2−/− mice had lower brain mRNA levels for proinflammatory factors compared with wild-type mice. Expression analysis demonstrated distinct changes in the content of synaptic proteins in wild-type mice, which were not observed in the NOS2−/− mice. Together, this data set outlines the importance of the NOS2 activation for long-term cerebral changes after severe sepsis.
Introduction
Sepsis-induced brain dysfunction receives increasing attention, since it has become evident that it directly causes brain damage and is well correlated with the rate of morbidity and mortality of intensive care unit patients (Sprung et al., 1990; Sharshar et al., 2004; Nguyen et al., 2006). Of note, up to 71% of septic patients develop septic encephalopathy (SE), a potentially irreversible acute cerebral dysfunction (Pine et al., 1983; Sprung et al., 1990; Wilson and Young, 2003).
SE is caused by systemic inflammation evoked by the immune response to bacterial lipopolysaccharide (LPS) or other endotoxic bacterial cell wall components in the absence of direct brain infection. The acute sepsis-induced syndrome is clinically characterized by slowing of mental processes, impaired attention, memory dysfunction, delirium, or coma. The pathogenesis of SE is likely to be influenced by various factors including the systemic and local generation of proinflammatory cytokines, activation of microglia, alterations of the cerebral microcirculation, neurotransmitter imbalances, and a negative impact of sepsis-induced peripheral organ failure (Wilson and Young, 2003).
Evidence from rodent models suggest that the peripheral administration of LPS either as single injection or given repeatedly affects learning and memory functions (Arai et al., 2001; Shaw et al., 2001; Sparkman et al., 2005). In line with these findings, LPS has been demonstrated to impair long-term potentiation (LTP), a key cellular process of learning and memory consolidation (Malenka and Nicoll, 1999), in vitro and in vivo (Commins et al., 2001; Jo et al., 2001; Hennigan et al., 2007). However, these investigations are likely to reflect the immediate effects of SE on brain functions, since the experimental paradigms applied were of rather acute nature, exposing animals or brain slices to LPS directly or just hours before the respective analysis. In contrast, the long-term consequences of sepsis on learning and memory have not been studied intensively in humans and rodent models until relatively recently (Barichello et al., 2005a,b; Hopkins and Jackson, 2006; Semmler et al., 2007).
Based on previous observations, which revealed sepsis-induced upregulation of the inducible isoform of nitric oxide synthase (NOS2) in the brain in vitro and in vivo (Wong et al., 1996; Suzuki et al., 1998; Semmler et al., 2005) and the fact that NOS2 inhibition prevented neuronal apoptosis in a model of acute sepsis (Semmler et al., 2005), we hypothesized that NO may also be involved in sepsis-induced long-term cognitive deficits.
Materials and Methods
Animals and experimental procedures
Wild-type C57BL/6 and NOS2 knock-out mice (Laubach et al., 1995) were both from The Jackson Laboratory. C57BL/6 and NOS2 knock outs were crossed resulting in NOS2 heterozygous mice. These mice were subsequentially crossed resulting in C57BL/6 wild-type and homozygous NOS2 knock-out mice, which were then used for the study. To induce sepsis, wild-type and NOS2−/− mice received a single intraperitoneal injection containing 5 mg/kg of LPS (0127:B8, Escherichia coli; Sigma) dissolved in 1 ml PBS or, as control, PBS only, at 3 months of age (Fig. 1A). A subset of mice was killed after 24 h to investigate brain mRNA levels of cytokines; the remaining animals were housed then in groups of four under standard conditions at a temperature of 22°C and a 12 h light/dark cycle with ad libitum access to standard food (Altromin) and tap water. Before behavioral studies, which were performed in the active phase of the animals, mice were housed for at least 2 weeks on a reversed light/dark cycle (lights off 8:00 A.M., on 8 P.M.). Animals were first tested in an open-field assessment, followed by an eight-arm radial maze (Fig. 1A). Animal care and handling was performed according to the declaration of Helsinki and approved by the local ethical committees.

Peripheral LPS administration causes sustained NOS2 expression in the brain of C57BL/6 mice. A, LPS treatment schedule including the time points for the different analyses. OF, Open field; RAM, radial arm maze. B, Western blot detection of NOS2 in the hippocampus (HC) and frontal cortex (FC) over the experimental period of 8 weeks. C, Comparison of NOS2 expression in LPS and vehicle (PBS)-injected mice at 1 and 8 weeks from whole-brain lysates. Densitometrical quantification verified a significant and sustained LPS effect on NOS2. Each lane represents a single animal (mean ± SEM, n = 6, student's t test, *p < 0.05; **p < 0.01). D, DAB immunostaining against NOS2 in an LPS-treated mouse (top). Scale bar, 50 μm. Representative confocal immunostaining of NOS2 and CD11b revealing colocalization (bottom). Scale bar, 10 μm.
