

Abstract
Spinal and bulbar muscular atrophy (SBMA) is an inherited neuromuscular disorder caused by a polyglutamine (polyQ) repeat expansion in the androgen receptor (AR). PolyQ-AR neurotoxicity may involve generation of an N-terminal truncation fragment, as such peptides occur in SBMA patients and mouse models. To elucidate the basis of SBMA, we expressed N-terminal truncated AR in motor neuron-derived cells and primary cortical neurons. Accumulation of polyQ-AR truncation fragments in the cytosol resulted in neurodegeneration and apoptotic, caspase-dependent cell death. Using primary neurons from mice transgenic or deficient for apoptosis-related genes, we determined that polyQ-AR apoptotic activation is fully dependent on Bax. Jun N-terminal kinase (JNK) was required for apoptotic pathway activation through phosphorylation of c-Jun. Expression of polyQ-AR in DP5/Hrk null neurons yielded significant protection against apoptotic activation, but absence of Bim did not provide protection, apparently due to compensatory upregulation of DP5/Hrk or other BH3-only proteins. Misfolded AR protein in the cytosol thus initiates a cascade of events beginning with JNK and culminating in Bax-dependent, intrinsic pathway activation, mediated in part by DP5/Hrk. As apoptotic mediators are candidates for toxic fragment generation and other cellular processes linked to neuron dysfunction, delineation of the apoptotic activation pathway induced by polyQ-expanded AR may shed light on the pathogenic cascade in SBMA and other motor neuron diseases.
Introduction
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease, is an X-linked disorder characterized by progressive degeneration of motor neurons in the spinal cord and brainstem (Kennedy et al., 1968). The cause of SBMA is a trinucleotide (CAG) repeat expansion in the androgen receptor (AR) gene (La Spada et al., 1991). At least eight other disorders, including Huntington's disease (HD), dentatorubral-pallidoluysian atrophy, and six forms of spinocerebellar ataxia (SCA), result from this type of mutation (Zoghbi and Orr, 2000; Nakamura et al., 2001). SBMA is unique among the polyQ diseases because expression of the disease gene alone is not sufficient to produce the phenotype. Transgenic models of SBMA strongly suggest that females are protected by low levels of androgen (Katsuno et al., 2002; Chevalier-Larsen et al., 2004; Sopher et al., 2004), making SBMA a sex-limited disorder. Together with results from Drosophila and in vitro studies, it appears that polyQ AR neurotoxicity requires ligand binding (Darrington et al., 2002; Takeyama et al., 2002; Walcott and Merry, 2002; Pandey et al., 2007). When androgen binds to its receptor, AR is released from protein chaperones, dimerizes, and translocates to the nucleus where it binds DNA and activates gene expression. An androgen antagonist that blocks AR-mediated gene expression, but fails to prevent nuclear translocation, cannot prevent disease in fly and mouse models of SBMA (Katsuno et al., 2002; Takeyama et al., 2002). Thus, nuclear localization of polyQ-AR is likely a critical step in the pathogenic process (La Spada and Taylor, 2003; Sopher et al., 2004).
Several polyQ-expanded proteins undergo site-specific proteolysis that promotes the toxicity of the mutant protein (Miyashita et al., 1997; Kobayashi et al., 1998; Wellington et al., 1998). The resulting proteolytic fragments are observed in affected patients and mouse models of polyQ disease (Paulson et al., 1997; Li et al., 1998a; Schilling et al., 1999a,b; Garden et al., 2002). Such proteolytic fragments produce significant cellular toxicity in model systems, under conditions where full-length polyQ protein has little impact (Ikeda et al., 1996; Cooper et al., 1998; Ellerby et al., 1999a). PolyQ-expanded AR undergoes proteolytic modification in vitro, and polyQ-expanded N-terminal AR fragments are toxic to tissue culture cells, including neuronal cells (Kobayashi et al., 1998; Merry et al., 1998; Ellerby et al., 1999b). Furthermore, numerous studies of polyQ disease proteins have implicated caspase cleavage in the pathogenic cascade (Goldberg et al., 1996; Kobayashi et al., 1998; Wellington et al., 1998; Ellerby et al., 1999a,b). In HD, polyQ-huntingtin with a putative caspase-6 cleavage site mutation is incapable of causing neurotoxicity in transgenic mice (Graham et al., 2006). Thus, while a role for apoptotic mediators in polyQ disease pathogenesis seems likely, the cascade of molecular events leading to apoptotic activation in the various polyQ diseases remains unknown.
Here we define the pathway by which polyQ-AR elicits apoptotic activation in motor neuron-like cells and primary neurons. We show that neuronal expression of N-terminal truncated polyQ-AR elicits intrinsic pathway-mediated neuronal apoptosis, initiated by Jun N-terminal kinase (JNK) and culminating in Bax activation. The c-Jun-N-terminal kinases (JNKs) are a pro-apoptotic subgroup of mitogen-activated protein kinases that mediate neuronal apoptosis in response to a variety of stimuli, including activation of the unfolded protein response and proteasome impairment, both of which have been implicated as consequences of polyQ toxicity (Nishitoh et al., 2002; Morfini et al., 2006). A principal target of JNK is the transcription factor c-Jun, which is activated by phosphorylation within its transactivation domain, leading to increased expression of target genes that initiate the apoptotic cascade. Two BH3-only proteins, Bim and DP5/Hrk, are activated downstream of JNK and c-Jun in the initiation of neuronal apoptosis (Inohara et al., 1997; Imaizumi et al., 1999; Harris and Johnson, 2001; Putcha et al., 2001; Whitfield et al., 2001; Imaizumi et al., 2004), and one of these BH3-only proteins, DP5/Hrk, has been implicated in motor neuron degeneration (Shinoe et al., 2001; Imaizumi et al., 2004). In this study, we demonstrate, for the first time, a role for DP5/Hrk-mediated, Bax-dependent intrinsic pathway apoptotic activation, initiated by JNK, in the polyQ motor neuron disease SBMA.
Materials and Methods
Cell culture, primary neuron culture, and transfection.
