

Abstract
We recently described a severe, potentially lethal, but treatment-responsive encephalitis that associates with autoantibodies to the NMDA receptor (NMDAR) and results in behavioral symptoms similar to those obtained with models of genetic or pharmacologic attenuation of NMDAR function. Here, we demonstrate that patients' NMDAR antibodies cause a selective and reversible decrease in NMDAR surface density and synaptic localization that correlates with patients' antibody titers. The mechanism of this decrease is selective antibody-mediated capping and internalization of surface NMDARs, as Fab fragments prepared from patients' antibodies did not decrease surface receptor density, but subsequent cross-linking with anti-Fab antibodies recapitulated the decrease caused by intact patient NMDAR antibodies. Moreover, whole-cell patch-clamp recordings of miniature EPSCs in cultured rat hippocampal neurons showed that patients' antibodies specifically decreased synaptic NMDAR-mediated currents, without affecting AMPA receptor-mediated currents. In contrast to these profound effects on NMDARs, patients' antibodies did not alter the localization or expression of other glutamate receptors or synaptic proteins, number of synapses, dendritic spines, dendritic complexity, or cell survival. In addition, NMDAR density was dramatically reduced in the hippocampus of female Lewis rats infused with patients' antibodies, similar to the decrease observed in the hippocampus of autopsied patients. These studies establish the cellular mechanisms through which antibodies of patients with anti-NMDAR encephalitis cause a specific, titer-dependent, and reversible loss of NMDARs. The loss of this subtype of glutamate receptors eliminates NMDAR-mediated synaptic function, resulting in the learning, memory, and other behavioral deficits observed in patients with anti-NMDAR encephalitis.
Introduction
Synaptic plasticity is thought to underlie mechanisms of memory, learning, and cognition. Central to these neurological functions is the proper synaptic localization and trafficking of the excitatory glutamate NMDA and AMPA receptors (Lau and Zukin, 2007; Shepherd and Huganir, 2007). The roles of these receptors at the synaptic and cellular levels have been established through animal models in which the receptors have been genetically or pharmacologically altered (Jentsch and Roth, 1999; Mouri et al., 2007). In humans, the role of these receptors in memory, learning, cognition, and psychosis comes from more indirect approaches, such as pharmacological trials [e.g., NMDA receptor (NMDAR) antagonists causing psychosis] (Gunduz-Bruce, 2009), and analysis of brain tissue from patients with Alzheimer's disease or schizophrenia in which several molecular pathways causing a downstream alteration of glutamate receptors are affected (Snyder et al., 2005; Hahn et al., 2006). We recently identified a disorder in which the extracellular domain of the NR1 subunit of the NMDAR is directly targeted by autoantibodies (Dalmau et al., 2007, 2008). Patients develop prominent psychiatric and behavioral symptoms, rapid memory loss, seizures, abnormal movements (dyskinesias), hypoventilation, and autonomic instability (Dalmau et al., 2007, 2008; Iizuka et al., 2008). In two series comprising 181 cases (Dalmau et al., 2008; Florance et al., 2009), there was a strong female predominance (ratio, 8.5:1.5) and the median age of the patients was 19 years (23 months to 75 years; 40% children). In 55% of the adults (less frequently in children), the disorder appears to be triggered by the presence of a tumor, mostly an ovarian teratoma that contains nervous system tissue and expresses NMDARs. Despite the severity of the symptoms, 75% of patients recover after receiving immunotherapy and, when appropriate, tumor removal, and 25% are left with memory, cognitive, and motor deficits, or, rarely, die of the disorder. The autoantibodies are present in patients' serum and CSF, the latter usually showing intrathecal synthesis and high antibody concentration (Dalmau et al., 2008; Florance et al., 2009). All patients' antibodies recognize the N-terminal extracellular domain of NR1 (amino acid residues 25–380), suggesting an antibody-mediated pathogenesis (Dalmau et al., 2008). Although patients' antibodies can cause a decrease in NMDAR cluster density, the underlying mechanisms remain poorly understood (Dalmau et al., 2008). Here, we report in vitro and in vivo studies that indicate the cellular mechanisms by which patients' antibodies lead to a reduction in surface and synaptic NMDAR density and function, likely underlying the learning, memory, and other behavioral deficits observed in patients with anti-NMDAR encephalitis.
Materials and Methods
Patients, NR1 antibodies, titers, and controls.
CSF and serum were obtained from randomly selected patients with anti-NMDAR encephalitis (supplemental Table 1, available at www.jneurosci.org as supplemental material) among a series of 320 cases. All patients had well characterized clinical manifestations of anti-NMDAR encephalitis, including at least four of the following features: prominent psychiatric symptoms, decreased level of consciousness, seizures, dyskinesias, autonomic instability, or hypoventilation. Antibodies to extracellular epitopes of the NR1 subunit of the NMDAR were demonstrated using three different assays, as reported previously (Dalmau et al., 2008): immunohistochemistry with rat and human brain, immunostaining of live, nonpermeabilized cultures of rat hippocampal neurons, and immunolabeling of HEK293 cells transfected with NR1 or NR1 and NR2 (forming NR1/2 heteromers). We previously reported that patients' NMDA receptor antibodies are IgG1 and IgG3, but not IgM (Tüzün et al., 2009); therefore, we will refer to purified antibodies from patients' serum as purified IgG. CSF from patients with high antibody titer was diluted so that the final titer used in experiments was within the range of undiluted CSF of many patients with this disorder (Dalmau et al., 2008).
Control serum or CSF samples were obtained from normal individuals and patients undergoing CSF analysis for a variety of disorders not associated with antibodies to the NMDAR; samples were randomly selected from 1500 cases negative for NR1 antibodies applying similar test and criteria as above.