Behavioral assessment
Open-field exploration.
Mice were placed into the center of a dimly lit (20–30 lux) chamber of the open-field apparatus (44 × 44 × 30 cm). Movements of the animals were tracked by an automatic monitoring system (TSE) for 15 min. Horizontal (distance traveled) and vertical activity (number of rears) were evaluated. The experiment was repeated on three consecutive days. Behavioral data were analyzed by two-way ANOVA (within factor, repetition; between factor, group) followed by Student–Newman–Keuls (SNK) post hoc test.
Eight-arm radial maze.
Learning and memory testing for each mouse was conducted in an eight-arm maze constructed of wood and elevated 50 cm from the floor. Each of the arms was 60 cm long and 6 cm wide and extended from an octagonal central platform, 10 cm across. Food cups, 1 cm deep, were placed 2 cm from the end of each arm. The testing room contained several visual cues outside of the maze and was dimly lit while sessions were in progress. Initially, the mice were trained for 3 d. During each training session, the mouse was placed on the center platform and allowed to move freely in the maze to obtain food pellets, which were presented in all the eight arms, for a period of 10 min. From day 4 on the mice were tested one session per day, for a total of 14 d. During the test sessions, four randomly selected arms were baited with 1 pellet of food each; the same arms were baited throughout the experiment. The mouse was allowed to move freely in the maze until it had collected the 4 food pellets or until 10 min had passed, whichever occurred first. Three parameters were evaluated: (1) re-entry into baited arms that had been visited during the session (working memory errors); (2) entries into unbaited arms (reference memory errors); (3) total arm entries needed for to collect all pellets in the maze. The task was considered learned when the working memory error was zero, and the average reference memory error was one or less than one in three successive sessions (Olton, 1987). Behavioral data were analyzed by two-way ANOVA (between factor, group; within factor, trial) followed by SNK test.
Small animal positron emission tomography
Positron emission tomography (PET) was performed on a 32-module quadHIDAC scanner (Oxford Positron Systems) dedicated to small animal imaging. The scanner has an effective resolution of 0.7 mm (full-width at half-maximum) in the transaxial and axial directions when using an iterative resolution recovery reconstruction algorithm (Schäfers et al., 2005).
Animals were anesthetized using isoflurane and 10 MBq of 18F-Fluordeoxyglucose (18F-FDG) in 100 μl saline were injected intravenously. After a 30 min interval, animals were placed in the PET scanner, and list mode data were acquired for 15 min. While being anesthetized, animals were placed on a heating pad to maintain normal body temperature and vital signs (heart rate, breathing, body temperature) were monitored. Acquisition data were reconstructed into a single-image volume with voxel size of 0.4 × 0.4 × 0.4 mm3.
Immunohistochemistry
Serial sagittal sections were cut (10 μm) from cryo-conserved hemispheres (Leica Cryostat CM 3050S), embedded in tissue-freezing medium (Jung/Leica Microsystems; #0201-08926) and mounted (Microscope Slides; #K0123b). After drying for 30 min at ambient temperature, for fixation slides were incubated in 4% paraformaldehyde (Roti Histofix; 4% #P087.4, Roth) for 20 min. Blocking of nonspecific binding was achieved by 1 h incubation in 5% normal goat serum (Linaris; #S-1000). Between the steps, slides were washed three times for 5 min in PBST. Immunostaining was performed overnight by incubation at 4°C with the following primary antibodies: (1) pAb rabbit-anti-glial fibrillary acidic protein (1:900 in 2% normal goat serum in PBST; DAKO; Z0334); (2) rat mAb #MCA 711 against murine CD11b (CD11b, 1:250; Serotec); and (3) mouse mAb #MAB 377 against neuronal nuclei (NeuN; 1:500; Millipore Bioscience Research Reagents). Afterward, slides were incubated with Alexa-Fluor 594 goat-anti-rat secondary antibodies hosted in goat for 1 h (1:800 in PBST; Invitrogen #A11036 and #A11020). Again, slides were washed with PBST between the steps. Finally, the slides were coverslipped in Mowiol 4-88 (Calbiochem/VWR #475904) and stored at −20°C in the dark until microscopy was performed. Double immunostaining for iNOS and CD11b was performed on cryofixed sections as described above. Sections were dried at ambient temperature for 1 h and then fixed in 4% PFA or methanol for 15 min at ambient temperature. After washing with PBS, the double staining was performed by adding first antibodies followed by overnight incubation at 4°C. The following antibodies were used: rat mAb #MCA 711 against murine CD11b (1:250; Serotec) rabbit pAb against iNOS, 32030 (1:150; Transduction Laboratories).