The AR-QN expression constructs and huntingtin exon-1/2-QN expression constructs (where N is the number of CAG repeats) have been described previously (Saudou et al., 1998; Taylor et al., 2003). MN-1 cells were cultured in 75 cm2 vented tissue culture flasks at 37°C in a humidified atmosphere containing 5% CO2 in DMEM (Invitrogen BRL) supplemented with 10% fetal bovine serum (v/v), 100 U/ml penicillin/100 μg/ml streptomycin and 2 mm l-glutamine. For Western blot and caspase activation assays, cells were seeded in six-well plates at a density of 300,000 cells per well at 24 h before transfection. We transfected MN-1 cells with 1 μg of plasmid DNA using Lipofectamine Plus following the manufacturer's protocol (Invitrogen). Cortical neurons were cultured from postnatal day 0 pups. Briefly, pups were decapitated into Hanks Media w/o Ca2+ and Mg2+, and cortices were dissected in Neurobasal-A (NBA) media supplemented with B27 and l-glutamine. After dissection, cortices were trypsinized for 25 min at 37°C and dissociated. Cells were plated at a density of 2.5 × 105 per well in 4-well chamber CC2 slides (Nalge-Nunc). After 3 d, neurons were cultured in NBA media supplemented with B27 and l-glutamine, and were transfected using Lipofectamine 2000 following the manufacturer's protocol (Invitrogen). Use of B27 supplement, instead of serum, assures primary cultures consisting of at least 90% neurons. Primary neurons were subjected to taxol treatment by replacing media with taxol-containing media at a concentration of 250 nm. All experiments and animal care were performed in accordance with University of Washington IACUC guidelines.
Western blot analysis.
Protein lysates were obtained by homogenizing cortices in sample buffer [160 mm Tris-HCl (pH 6.9), 4% SDS, 200 mm dithiothreitol, 20% glycerol, 0.004% Bromophenol blue] at a ratio of 1:10–1:20 (w/v). MN-1 cells were harvested by scraping in ice-cold lysis buffer (68 mm sucrose, 200 mm mannitol, 50 mm KCl, 1 mm EGTA, 1 mm EDTA, 1 mm DTT and 1× Complete Protease Inhibitor (Boehringer Mannheim). Cell membranes were disrupted with 80 strokes of a Wheaton dounce homogenizer (B-type pestle) and centrifuged at 4°C at 800 × g. The nuclear pellets were lysed in a buffer consisting of 150 mm NaCl, 6 mm Na2HPO4, 4 mm NaH2PO4, 1.2 mm EDTA, 1% NaDOC, 0.5% Triton-X, 0.1% SDS, and Complete Protease Inhibitor. Primary cortical neurons were harvested in radioimmunoprecipitation assay lysis buffer (10 mm Tris, 0.1% SDS, 1% SDOC, 0.01% TX-100, 150 mm NaCl) and homogenized by passing 5× through a 26.5 gauge syringe. Protein lysates (50 μg) were run on a 10% Bis-Tris gel (Invitrogen) and transferred to PVDF membranes (Millipore) using a semidry transfer system (Invitrogen). The membranes were blocked with 0.05% Tween 20, Tris-buffered saline (T-TBS) containing 5% nonfat dried milk at 4°C overnight, and then probed with phospho-c-Jun antibody (Ser63) II (1:1000, Cell Signaling), anti-AR N20 (1:1000, Santa Cruz), or anti-PUMA antibody (1:1000, Cell Signaling) in T-TBS containing 5% nonfat dried milk at room temperature (RT) for 1 h. After washing, membranes were incubated with HRP-conjugated anti-rabbit IgG (1:2000, Santa Cruz) in T-TBS for 1 h at RT. Blots were stripped and reprobed with mouse anti-actin antibody (1:5000, (Millipore Bioscience Research Reagents/Millipore), washed, and incubated in anti-mouse-HRP antibody (GE Healthcare). After treatment with ECL chemi-luminescence (NEN), the membranes were visualized by autoradiography. Band intensities were calculated using NIH ImageJ freeware. All immunoblots were performed in duplicate or triplicate.
FACS-assisted cell viability assay.
At indicated time points, cells were harvested with trypsin, gently pelleted by centrifugation and resuspended in PBS with 1% serum on ice at a concentration 106/ml. The cells were stained with propidium iodide 1 μg/ml (PI, Sigma), gently vortexed, and incubated for 15 min at RT in the dark. 20,000 nongated events were acquired for each sample (Beckman Coulter FACS Calibur instrument; CellQuest software package for analysis). Results were expressed as a percentage of green fluorescent protein (GFP)/PI double-positive (nonviable, transfected) cells relative to total GFP positive (all transfected) cells.
Caspase assay.
At the indicated time points, cells were harvested and lysed in 300 μl of buffer consisting of 10 mm Tris pH 7.3, 10 mm NaH2PO4, 150 mm NaCl, and 1% Triton X-100 and stored at −80°C. Caspase-3 activity was determined by incubating 100 μg of lysate with 50 μm fluorogenic substrate Ac-DEVD–AFC (Biosource International) in a total volume of 200 μl of assay buffer (20 mm HEPES, pH 7.4, 100 mm NaCl, 1 mm EDTA, 0.2% CHAPS, 20% glycerol, 10 mm DTT) in the dark for 2 h at 37°C. Substrate cleavage was detected using Cytofluor II Fluorescence multi-well plate reader (Perspective Biosystems) with excitation and emission wavelengths of 420 and 520 nm, respectively.
DNA laddering assay.
To detect apoptosis-induced fragmentation of DNA, 106 cells were harvested and genomic DNA was extracted using the DNeasy Kit (Qiagen). Genomic DNA (500 ng) was ligated to 12-mer adapter oligonucleotides (ApoAlert LM-PCR Ladder Assay Kit, Clontech) as described in the manufacturer's protocol. Following ligation, samples were subjected to 15 – 20 cycles of PCR amplification (72°C for 8 min, followed by thermal cycling between 94°C for 1 min and 72°C for 3 min) using Advantage cDNA Polymerase Mix (Clontech). Reaction products were electrophoresed on a 1.5% agarose/ethidium bromide gel.
Immunocytochemistry and primary neuron toxicity assays.
Neurons were fixed with 4% paraformaldehyde (PFA) for 10 min at RT, washed with PBS, counterstained with Hoescht dye, and coverslipped. For the toxicity studies, cells were fixed in 4% PFA, permeabilized with 0.25% Triton X-100, blocked with Pro-Block, then incubated overnight in rabbit anti-active caspase-3 (1:100, Cell Signaling) and mouse-anti MAP2 (1:200, Millipore Bioscience Research Reagents). Cells were then incubated in anti-rabbit 594 (1:1000, Invitrogen) and anti-mouse 647 (1:1000, Invitrogen) and counterstained with DAPI. Note that although GFP and AlexaFluor 647 (Cy5) labeling both appear green on fluorescent images, different filters allow us to differentiate them with four-channel imaging. For quantification of c-Jun-P positive neurons, we fixed neurons at 18 h after transfection and incubated overnight with anti-PhosphoSer73-c-Jun (1:1000, Cell Signaling). JNK Inhibitor II (Cal Biochem) was reconstituted in DMSO and added to primary neuron culture media to yield a final concentration of 10 μm (Donovan et al., 2002), while JNK Inhibitor III (Cal Biochem) was reconstituted in DMSO and added to primary neuron culture media to yield a final concentration of 5 or 20 μm (Holzberg et al., 2003). JNK Inhibitor-containing media or DMSO-only containing media was used to replace primary neuron culture media, and active caspase-3 and MAP2 immunoreactivity were measured 24 h after media replacement. Fluorescence was analyzed on a Zeiss inverted fluorescence microscope and image analysis was done with SLIDEBOOK software. All experiments were done in triplicate or quadruplicate, and ≥50 neurons were quantified per condition in each run.