The goat secondary antibodies (dichlorotriazinylaminofluorescein-conjugated anti-rabbit 1:150; Texas Red-conjugated anti-rat 1:80; Jackson Immuno Research Laboratories) were applied sequentially after washing in PBS. Negative controls included nonspecific IgG instead of primary antibodies, preabsorption with respective cognate peptides (150–200 μg of peptide/ml of antibody-working solution), and omission of the secondary antibody.
Confocal laser-scanning microscopy
Double-labeled specimens were analyzed with a confocal laser-scanning microscope (Multiprobe 2001; Invitrogen) equipped with an Ar/Kr laser with balanced emission at 488, 568, and 647 nm. Images were acquired at using a 40× objective. To achieve an optimal signal-to-noise ratio for each fluorophore, sequential scanning with 568 and 488 nm was used. Images were processed using ImageSpace 3.10 software (Invitrogen) on a Silicon Graphics power series 310GTX work station. Original section series were subjected to Gaussian filtration to reduce noise and enhance weakly but specifically labeled parts. Original and filtered sections were projected on one plane using a maximum-intensity algorithm and in some cases using depth-coding and surface-rendering algorithms.
Quantification of immunohistochemistry
For quantitative image analysis of hippocampal immunostaining, serial sagittal sections of one hemisphere were collected (lateral position +0.5 to +2.25 from bregma). GFAP, CD11b, and NeuN immunostaining were evaluated on sagittal brain sections of six animals from each group. For each animal, antigens were detected in 10 parallel sections having a distance of 70 mm to each other and showing both the hippocampus and cortex. In each section, staining in the hippocampus and the frontal cortex were evaluated. The stained area was determined and given as percentage of the respective brain region. All images were acquired on BX-61 microscope (Olympus), equipped with a digital camera (F-View II; Olympus), with identical exposure time. Image analysis was performed using Cell P (Olympus). For each animal, average values from all sections were determined. Data were analyzed by one-way ANOVA and Tukey's post hoc tests using SYSTAT software (Systat).
Real-time PCR
Neocortical, hippocampal, and cerebellar tissue was dissected from one hemisphere on a cold glass plate. RNA was extracted from these tissues using Trizol (Life Technologies Invitrogen). Total RNA was quantified spectrophotometrically and reverse transcribed using the RevertAid First Strand cDNA Synthesis kit (Fermentas) according to the manufacturer's instructions. PCR was performed as described previously (Heneka et al., 2002) using the following primers: TNFα forward 5-GCA CAG AAA GCA TGA TCC GC-3 and TNFα reverse 5-TGT CTT TGA GAT CCA TGC CG-3, Il-1β forward 5-CCTGTGTAATGAAAGACGGC-3 and Il-1β reverse 5-AAGGGA GCTCCTTCACA TGC-3, RANTES forward 5-GTG CCC ACG TCA AGG AGT ATT TCT-3 and RANTES reverse 5-GAC CGA GTG GGA GTA GGG GAT TAC-3, GFAP forward 5-TCC GCG GCA CGA ACG AGT C-3 and GFAP reverse 5-CAC CAT CCC GCA TCT CCA CAG TCT-3, GAPDH forward 5-TCA CCA GGG CTG CCA TTT GC-3 and GAPDH reverse 5-GAC TCC ACG ACA TAC TCA GC-3. PCR was performed on cDNA generated from individual animals in each group. PCR conditions were 35 cycles of denaturation at 95°C for 30 s, annealing at 63°C for 45 s, and extension at 72°C for 45 s using a thermal cycler (Biometra Personal T; Biometra). PCR products were separated by agarose gel electrophoresis and detected using an AlphaInotech imaging system, and band intensities determined using ImageJ. Data were analyzed by ANOVA with Tukey's post hoc test (Systat).
Synaptosomal preparation
Whole-brain hemispheres were homogenized in 9 volumes of 50 mm Tris-acetate, pH 7.4, 5 mm NaF, 2 mm Na3VO4, 5 mm Na4P2O7, 1 mm PMSF, protease inhibitor mixture (1:500; Sigma) by 15 slow up-and-down strokes using a Potter-Evehjem homogenizer at 700 rpm (Fisher Scientific; clearance 0.15 mm). Fractions of 1 ml were centrifuged in 1.5 ml tubes at 800 × g for 5 min. The supernatant was centrifuged at 16,000 × g for 20 min. The resulting pellet was resuspended in 300 ml homogenization buffer and loaded on a gradient consisting of 300 ml 1.4 m sucrose and 500 ml 1.0 m sucrose. The gradient was centrifuged at 9100 g for 10 min without brake. The synaptosomes were collected from the 1.4 m sucrose layer with a Pasteur pipette. Synaptosomes were suspended in 0.4 m sucrose, pelleted at 16,000 × g for 20 min and resuspended in RIPA for protein determination and Western blot.
Western blotting
Synaptosomal proteins (20 μg) were separated in 4–12% NuPAGE gels, transferred to Immobilon-P polyvinylidene fluoride membranes and incubated with antibodies directed against a set of presynaptic and postsynaptic protein targets including synaptotagmin (Sigma; S2177, 1:500), synaptobrevin (Synaptic Systems; #104211, 1:500), munc-18 (BD Biosciences; #610336, 1:500), PSD-95 (Cell Signaling Technologies; #2507, 1:1000), synaptophysin (Millipore Bioscience Research Reagents; #MAB5258, 1:500), and CaMKII (Santa Cruz; #sc-9035, 1:500). To visualize immunoreactions, blots were incubated with ECL reagent (GE Healthcare, Pharmacia), and digital images were obtained with the ChemiDoc System (Bio-Rad). Signal intensities were determined using ImageJ. Data were analyzed by ANOVA and Tukey's post hoc tests (Systat).
Results
Peripheral administration of LPS at 3 months rapidly lead to signs of sickness behavior including reduced exploration, grooming, decreased motor activity, and anhedonia without an obvious difference between NOS2−/− and wild-type mice. In line with this, two animals in each group (wild type and NOS2−/−) died in response to LPS challenge within the first 24 h (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material); thereafter, no further deaths occurred, and all animals survived until the end of the experiment. LPS injection caused a sustained increase of NOS2 expression which is detectable in the hippocampus and frontal cortex of wild-type mice after 2 months (Fig. 1B).
When compared with vehicle injection, LPS did significantly increase NOS2 expression at 1 and 8 weeks after the initial immune challenge (Fig. 1C). Immunohistologically, the majority of the NOS2-positive cells were found in close vicinity to blood vessels at both time points (Fig. 1D). Double immunostaining and subsequent confocal analysis revealed that most NOS2-positive cells were also immunoreactive for CD11b, suggesting that NOS2 was expressed by cells from the myeloid lineage and most likely by microglia or invaded macrophages (Fig. 1D).
Since previous observations (Semmler et al., 2005, 2007) suggested a role of NOS2 expression in the brain's response to endotoxemia, we aimed to determine whether nitric oxide is involved in LPS causing long-term impairment of memory and cognition after a single LPS challenge. Thus, wild-type and NOS2−/− mice that had previously been challenged with LPS or control vehicle were analyzed at 2 month by first open-field analysis and afterward by radial arm maze test.
Open-field testing assessed general motor behavior of the mice including inactivity time, corner time, center time as well as rearing, grooming, urination, and defecation. While there was a general tendency of the NOS2−/− mice to spend less time in the corner and more in the center, this phenomenon did not reach the level of statistical significance and was not influenced by LPS administration. Likewise, all other parameters did not show differences (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
For the eight-arm maze test, the behavioral parameters included the total number of trials needed to complete the task, working, and reference memory errors. Wild-type mice and NOS2−/− mice did not show any differences when treated with control vehicle. LPS treatment resulted in a worse performance on all behavioral measures examined in wild-type mice, which was not observed in NOS2−/− animals (Fig. 2). This result suggests that NOS2-derived nitric oxide generation induced by peripheral administration of LPS contributes to sustained memory dysfunction.

LPS-induced long-term memory impairment. Radial arm maze test: wild-type mice (wt) challenged with LPS (wt+LPS) needed more trials and committed more errors during the test compared to NOS2−/− (NOS2−/− +LPS). A, Total trials (mean ± SEM, n = 10–12, ANOVA, F = 5.73, Tukey post hoc analysis, *p < 0.05; ***p < 0.001 wt+LPS vs wt, NOS2−/−, NOS2−/− +LPS). B, Working memory errors (mean ± SEM, n = 10–12, ANOVA, F = 3.56, Tukey post hoc analysis, *p < 0.05 wt+LPS vs wt, NOS2−/−, NOS2−/− +LPS). C, Reference memory errors (mean ± SEM, n = 10–12, ANOVA, F = 10.80, Tukey post hoc analysis, ***p < 0.001 wt+LPS vs wt, NOS2−/−, NOS2−/− +LPS).