RNA interference constructs.
The lentiviral vector, pRRLsin-cppt-pgk-wpre-GFP (Add-gene plasmid #12252) was modified to express a short hairpin sequence targeting either the murine DP5 or Bim mRNA. Briefly, the WPRE promoter and GFP sequence were replaced with a CMV promoter and eCFP sequence from the plasmid pECFP-C1 (Clontech) by cloning into unique XcmI and SalI sites. The mouse U6 promoter and shRNA sequence were then PCR subcloned 5′ to the CMV/eCFP sequence.
Real-time RT-PCR.
Cells were harvested and total RNA purified (Qiagen). Quantification of DP5, Bim, or Puma RNA expression in primary cortical neurons or Neuro2a cells was done, using murine-specific TaqMan Assay-on-Demand primers and probe (ABI), and normalizing to eukaryotic 18S ribosomal RNA, according to the manufacturers' instructions (ABI). All experiments were done in triplicate.
Statistical analysis.
All errors bars shown in the Figures are SEM. All data were prepared for analysis with standard spreadsheet software (Microsoft Excel). Statistical analysis was done using Microsoft Excel or the VassarStats website (http://faculty.vassar.edu/lowry/VassarStats.html). For ANOVA analysis involving multiple sample comparisons, we performed post hoc testing to discriminate significance relationships.
Results
N-terminal truncation products occur in the cytosol in SBMA cells and motor neurons
We previously generated AR YAC CAG100 transgenic mice that strikingly recapitulate the late-onset, gender-dependent, neurogenic muscular atrophy phenotype of SBMA patients (Sopher et al., 2004). To determine whether an N-terminal AR truncation fragment is produced in this highly representative SBMA mouse model, we performed Western blot analysis on brain protein lysates obtained from presymptomatic AR YAC CAG100 transgenic mice and age-matched AR YAC CAG20 and nontransgenic controls. Immunoblotting with an anti-AR antibody, directed against the N terminus, revealed an ∼65 kDa truncation fragment in AR YAC CAG100 mice, but no truncation fragment was detected in AR YAC CAG20 mice (Fig. 1A). Fractionation of HEK293T cells expressing AR112 protein revealed that proteolytic cleavage of AR is ligand-dependent, and that an N-terminal fragment of ∼65 kDa is present in the cytosol as well as the nucleus (Fig. 1B). To establish whether the N-terminal truncated AR protein was expressed in motor neurons, we performed immunohistochemistry (IHC) upon lumbar spinal cord sections from SBMA AR100 mice, and detected AR protein in motor neuron perinuclear cytosol (Fig. 1C,D), and nuclei (Sopher et al., 2004), with an antibody that recognizes the N-terminal region of AR. Immunostaining with a C-terminally directed anti-AR antibody, however, did not reveal immunoreactivity in the perinuclear cytosol (data not shown). This observation is consistent with a recent study of a different SBMA mouse model in which insoluble, polyQ-expanded AR protein derived from brain extract consisted of a similarly sized N-terminal truncated AR protein (Li et al., 2007). In that study, the production of N-terminal AR truncation fragments correlated with neurological disease progression (Li et al., 2007).

SBMA transgenic mice express an N-terminal AR truncation product in the cytosol. A, Western blot analysis of cortex. Protein lysates from 12-month-old AR YAC CAG100 transgenic mice (AR100), nontransgenic littermates (NTg), and AR YAC CAG20 transgenic controls (AR20) were immunoblotted with AR antibody N20. Full-length AR100 and AR20 protein is detected (arrows). An ∼65 kDa truncation product (asterisk) is detected in the AR100 mice, but no truncation fragment is present in the AR20 mice. Arrowhead indicates a cross-reactive band that serves as a loading control. The 65 kDa N-terminal truncation fragment in AR YAC CAG100 mice is also present in spinal cord lysates (data not shown). B, Fractionation of AR truncation fragments. We transfected HEK293T cells with an AR112 expression construct and cultured the transfected cells in either the presence or absence of the AR ligand, dihydrotestosterone (DHT). Ligand binding elicited AR truncation that was more prominent in the cytosol (Cyto) than in the nucleus (Nucl), and arrowhead indicates an AR N-terminal truncation product of comparable size to the fragment detected in vivo. We confirmed integrity of the fractions by reprobing immunoblots with compartment-specific antibodies (data not shown). C, D, IHC analysis suggests accumulation of AR in perinuclear cytosol. Lumbar cord sections from 4-month-old nontransgenic mice (C) and AR YAC CAG100 transgenic mice (D) were stained with NeuN to label neurons (green; left), N-terminally directed anti-AR antibody N-20 (red; middle), and DAPI (blue). Perinuclear staining (arrows) is apparent in the merged images (right) of motor neurons in AR YAC CAG100 spinal cord sections (D), but not in nontransgenic controls (C). There was no perinuclear AR staining in AR YAC CAG20 transgenic mice (data not shown).
Truncated polyQ-expanded AR induces apoptosis in MN-1 cells and in primary neurons
In light of the evidence for the production and pathogenic nature of N-terminal truncated AR protein, we engineered AR expression constructs, terminating within a region of putative proteolytic cleavage (Ellerby et al., 1999b), to approximate the size of the N-terminal AR fragments observed in SBMA patient tissue and SBMA mice, and tagged them with GFP (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Transfection of N-terminal truncated AR expression constructs into the motor neuron-derived cell line, MN-1, yielded polyQ length-dependent toxicity (Fig. 2A). The toxicity induced by polyQ-expanded AR was readily observed by fluorescence microscopy, as cells transfected with polyQ-expanded AR became progressively rounded and ultimately lifted off the bottom of the flask. Examination of living cells, transfected with Nt-AR112-GFP and stained with Hoechst 33342, revealed numerous inclusion-containing cells with condensed, fragmented nuclei (data not shown). To verify that polyQ-expanded AR toxicity in culture was leading to apoptotic cell death, we examined transfected MN-1 cells for evidence of endonuclease-mediated laddering of chromosomal DNA and activation of caspase-3, two biochemical hallmarks of apoptotic cell death. MN-1 cells expressing Nt-AR112-GFP displayed DNA laddering that was first detectable 48 h after transfection, and peaked 96 h after transfection (Fig. 2B). Induction of DNA laddering did not occur after transfection of Nt-AR19-GFP, indicating that apoptotic activation was polyQ length-dependent (Fig. 2B). Apoptotic cell death is consummated in large part by activation of the caspase cascade. Caspase-3 is an executioner caspase whose ultimate activation may be elicited by a wide variety of toxic insults (Stennicke and Salvesen, 1998). Transfection of MN-1 cells with Nt-AR112-GFP yielded polyQ length-dependent activation of caspase-3, with a peak ∼72 h, correlating well with the time course of DNA laddering and loss of cell viability (Fig. 2C).