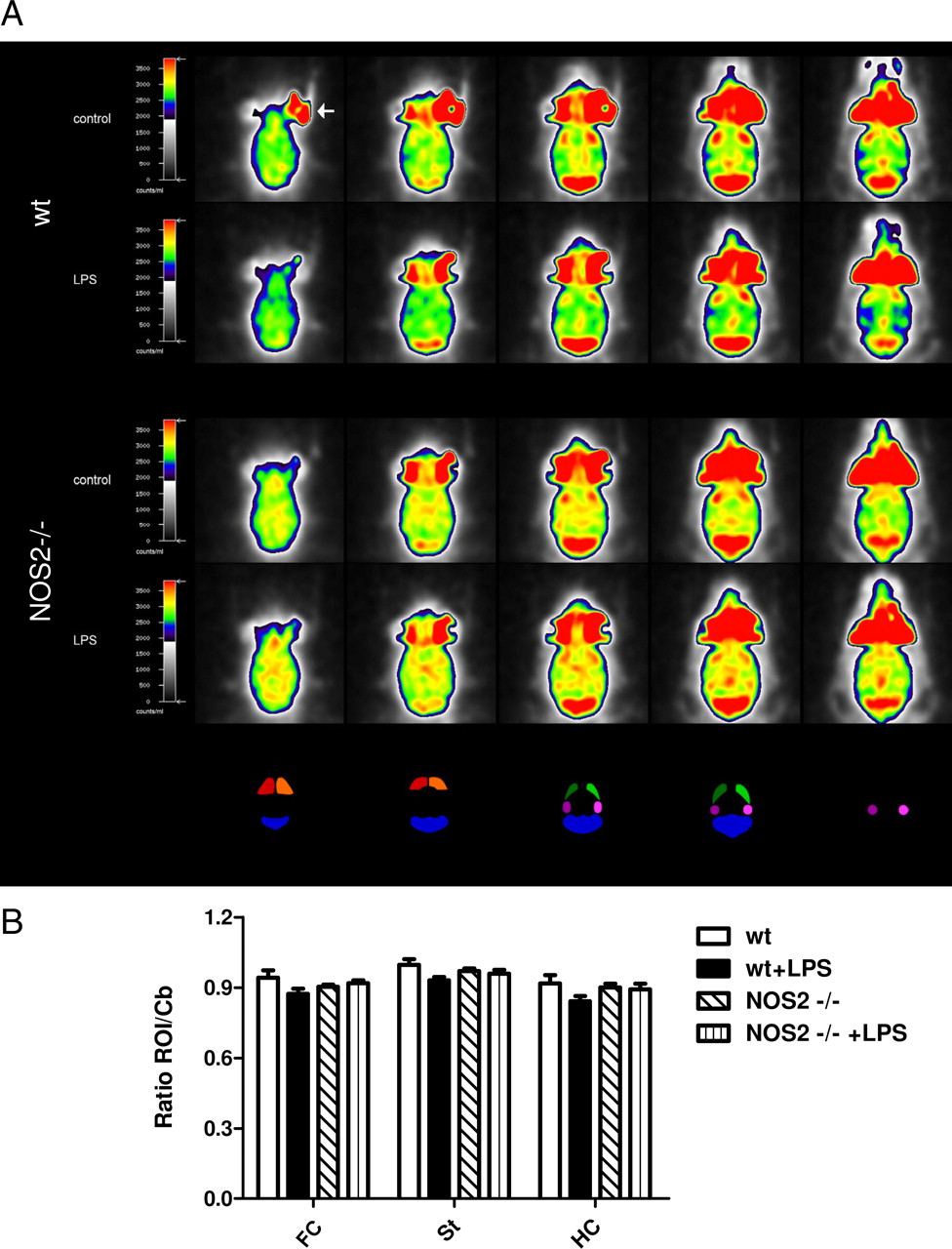
Since we have previously shown that acute sepsis can affect cerebral glucose utilization (Semmler et al., 2008), 18F-FDG–PET analysis was performed in the hippocampus, cortex, and striatum to determine whether the observed behavioral changes would be accompanied by alterations of cerebral glucose uptake (Fig. 3). Although there was a consistent tendency of LPS to reduce cerebral glucose utilization in all brain areas evaluated, this did not reach the level of statistical significance (p < 0.058, ANOVA followed by Tukey test). Of note, there was no such tendency in NOS2−/− mice.

Analysis of cerebral glucose uptake 2 months after sepsis. A, Shown are five representative transversal 18F-FDG–PET brain slices of wild-type and NOS2−/− mice treated with vehicle or LPS. Corresponding region-of-interest (ROI) masks are displayed below. B, Quantification of 18F-FDG uptake (relative to the cerebellum) did not reveal any significant differences between groups in all brain areas examined including the frontal cortex (FC), the striatum (St), and hippocampus (HC). The arrow exemplarily indicates noncerebral uptake of 18F-FDG in retro-orbital Harderian glands routinely seen with all PET studies (mean ± SEM, n = 5).
To assess the degree of morphological changes in the hippocampus CD11b, GFAP, and NeuN was immunohistochemically assessed in serial sections with a defined intersection distance. At 2 months after sepsis, there were no changes in the area fractions of NeuN or GFAP immunopositive stainings (Fig. 4B) nor in the number of NeuN- or GFAP-positive cells (data not shown), suggesting that LPS-induced sepsis did not result in overt neuronal loss or sustained astroglial activation. However, LPS treatment showed a sustained increase in CD11b immunoreactivity in wild-type mice, which was not observed in NOS2−/− mice (Fig. 4A,B), indicating that NOS2-derived nitric oxide contributes to the neuroinflammatory reaction and long-term microglial changes after peripheral LPS administration.

LPS induced a sustained activation of microglia and inflammatory gene transcription 2 months after sepsis. A, CD11b immunohistochemistry demonstrates sustained and focal microglial activation in the frontal cortex in response to LPS administration (mean ± SEM, n = 10, ANOVA, F = 8.71, Tukey post hoc analysis, *p < 0.05). B, Quantification of NeuN and GFAP immunostaining 2 months after sepsis (mean ± SEM, n = 10, ANOVA, F = 2.98 for GFAP and F = 1.3 for NeuN). C, RT-PCR analysis of TNFα and Il-1β transcripts in wild-type (wt) and NOS2−/− mice in the frontal cortex (FC), hippocampus (HC), and the cerebellum (Cb) (mean ± SEM, n = 5, ANOVA, F = 16.13 for FC, F = 3.67 for HC and F = 6.62 for CB, Tukey post hoc analysis, *p < 0.01 wt or NOS2−/− vs wt+LPS, ##p < 0.01 NOS2−/− vs NOS2−/− + LPS). D, RT-PCR analysis of Il-1β transcripts in wt and NOS2−/− mice in the FC, HC, and the Cb (mean ± SEM, n = 5, ANOVA, F = 7.36 for FC, F = 1.72 for HC and F = 0.43 for CB, Tukey post hoc analysis, *p < 0.01 wt or NOS2−/− vs wt+LPS, ##p < 0.01 NOS2−/− vs NOS2−/− + LPS).
To further characterize this chronic microglia activation, we determined mRNA levels of various cytokines and chemokines including TNF-α, IL-1β (Fig. 4C,D), RANTES, and GFAP (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) in the frontal cortex, the hippocampus, and the cerebellum. LPS challenge of wild-type mice caused a significant increase in mRNA levels of TNF-α, IL-1β, RANTES in the cortex and in tendency in the hippocampus, too (Fig. 4C,D). LPS also lead to a significant increase of TNF-α mRNA in the cerebellum. Importantly, NOS2−/− showed a pronounced decrease of TNF-α, IL-1β, RANTES mRNA levels in the cortex, hippocampus and cerebellum, indicating the NOS2-dependent NO generation is involved in inflammatory gene regulation or alternatively in neuronal or glial damage that accounted for the sustained neuroinflammatory reaction (Fig. 4C; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Supporting the immunohistochemical results, GFAP did not show any significant change between groups and treatments in all brain areas examined. Importantly, we did not find any difference in hippocampal or cortical mRNA levels of TNF-α and IL-1β at 24 h after LPS injection (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), suggesting that in both animal groups, NOS2−/− and wild-type mice, these brain regions were similarly affected by the peripheral LPS administration.
Because the immunohistochemical results suggested that neuronal loss does not account for the observed behavioral deficits in response to LPS, we analyzed changes of a number of presynaptic and postsynaptic proteins, known to be critical for spatial memory formation. LPS treatment of wild-type mice increased the synaptotagmin protein levels in synaptosomal lysates, while the same stimulus significantly reduced PSD-95, synaptophysin, munc-18, and CaMKII. LPS-challenged NOS2−/− mice were protected from LPS-induced reductions of synaptotagmin, synaptophysin, munc-18, and CaMKII, when compared to NOS2−/− controls. Importantly, the LPS-induced decrease of PSD-95 was more pronounced in wild-type than in NOS2−/− mice (Fig. 5). Altogether, these data suggest that a single LPS exposure leads to distinct changes at the synaptic level which is detectable 2 months afterward. Since NOS2 deficiency provides protection from these synaptic alterations, it is likely that nitric oxide is a mediator of LPS-induced synaptic changes.