Expression of N-terminal polyQ AR fragment in MN-1 cells induces caspase-mediated apoptosis via intrinsic pathway. A, FACS-assisted cell viability assay demonstrates polyQ length-dependent cytotoxicity of N-terminal AR fragment (*p < 0.01; Student's t test). B, AR112 truncation fragment induces apoptotic nuclear fragmentation, reaching a peak at 96 h after transfection. Bax induces nuclear fragmentation with more rapid kinetics, peaking at 48 h. Untransfected (UT) cells and AR19-transfected cells do not show nuclear fragmentation. C, AR112 truncation fragment induces caspase-3 activation reaching a peak at 72 h after transfection (*p < 0.01; Student's t test). Bax induces caspase-3 activation with more rapid kinetics, peaking at 24 h. D, Fluorogenic substrate assay demonstrates activation of caspase-9 in AR112-transfected MN-1 cells peaking 72 h after transfection (*p < 0.01; Student's t test). Caspase-9 is activated in Bax-transfected cells peaking 24 h after transfection. E, Western blot analysis reveals prominent cleavage of pro-caspase-9 in AR112-transfected cells 72 h after transfection to yield considerable generation of active caspase-9. F, Dominant-negative caspase-9 expression rescues polyQ-length-dependent toxicity of N-terminal truncated AR112. MN-1 cell death, as measured by propidium iodide uptake, is significantly improved when dominant negative caspase-9 is cotransfected with the AR112 truncation fragment (*p < 0.01; Student's t test).
To explore the pathway upstream of caspase-3 activation, we measured the activity of two initiator caspases, caspase-8 and caspase-9, using specific fluorogenic substrates. Nt-AR112-GFP led to activation of caspase-9, but not caspase-8 (Fig. 2D; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Consistent with these observations, we found that the caspase-9 inhibitor zLEHD-fmk attenuated caspase-3 activation following transfection of MN-1 cells with Nt-AR112-GFP, but the caspase-8 inhibitor zIETD-fmk did not (data not shown). Activation of caspase-9 in the setting of truncated AR112 toxicity was further confirmed by Western blot analysis, which revealed that a 48 kDa pro-caspase-9 precursor was processed into the active caspase-9 fragment (Fig. 2E). When we coexpressed a dominant negative version of caspase-9 (Richter et al., 2001), we observed a marked reduction in toxicity in MN-1 cells expressing Nt-AR112-GFP (Fig. 2F), further confirming that caspase-9 was mediating AR polyQ length-dependent apoptotic cell death.
To examine the toxicity of N-terminal truncated polyQ-expanded AR fragment in postmitotic neurons, we derived primary mouse cortical neurons from neonatal C57BL/6J pups and, using standard lipofection, we transfected primary cortical neurons with Nt-AR19-GFP and Nt-AR112-GFP CMV expression constructs. GFP tagging allowed us to track the fate of individual cortical neurons expressing truncated AR protein products. Using this approach, we found that truncated AR112 accumulated in the perinuclear cytosol, while truncated AR19 was diffusely distributed throughout the cytosol and neurite processes (Fig. 3A,B). Cortical neurons expressing Nt-AR112-GFP demonstrated nuclear condensation, loss of microtubule-associated protein 2 (MAP2) staining, and caspase-3 activation, all by 24 h after transfection (Fig. 3B,C). These findings suggested a polyQ length-dependent induction of apoptotic cell death, as we found that nuclear morphological changes indicative of cell death correlated with caspase-3 activation for ∼98% of assayed neurons (data not shown). When we expressed full-length AR112 in primary cortical neurons, we did not observe appreciable neurite degeneration or apoptotic cell death at 24 h after transfection in the presence of dihydrotestosterone, an AR ligand that favors translocation into the nucleus (Fig. 3D). Hence, nuclear localization was observed, and although neurite degeneration and apoptotic cell death did ultimately occur at 60 h after transfection, this was only after polyQ-expanded AR had relocalized to the cytosol (data not shown). To confirm that localization to the cytosol is required for neurite degeneration and apoptotic cell death, we fused a nuclear localization signal (NLS) to the N terminus of the truncated AR coding sequence. Cortical neurons transfected with NLS-AR110 showed nuclear accumulation of AR protein, but did not display neurite degeneration or apoptotic cell death, unlike cortical neurons transfected with unmodified truncated AR112 expression constructs (Fig. 3E).
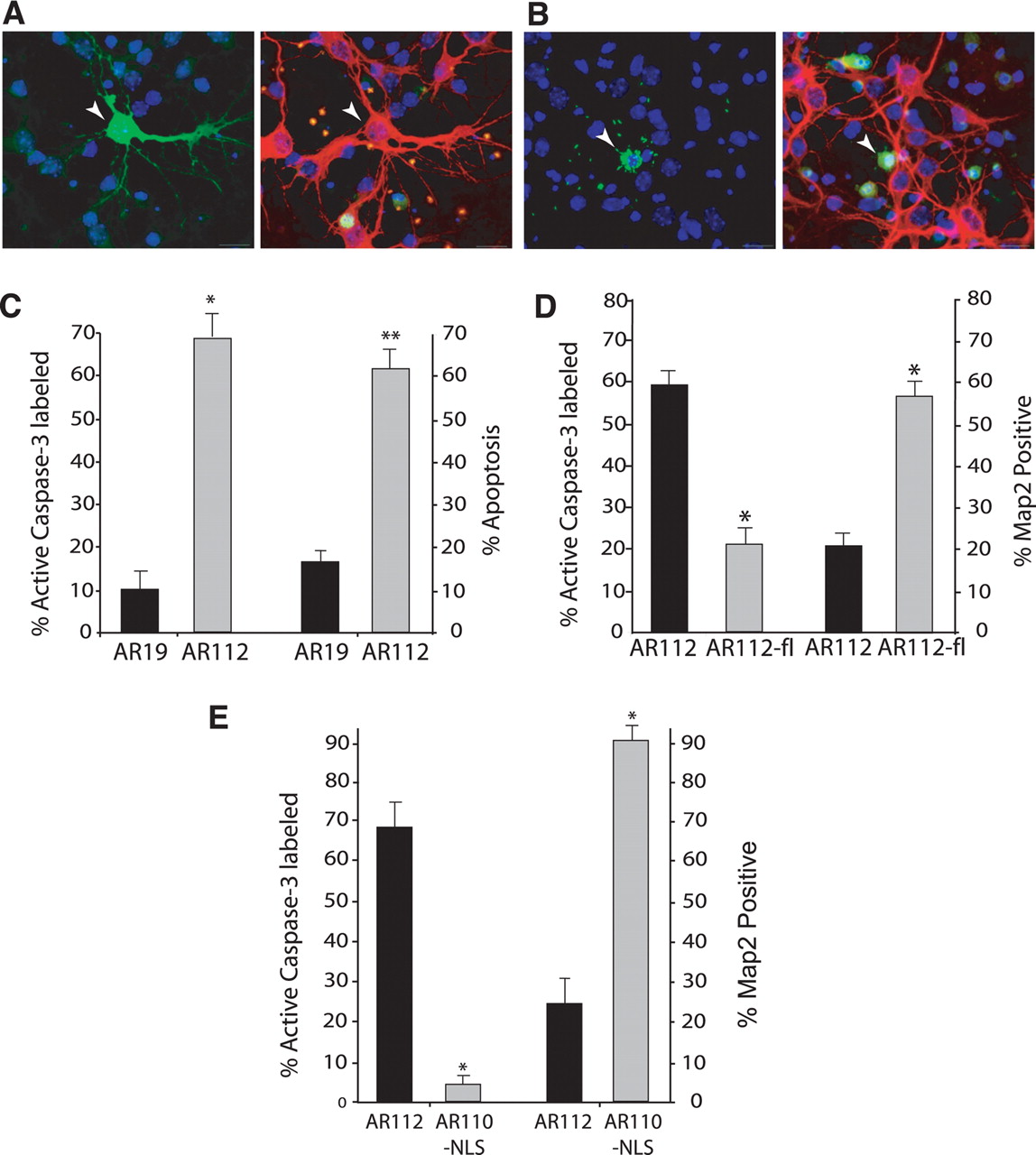