Alterations of synaptic key proteins in wild-type and NOS2−/− mice at 2 months after sepsis. A, Synaptosomal preparations from the whole-brain hemispheres if individual mice were separated by SDS gel electrophoresis and immunoblotted for the synaptic proteins synaptotagmin, synaptobrevin, munc-18, PSD-95, synaptophysin and CaMKII. B, Densitometrical analysis of the immunoblots from A (n = 4, mean ± SEM, ANOVA, F = 19.06 for synaptotagmin, F = 10.77 for synaptobrevin, F = 10.5 for munc18, F = 119.0 for PSD-95, F = 12.14 for synaptophysin, F = 18.68 for CaMKII, Tukey post hoc analysis *p < 0.05, **p < 0.01, ***p < 0.001).
Discussion
Sepsis-induced encephalopathy (SE) may lead to cognitive deficits which can persist for months or years (Hopkins et al., 1999; Rothenhäusler et al., 2001; Hopkins and Jackson, 2006). Among several pathogenic mechanisms, circulation of bacterial cell wall components, peripheral release of inflammatory mediators as well as the stimulation of innate immune mechanisms within the brain are considered to contribute to SE. Additionally, the negative impact of peripheral organ failure in response to sepsis is likely to affect brain function.
In rodent models, sepsis induced by peripheral injection of LPS has been shown to activate microglial cells (Buttini et al., 1996) and thus mimics the microglia activation observed in brains from patients who died from sepsis (Lemstra et al., 2007). LPS injection can therefore be used to model the effect of a defined concentration of this immunostimulant and the subsequent induction of neuroinflammatory events. In the present study, a single injection of a high dose of LPS caused sustained microglial activation, which was detectable up to 8 weeks after the initial immune challenge. Similarly, persistent neuroinflammation has been described in a model of chronic intracerebroventricular LPS infusion in rats, where microglia activation was observed at 37 d after cessation of LPS exposure (Hauss-Wegrzyniak et al., 2000). Although there are important differences between both models, these findings collectively support the hypothesis that LPS can lead to a long-lasting activation of the innate immune system, independently of the route of exposure. Importantly, this neuroinflammatory reaction was accompanied by a significant increase of NOS2 expression, predominantly located in the vicinity of blood vessels.
LPS administration in rodents leads rapidly to behavioral changes characterized by a reduction of food and water intake, less social interactions, decreased locomotor activity and exploration, summarized as “sickness behavior.” In the present study, there was no obvious difference between the experimental groups regarding this phenomenon. However, this initial sickness behavior was not quantified, and a more detailed analysis of this very initial phase may have detected more subtle changes. Nevertheless, the number of LPS-induced deaths, as well as the transcription of two key cytokines, TNF-α and IL-1β in the hippocampus and frontal cortex, was not different between NOS2-deficient and wild-type mice. Several studies suggest that peripheral LPS administration also causes learning and memory impairment in mice (Arai et al., 2001; Sparkman et al., 2005). While in these studies the LPS administration was performed daily just hours before the behavioral tests, a further study found that a single injection of LPS was sufficient to cause persistent spatial memory deficits which were detectable over the entire test period of 11 d (Shaw et al., 2001). Extending these observations, the present study investigates locomotion and spatial memory changes at 2 months after LPS treatment, a time point where no symptoms of sickness behavior were detectable anymore. Mice immunochallenged with LPS showed impaired spatial memory performance in the eight-arm radial maze as compared to vehicle-treated controls. Based on the observation that LPS administration caused sustained induction of NOS2 expression in the brain and the previously reported in vivo detection of nitric oxide by electron paramagnetic resonance in the brain of LPS-treated rats (Suzuki et al., 1998), we used NOS2-deficient mice to address the functional role of inflammation-induced nitric oxide generation. Importantly, NOS2 deficiency protected mice from LPS-induced memory dysfunction, suggesting that NOS2-derived nitric oxide contributes to neurocognitive changes induced by inflammation. This assumption is further supported by the observation that inhibition of NOS2 by aminoguanidine attenuated learning and memory deficits in a four-vessel occlusion model of cerebral ischemia (Mori et al., 2001). In line with this, it has been shown that hippocampal LTP, a major indicator of synaptic efficacy underlying learning and memory formation (Malenka and Nicoll, 1999; Malenka and Bear, 2004), was reduced by LPS in vitro and in vivo (Cunningham et al., 1996; Commins et al., 2001; Jo et al., 2001). Importantly, inhibition of NOS2 has been found to prevent inflammatory suppression of LTP in similar models (Mori et al., 2001; Togashi et al., 2001; Wang et al., 2004). Together, these data suggest that inflammation-induced, NOS2-derived nitric oxide can cause learning and memory dysfunction.