Expression of AR truncation fragment in the cytosol results in neurite degeneration and apoptotic cell death in a primary cortical neuron model of AR-polyQ neurotoxicity. A, B, Here, we see representative images of AR-transfected neurons using four-channel imaging: DAPI (blue), GFP (green, left), Cy3-labeled MAP2 (red), and Cy5-labeled active caspase-3 (green, right). A, Transfection of AR19 does not cause toxicity. Left, Here we see GFP-tagged AR19 expression in a transfected neuron (arrowhead), demonstrating the presence of AR in the cytosol and neuritic processes. Right, Labeling of MAP2 (red) reveals normal, healthy neurites in the AR19-expressing neuron, and a nucleus that lacks caspase-3 activity (green). B, Transfection of AR112 produces neurotoxicity. Left, Neuron expressing GFP-tagged AR112 (arrowhead) displays nuclear condensation and fragmentation. Right, AR112-expressing cortical neuron displays no MAP2 staining (red), but does label positive for active caspase-3 (green). C, Comparison of AR19-expressing cortical neurons to AR112-expressing cortical neurons reveals marked caspase-3 activation and frequent apoptotic nuclear morphology in neurons expressing polyQ-expanded AR (*p < 0.01, **p < 0.001; ANOVA). D, Primary neurons were transfected with Nt-AR112-GFP (AR112) or with the GFP-tagged full-length AR112 (AR112-fl). Primary neurons were cultured in 10 μm DHT to permit ligand activation of AR112-fl. Quantification of caspase-3 activation and MAP immunoreactivity indicates that AR112-fl does not produce significant neurotoxicity (*p < 0.05; ANOVA). All results were obtained at 24 h after transfection for A–E. E, Targeting polyQ-expanded, N-terminal truncated AR to the nucleus does not produce caspase activation, cell death, or neurite degeneration at 48 h after transfection (*p < 0.01; ANOVA). There was also no toxicity at 24 h after transfection (data not shown).
The Bcl-2 family of apoptosis regulators mediates AR112-induced neuronal apoptosis
Activation of caspase-9 is mediated by the intrinsic pathway in which increased mitochondrial membrane permeability is induced by changes in the bioenergetics state of the cell, shifts in Ca2+ concentration, or alterations in the ratio of pro-apoptotic to anti-apoptotic members of the Bcl-2 family of proteins (Li et al., 1997; Yuan and Yankner, 2000). The release of pro-apoptotic factors, including cytochrome c and Smac/Diablo, from the mitochondrial intermembrane space promotes the formation of the Apaf-1 apoptosome that cleaves pro-caspase-9. Selective activation of caspase-9 by polyQ-expanded AR suggested that apoptosis was mediated by the intrinsic pathway. This hypothesis was supported by the observation that over-expression of Bcl-2 markedly ameliorated the toxicity of Nt-AR112-GFP in MN-1 cells (Fig. 4A). To determine whether Nt-AR112-GFP toxicity in primary neurons also involved the intrinsic pathway, we obtained transgenic mice overexpressing Bcl-2 under the control of the neuron specific enolase promoter (Martinou et al., 1994). We transfected primary cortical neurons from Bcl-2 overexpressing mice or from wild-type littermate controls with Nt-AR112-GFP. Bcl-2 overexpression provided nearly complete protection from AR112 neurotoxicity (Fig. 4B), suggesting that neurodegeneration in this model is mediated by activation of pro-apototic Bcl-2 family members. The pro-apoptotic Bcl-2 family member Bax mediates neuronal apoptosis in response to a wide variety of stimuli, and expression of polyQ-expanded proteins has been reported to upregulate Bax in vitro, although a role in polyQ neurotoxicity has never been shown (Chou et al., 2006; Wang et al., 2006). To test the possibility that Bax mediates polyQ-induced neurodegeneration, we cultured primary cortical neurons from Bax null mice (Knudson et al., 1995). Bax null neurons were completely resistant to AR112 neurotoxicity, demonstrating that apoptosis induced by truncated polyQ-expanded AR requires Bax and is therefore mediated by the intrinsic pathway (Fig. 4C–E).

AR112 truncation fragment expression activates apoptosis via the intrinsic pathway. A, Bcl-2 protects MN-1 cells against AR112-induced cell death (*p < 0.01; Student's t test). B, Primary cortical neurons derived from Bcl-2 transgenic mice are strongly protected against AR112 neurotoxicity (*p < 0.01; ANOVA). C, Primary cortical neurons derived from Bax null mice are fully protected from AR112 neurotoxicity (*p < 0.01; ANOVA). D, E, Here we see representative images of AR-transfected neurons using four-channel imaging: DAPI (blue), GFP (green, left), Cy3-labeled active caspase-3 (red), and Cy5-labeled MAP2 (green, right). D, Transfection of Nt-AR112-GFP into Bax null neurons does not cause toxicity. Left, We see a neuron expressing AR112. At 24 h after transfection, no active caspase-3 (red) is detected in the AR112-transfected neuron (middle). Cy5 (green) labeling of MAP2 reveals normal, healthy neurites in this AR112-expressing neuron (right). E, Transfection of Nt-AR112-GFP into Bax+/− neurons does produce neurotoxicity. Left, We see a neuron expressing AR112. At 24 h after transfection, immunostaining for active caspase-3 (red) strongly labels this AR112-expressing neuron (middle). While numerous nontransfected cortical neurons exhibit healthy MAP2-positive processes (Cy5, green), the AR112-expressing cortical neuron displays no MAP2 staining (right). In all cases, nuclear condensation indicative of neuron cell death corresponded to caspase-3 activation in these experiments (data not shown).
Jun N-terminal kinase activation of c-Jun mediates AR112 neuronal apoptosis
We examined the phosphorylation status of c-Jun in MN-1 cells expressing truncated AR112, and documented production of phospho-c-Jun (Fig. 5A). Accumulation of phospho-c-Jun before onset of detectable apoptosis suggests a role for this transcription factor in mediating polyQ-expanded AR toxicity. To test this hypothesis, MN-1 cells were cotransfected with Nt-AR112-GFP and dominant-negative forms of c-Jun. TAM67 is a dominant negative mutant of c-Jun that was generated by removal of the transactivation domain (Chiariello et al., 2000). Coexpression of Nt-AR112-GFP with increasing ratios of TAM67 led to reduced cytotoxicity (Fig. 5B). SID-c-Jun is another potent dominant-negative mutant generated by replacing the transactivation domain with the Sin3A-binding domain of Mad (Iavarone et al., 2003). SID-c-Jun yielded a similar reduction in Nt-AR112-GFP cytotoxicity (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). These observations suggested that c-Jun-mediated transcription contributes to polyQ-expanded AR neurotoxicity. We then examined the role of JNK and c-Jun phosphorylation in our primary neuron model, and found that truncated AR112, but not AR19, yielded rapid accumulation of phospho-c-Jun; similar to neurons treated with taxol, a drug that activates the JNK pathway (Fig. 5C–E). Phosphorylation of c-Jun occurred at the 18 h time point, well before loss of MAP2 staining, activation of caspase-3, or nuclear condensation. To determine whether JNK activity is required for AR112-induced apoptosis in primary cortical neurons, we pretreated cultures with the JNK inhibitor SP600125 at a concentration specific for JNK (10 μm) (Bennett et al., 2001; Donovan et al., 2002; Bogoyevitch et al., 2004). Pretreatment with the SP600125 kinase inhibitor prevented neurite degeneration, caspase-3 activation, and apoptotic cell death in AR112-transfected neurons (Fig. 5F). To confirm that this effect was JNK-dependent, we repeated this experiment with a different JNK inhibitor, and again noted significant reductions in caspase-3 activation, neurite degeneration, and apoptotic cell death in AR112-transfected neurons (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). These results indicated that truncated polyQ-expanded AR activates JNK leading to phosphorylation of c-Jun, which then initiates a Bax-dependent apoptotic cascade. To determine the broader relevance of our findings to polyQ neurodegeneration, we obtained N-terminal truncated huntingtin (htt) expression constructs and transfected them into Bax null and control primary cortical neurons. We noted polyQ-htt length-dependent caspase-3 activation and loss of MAP2 immunoreactivity that was almost completely prevented by the absence of Bax (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material). N-terminal truncated polyQ-expanded htt expression also elicited marked phosphorylation of c-Jun (supplemental Fig. 3D, available at www.jneurosci.org as supplemental material).

JNK activation of c-Jun mediates polyQ-AR neurotoxicity. A, Western blot analysis for activated (serine-63-phosphorylated) c-Jun in nuclear extracts isolated from untransfected MN-1 cells (UT), AR19-expressing MN-1 cells (AR19), AR112-expressing MN1 cells (AR112), or staurosporine-treated MN-1 cells (sts; positive control). TATA-binding protein (TBP) served as the loading control. The ratio of c-Jun-P(S63):TBP is markedly increased for AR112-expressing MN-1 cells (6.5×) and staurosporine-treated MN-1 cells (14.1×). B, FACS-assisted cell viability assay demonstrates that coexpression of c-Jun dominant-negative TAM67 protects MN-1 cells from AR112 cytotoxicity (*p < 0.01, **p < 0.001; Student's t test). C, Taxol, a positive control for JNK activation of c-Jun, yields numerous anti-c-Jun-P-positive neuron nuclei at 18 h after treatment (DAPI, blue; c-Jun-P, red). D, Primary neurons expressing the AR19 truncation fragment do not display activation of c-Jun-P at 18 h after treatment (AR19-GFP, green; DAPI, blue; c-Jun-P, red). E, Primary neurons expressing AR112 truncation fragment display activation of c-Jun-P at 18 h after treatment (AR112-GFP, green; DAPI, blue; c-Jun-P, red). F, Primary neurons treated with the JNK inhibitor SP600125 are protected from AR112 neurotoxicity (*p < 0.01, **p < 0.001; ANOVA). In all cases, nuclear condensation indicative of neuron cell death corresponded to caspase-3 activation in these experiments (data not shown).
Truncated AR112 apoptotic activation is mediated by DP5/Hrk
To assess the role of BH3-only proteins in mediating apoptosis induced by polyQ-expanded AR, we obtained Bim knock-out mice and DP5/Hrk knock-out mice (Putcha et al., 2001; Imaizumi et al., 2004). We transfected cortical neurons from DP5 null mice and DP5 heterozygous littermates with Nt-AR19-GFP and Nt-AR112-GFP, quantified caspase-3 activation in AR-expressing neurons, and found that absence of DP5/Hrk provided a partial, yet highly significant degree of protection against AR112-induced apoptosis (Fig. 6A). While the degree of protection afforded by the absence of DP5/Hrk was only partial, it did approximate the level of protection observed in other single BH3-only knock-out neuron studies (Putcha et al., 2001; Imaizumi et al., 2004). We then performed a similar experiment, this time using primary neurons from Bim null mice, but observed no protective effect against AR112 neurotoxicity (Fig. 6B). As complete absence of a BH3-only protein in knock-out mice may result in compensatory upregulation of other related BH3-only proteins, we reasoned that acute knock-down experiments were warranted. To evaluate the role of DP5 and Bim in polyQ-AR neurotoxicity, we derived plasmid-based U6 promoter-driven DP5 shRNA and Bim shRNA knock-down constructs. After validating these constructs in neuro2a cells (supplemental Fig. 4A,B, available at www.jneurosci.org as supplemental material), we cotransfected primary cortical neurons with Nt-AR112-GFP and the DP5 shRNA knock-down construct or the Bim shRNA knock-down, using empty vector as a negative control in each case. DP5 knock-down yielded an even greater degree of protection from polyQ-AR neurotoxicity (Fig. 6C), while Bim knock-down again provided no protection against polyQ-AR neurotoxicity (Fig. 6D).