Immunostimulated nitric oxide production has been shown to exert detrimental effects by a variety of mechanisms including the induction of apoptosis, inhibition of mitochondrial respiration, protein nitrosylation, and potentiation of excitotoxicity (Hewett et al., 1994; Nicotera et al., 1999; Moncada and Bolaños, 2006; Brown, 2007; Calabrese et al., 2007). More specifically, nitric oxide has been shown to affect neighboring cell survival through DNA damage, poly-ADP ribose synthetase activation, and subsequent energy depletion (Zhang et al., 1994). Furthermore, NO is able to cause p53 accumulation (Forrester et al., 1996) and mediates the release of apoptosis-inducing factor evoked by systemic LPS administration (Czapski et al., 2007). The effects of nitric oxide and reactive nitrogen species on mitochondrial function are complex and include the reversible inhibition of cytochrome oxidase inhibition of complex I and II, the induction of mitochondrial permeability transition, and production of radical oxygen species from mitochondria (Brown and Borutaite, 2004). Using a higher concentration of LPS, we previously observed neuronal cell death within the hippocampus (Semmler et al., 2005, 2007). However, we did not detect neuronal loss which could account for the observed behavioral phenotype. In keeping with this, no significant change of cortical glucose utilization was detectable at 2 months after LPS administration. This further substantiates that neither overt neuronal loss nor altered neuronal metabolism mediates the impaired learning and memory behavior in response to LPS treatment.
In contrast, analysis of the brain innate immune system revealed a subtle but sustained activation of microglia that was detected in wild-type mice but not in NOS2-deficient animals and was paralleled by a distinct regulation of cytokine mRNA levels, including those of TNFα and IL-1β mRNAs. It remains unclear whether nitric oxide directly affected the transcription of the TNFα and IL-1β genes as previously described (Kröncke, 2003) or caused cellular damage which in turn accounted for the sustained activation of microglia and cytokine transcription. Given the reported negative impact of TNFα and IL-1β on learning and memory function, as well as on LTP (Tancredi et al., 1992; Oitzl et al., 1993; Gibertini et al., 1995), the reduced levels of these cytokines in NOS2-deficient mice may partly explain the observed protection. As a further possibility, NOS2 deficiency has been shown to generally protect from septic shock and organ failure (Wei et al., 1995), and thus, a reduced level of peripheral toxins and cytokines could also contribute to neuroprotection.
Based on the many findings that synapses are particularly vulnerable in neurodegenerative conditions, we next studied the effects of LPS and NOS2 deficiency on the synaptosome and more precisely on the regulation of synaptic proteins, shown to have key functions for learning and memory. LPS induced distinct alterations at the synaptic protein level, and more specifically a significant reduction of PSD-95, CaMKII, and synaptophysin may also contribute to the observed memory deficits, given the role of these proteins in synaptic plasticity (Silva et al., 1992; O'Brien et al., 1998; Janz et al., 1999; Fukunaga and Miyamoto, 2000). Interestingly, a previous in vitro study found that microglial-derived nitric oxide impairs the anterograde axonal transport of synaptophysin, a mechanism which may be involved in the significant reduction of synaptophysin observed in the present study (Stagi et al., 2005).
The reduction of PSD-95 is of particular importance, since this protein is a central player at the postsynaptic density by scaffolding specialized large membrane complexes consisting in the case of PSD-95 of NMDA, neuroligin, and β-adrenergic receptors. In line with the findings presented in this study, mice lacking PSD-95 show a severely impaired spatial memory performance (Migaud et al., 1998; Kim and Sheng, 2004).
Importantly, NOS2 deficiency protected from these synaptic changes. It remains unclear by which molecular mechanisms nitric oxide causes the observed synaptic alterations and moreover if those are ultimately responsible for the observed phenotype. It can, however, be speculated that NOS2 deficiency protects from synaptic alterations by interfering with more than one detrimental pathomechanism.
Together, this study highlights the impact of severe sepsis on brain function and reveals that sepsis can cause persistent learning and memory deficits in mice. Sustained microglial activation may represent a valuable therapeutic target in patients suffering from severe sepsis. To exclude compensatory responses due to NOS2 deficiency and to test a potential therapeutic benefit, future studies should include treatment protocols with NOS2-selective inhibitors. In particular, inflammatory-induced and nitric oxide-mediated alterations of synapses should be further investigated to unravel the underlying molecular mechanisms, since specific protection of synapses from immunostimulated changes may prove to be beneficial.
Footnotes
-
This study was supported by a grant from the Deutsche Forschungsgemeinschaft to M.T.H. (HE 3350/4-1, 4-2) and the Interdisciplinary Centre for Clinical Research Münster, core unit Small Animal PET (S.H., M.S.).
- Correspondence should be addressed to Dr. Michael T. Heneka, Department of Neurology, Clinical Neuroscience, Sigmund-Freud-Strasse 33, 53127 Bonn, Germany. michael.heneka{at}ukb.uni-bonn.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}