Absence or knock-down of DP5, but not Bim, protects against AR112 neurotoxicity. A, DP5 null cortical neurons are protected against AR112 neurotoxicity (*p < 0.01, **p < 0.001; ANOVA). B, Bim null status does not protect primary cortical neurons from AR112 neurotoxicity (p = 0.88, p = 0.20; ANOVA). C, We performed cotransfection of wild-type primary cortical neurons with Nt-AR112 and a DP5 shRNA expression construct (DP5 k.d.) or empty vector (EV). Knock-down of DP5 significantly protected AR112-expressing neurons from neurotoxicity (*p < 0.05; ANOVA). D, Similar cotransfections of wild-type primary cortical neurons with Nt-AR112 and a Bim shRNA expression construct (Bim k.d.) yielded no protection against AR-polyQ neurotoxicity (p = 0.38, p = 0.36; ANOVA). E, Real-time RT-PCR analysis of DP5 gene expression as a ratio of β-actin gene expression is shown for wild-type (Wt), Bim heterozygous null (Bim+/−), and Bim homozygous null (Bim−/−) neurons. Expression of DP5 is significantly increased in Bim−/− neurons compared with either Bim+/− or Wt neurons (*p < 0.05; two-tailed t test). F, Bim–DP5 double knock-out (DKO) neurons do not protect against AR112 neurotoxicity (p = 0.26, p = 0.29; ANOVA).
As JNK activation results in the transactivation of both Bim and DP5, we decided to measure the expression level of DP5 in Bim null neurons and in Bim knock-down neurons. We observed significant upregulation of DP5 in Bim null neurons (Fig. 6E), and marked upregulation of DP5 in Bim knock-down neurons (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material), suggesting that compensation by DP5 may account for the lack of Bim null protection. Analysis of Bim expression in DP5 null cortical neurons, however, did not reveal compensatory upregulation of Bim (data not shown). Compensatory upregulation of DP5 in Bim null cortical neurons thus led us to derive double knock-out (DKO) mice null for both Bim and DP5/Hrk. When we transfected DKO cortical neurons with Nt-AR112-GFP expression constructs and scored AR112-expressing neurons for caspase-3 activation and loss of MAP2 immunoreactivity, we found no evidence for protection against AR112-mediated neurotoxicity (Fig. 6F). To determine why DKO cortical neurons could not protect against polyQ-AR stress, we examined the expression levels of other BH3-only proteins. We documented marked induction of Puma in stressed DKO neurons (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material). Indeed, induction of Puma at the protein level was apparent in Bim null cortical neurons (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). Evaluation of taxol-induced Noxa activation in Bim null and DKO neurons yielded similar results (data not shown), supporting pronounced compensation of multiple BH3-only proteins in Bim null and DKO mice.
Discussion
One important theme in neurodegeneration has been the recognition of protein misfolding as a common feature (Taylor et al., 2002). Accumulation of misfolded proteins (or peptide fragments thereof) has been linked to endoplasmic reticulum (ER) stress in such disorders (Kouroku et al., 2002; Nishitoh et al., 2002; Rao et al., 2004). In the polyQ diseases, inadequate degradation of misfolded proteins or peptide fragments by the ubiquitin-proteasome system may represent an early step in disease pathogenesis, and likely sets the stage for ER stress activation (Sherman and Goldberg, 2001). Although nuclear localization of polyQ disease proteins appears to be an essential step in disease pathogenesis for HD, SBMA, and SCA1 (Skinner et al., 1997; Saudou et al., 1998; Katsuno et al., 2002), the pathogenic cascade may also involve stress activation pathways in the cytosol (Lindholm et al., 2006; Sekine et al., 2006). To determine the series of molecular steps culminating in cell death in our AR-polyQ models, we began by characterizing the terminal events. Condensed nuclear morphology, DNA laddering, and caspase-3 activation indicated that cellular demise was apoptotic. Apoptosis may be triggered by binding of “death receptors” to activate the caspase-8 dependent extrinsic pathway, or by an intrinsic Bax-dependent pathway culminating in caspase-9 activation (Yuan and Yankner, 2000). Our results revealed that AR polyQ length-dependent cell death is mediated by the intrinsic pathway. Indeed, transfection of N-terminal AR-polyQ into Bax null neurons yielded complete protection against AR polyQ length-dependent apoptotic cell death. Previous studies of polyQ neurotoxicity have yielded conflicting data, as one group detected Bax activation in cerebellar neurons expressing either polyQ-expanded ataxin-3 or ataxin-7 (Chou et al., 2006; Wang et al., 2006), while another group reported that isolated polyQ expansion tracts produce nonapoptotic, Bax-independent neurotoxicity in cultured cerebellar neurons (Moulder et al., 1999). Our results, however, indicate that Bax is essential for apoptotic activation in polyQ neurodegeneration, albeit for truncated AR and htt proteins.
As polyQ-expanded proteins are known to elicit ER stress (Kouroku et al., 2002; Nishitoh et al., 2002; Rao et al., 2004), and ER stress has been coupled to JNK activation (Urano et al., 2000), we reasoned that AR polyQ length-dependent cell death would be JNK-dependent, and noted significant protection against AR polyQ neurotoxicity with two different JNK inhibitors (Bennett et al., 2001; Donovan et al., 2002; Holzberg et al., 2003). To delineate the pathway by which AR-polyQ activates apoptosis, we considered how JNK activation could produce Bcl-2 inhibition in neurons. The balance of pro-apoptotic and anti-apoptotic activities of the Bcl-2 family of proteins constitutes a critical checkpoint regulating apoptosis. The “BH3-only” members of the Bcl-2 family are initiators of programmed cell death and stress-induced apoptosis mediated by the mitochondrial pathway (Huang and Strasser, 2000). The BH3-only proteins function by antagonizing anti-apoptotic Bcl-2, Mcl-1, and Bcl-xL, leading to Bax and/or Bak-dependent loss of mitochondrial membrane integrity. As Bim and DP5/Hrk are activated downstream of JNK and c-Jun in neuronal apoptosis (Inohara et al., 1997; Imaizumi et al., 1999; Harris and Johnson, 2001; Putcha et al., 2001; Whitfield et al., 2001; Imaizumi et al., 2004), we hypothesized that AR-polyQ neuron cell death might be mediated by these BH3-only proteins. DP5/Hrk is a particularly attractive candidate, as it has been implicated in neuron death both in DP5 null mice and in human ALS patients (Imaizumi et al., 1997; Shinoe et al., 2001; Imaizumi et al., 2004). DP5 null status conferred significant protection against AR-polyQ neurotoxicity, akin to the level of protection afforded in other stress paradigms performed in single BH3-only knock-out neurons (Putcha et al., 2001; Imaizumi et al., 2004). Absence of Bim, however, did not yield protection against AR-polyQ in primary neurons. We attributed this lack of Bim protection to compensatory upregulation of other BH3-only proteins, as we documented increased DP5/Hrk expression in both Bim null neurons and in acute Bim shRNA knock-down experiments. Interestingly, Bim – DP5 DKO cortical neurons also could not protect against AR polyQ stress. Although the basis for this lack of protection was unclear, significant compensatory upregulation of yet another BH3-only protein, Puma, may account, at least in part, for this observation. Markedly pronounced compensation of BH3-only proteins in Bim null neurons is consistent with an emerging view of Bim as a key regulator of intrinsic pathway activation and as a sensor of ER stress (Puthalakath et al., 2007; Willis et al., 2007); hence, a role for Bim in mediating polyQ-AR apoptotic activation cannot be excluded by this study. Nonetheless, our results clearly implicate DP5/Hrk in AR polyQ length-dependent apoptotic activation, suggesting DP5/Hrk upregulation could be involved in SBMA disease pathogenesis. To our knowledge, this investigation is the first to directly document a role for a BH3-only protein in polyQ neurodegeneration.
In this study, we found that expression of an N-terminal AR fragment produced polyQ length-dependent neurodegeneration and cell death in MN-1 motor neuron-like cells and in primary neurons. While the role of N-terminal truncation fragments in some polyQ diseases may be unclear, evidence for the existence of pathogenic N-terminal truncation fragments in SBMA is strong. The pathologic hallmark of SBMA is the presence of both cytoplasmic and nuclear AR inclusions that are detectable in motor neurons of the brainstem and spinal cord, neurons of the dorsal root ganglia, and peripheral tissues including the skin, testis and other visceral organs (Li et al., 1998a,b; Adachi et al., 2005). These inclusions are detected by antibodies that recognize the N terminus of the AR protein, but not by antibodies against the middle or C terminus, suggesting that the C terminus of AR is truncated or masked. Mouse and Drosophila models of SBMA in which full-length, polyQ-expanded AR is expressed are also characterized by AR inclusions recognized by antibodies against the N terminus of AR (Takeyama et al., 2002; Walcott and Merry, 2002; Chevalier-Larsen et al., 2004). Extraction of AR protein from affected tissues in these animals, followed by immunoblotting, demonstrates the accumulation of N-terminal fragments that correlate with disease progression (Takeyama et al., 2002; Li et al., 2007). These results strongly suggest that AR truncation, rather than epitope masking, is the basis for detection of N-terminal AR epitopes in SBMA patient tissue and animal models. Moreover, there is extensive evidence that an N-terminal truncation product mediates the cytotoxicity observed in SBMA (Kobayashi et al., 1998; Merry et al., 1998; Ellerby et al., 1999b; Li et al., 2007).
To assess the relevance of our findings to other polyQ diseases involving “toxic fragments,” we transfected N-terminal truncated polyQ-huntingtin (htt) into primary cortical neurons from wild-type and Bax null mice. N-terminal polyQ-htt protein accumulated in the cytosol, yielded caspase-3 activation and neurodegeneration, and was Bax-dependent, akin to polyQ-AR. In HD, the necessity of proteolytic cleavage for disease pathogenesis is supported by absence of neurodegeneration in HD YAC transgenic mice expressing htt-128Q with a caspase cleavage site mutation at amino acid position 586 (Graham et al., 2006). Subsequent studies have suggested that htt cleavage is mediated by caspase-6 in the nucleus, and that htt truncation fragments move out of the nucleus into the cytosol (Warby et al., 2008). In SCA3, proteolytic cleavage of polyQ-ataxin-3 enhances misfolding and neurotoxicity in neuroblastoma cells and transgenic mice by yielding a C-terminal polyQ-ataxin-3 fragment (Haacke et al., 2006; Colomer Gould et al., 2007), but the cleavage enzyme, subcellular site of cleavage, and cellular toxicity process remain unknown.
An unresolved question in the neurodegenerative disease field is the contribution of apoptosis to neuron dysfunction and cell death. While considerable evidence exists for prominent activation of apoptotic pathways in degenerating neurons from patients with neurodegenerative disorders (for review, see Mattson, 2000; Yuan and Yankner, 2000), evidence for neuron death mediated by apoptosis is scant in both patient material and animal models (Turmaine et al., 2000; Conforti et al., 2007). Why is this the case? As postmitotic neurons wired into complex synaptic pathways are difficult, if not impossible, to replace, many “brakes,” including anti-apoptotic factors such as XIAP and NAIP, prevent the loss of neurons in the face of apoptotic activation (Perrelet et al., 2002). Indeed, modifications of classic apoptosis pathways may explain the cell death process in neurons (Sperandio et al., 2000; Okada and Mak, 2004). However, even if diseased neurons do not die by classic apoptosis, activation of apoptotic mediators could play a crucial role in the pathogenic cascade. For example, elaboration of caspases and other proteolytic enzymes downstream of the JNK activation pathway may be an early and crucial step in the production of misfolded toxic peptides. In AD and HD, mouse models expressing disease mutations do not become affected when disease proteins contain second site mutations at putative caspase cleavage sites (Galvan et al., 2006; Graham et al., 2006). Other in vivo studies in mice similarly point to a contribution of active caspases and other apoptotic mediators in disease pathogenesis (Ona et al., 1999; Yuan and Yankner, 2000). In ALS, crossing SOD1 mutant mice with Bcl-2-overexpressing transgenic mice significantly retards disease progression (Kostic et al., 1997). Our findings support the view that apoptotic activation contributes to SBMA disease pathogenesis. Furthermore, the delineation of the JNK–c-Jun-phosporylation–DP5–Bcl-2–Bax pathway in AR polyQ neurotoxicity provides us with a framework to investigate how these events intertwine with ligand dependency, proteolytic cleavage, and subcellular localization. If cross talk does exist between this pathway and key processes of AR polyQ neurotoxicity, then rational decisions about how to pursue therapy for SBMA should be possible.
Footnotes
-
This work was supported by National Institutes of Health Grants NS41648 (A.R.L.), NS35533 (R.S.M.), NS40251A (L.M.E.), and NS53825 (J.P.T.); intramural National Institute of Neurological Disorders and Stroke funds (K.H.F.); and grants from the Muscular Dystrophy Association (A.R.L., J.P.T., and L.M.E.). We thank S. Finkbeiner, R. Youle, S. Gutkind, C. Duckett, E. Johnson, and G. Nunez for providing reagents or mice.
- Correspondence should be addressed to Dr. Albert R. La Spada, Department of Laboratory Medicine, University of Washington Medical Center, Box 357110, Room NW 120, Seattle, WA 98195-7110. laspada{at}u.washington.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}