Antibody titers from patients and controls were determined by ELISA (Dalmau et al., 2008).
Preparation of patient and control CSF and IgG.
Patient or control CSF and serum were collected, filtered, and kept frozen until use. CSF from individual patients with high NMDAR antibody titer was diluted 1:15–60 to treat neurons in vitro and used undiluted for in vivo experiments. In some experiments, patient IgG antibodies were purified from serum with protein A/G-Sepharose columns and used to treat neurons. To prepare patient and control IgG, 2 ml of serum was incubated with a 1 ml of bio-spin chromatography column (Bio-Rad) of protein A/G-Sepharose beads (50:50) for 30 min on an orbital shaker at 4°C. After three washes with PBS, the sample was eluted with 100 mm glycine, pH 2.5, and neutralized with Tris-HCl, pH 8.0, dialyzed against PBS, concentrated in stock solutions of 20 mg/ml, and stored at −80°C. IgG concentration (∼1 mg/ml) and pH (7.4) was adjusted before use. Each IgG preparation was tested for antibody reactivity by staining human or rat brain sections or HEK cells expressing NR1/NR2 heteromers of the NMDAR as previously described (Dalmau et al., 2007, 2008). Both patients' CSF and IgG decreased surface and total NMDARs to the same extent (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Cell culture and patient antibody treatment.
Briefly, isolated rat hippocampi were placed in Ca2+-free HBSS (HBSS; Invitrogen) containing 1% papain for 20 min, triturated in Basal Media Eagle (Invitrogen) supplemented with B-27 (Invitrogen) and plated at 100,000 or 400,000 (for biotinylation) cells per milliliter in Neural Basal (NB) (Invitrogen) supplemented with 10% FBS (HyClone), B-27, 1% penicillin and streptomycin (Invitrogen), and 1% l-glutamine (Invitrogen) on poly-l-lysine-coated (Sigma-Aldrich) coverslips in 24-well plates. Culture media was changed to NB supplemented with B27 at 4 d in vitro (div). Cells were maintained at 37°C, 5% CO2, 95% humidity; medium was changed weekly. Neurons were treated with CSF or IgG from individual patients or controls for 1 d beginning at 14 d in vitro; in some experiments, neurons were treated for 3 or 7 d beginning at 14 d in vitro.
Immunostaining for presynaptic and postsynaptic components, confocal imaging, and image analysis.
To stain surface NMDAR clusters, control or treated neurons were washed in Neurobasal plus B27 and incubated with patient CSF containing anti-NR1 antibodies for 30 min, washed and incubated with fluorescently conjugated anti-human secondary antibodies for 30 min, and washed in PBS. Neurons were then fixed in 4% paraformaldehyde, 4% sucrose in PBS, pH 7.4, for 15 min, permeabilized with cold 0.25% Triton X-100 for 5 min, and blocked in 5% normal goat serum (Invitrogen) for 1 h at room temperature (RT). Additional immunostaining was performed with various combinations of primary antibodies: to label glutamate receptors, anti-NR1 against the intracellular C terminus (1:1000; Millipore Bioscience Research Reagents), anti-GluR1 (1:10; Calbiochem) or anti-GluR2 (1:100; Millipore Bioscience Research Reagents); to label postsynaptic densities, PSD-95 (1:500; Bioaffinity Reagents); to label dendrites, mouse anti-MAP2 (1:1000; gift from Dr. V. Lee, University of Pennsylvania, Philadelphia, PA); to label presynaptic terminals, mouse anti-SV2 (1:200; Developmental Studies Hybridoma Bank), guinea pig anti-VGLUT 1 (1:1000; Millipore Bioscience Research Reagents), or mouse anti-Bassoon (1:400; Assay Designs). Antibodies were visualized after staining with the appropriate fluorescently conjugated secondary antibodies (1:200; Jackson ImmunoResearch).
Images were obtained using a confocal microscope (Leica TCS SP2). Images were thresholded automatically using iterative segmentation (Bergsman et al., 2006), and the number and area of individual immunostained presynaptic or postsynaptic clusters were determined using interactive software (custom-written ImageJ macros). Clusters with pixel overlap of presynaptic and postsynaptic markers were considered colocalized and thus synaptic (Krivosheya et al., 2008).
Biotinylation of surface proteins and analysis by Western blot.
Neurons were treated with 1 μg to 1 mg/ml IgG for 1 d, washed with PBS supplemented with 0.1 mm CaCl2 and 1 mm MgCl2 (rinsing buffer), and incubated for 30 min at 4°C with 1 mg/ml Sulfo-NHS-Biotin (Thermo Fisher Scientific) in rinsing buffer. Neurons were then washed with rinsing buffer plus 100 mm glycine (quenching buffer), incubated in quenching buffer for 30 min at 4°C to quench excess biotin, and then lysed in RIPA buffer [150 mm NaCl, 1 mm EDTA, 100 mm Tris-HCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, pH 7.4, supplemented with 1:500 protease inhibitor mixture III (Calbiochem)] at 4°C for 1 h. Lysates were cleared of debris by centrifugation at 12,400 × g for 20 min. An aliquot of the remaining supernatant was taken for the lysate fraction, and a second aliquot was incubated with avidin-linked agarose beads (Immobilized Monomeric Avidin; Thermo Fisher Scientific) overnight at 4°C. After centrifugation, the supernatant was removed and the beads (surface fraction) were washed with 1× RIPA buffer, 2× high-salt wash buffer (500 mm NaCl, 5 mm EDTA, 50 mm Tris, 0.1% Triton X-100, pH 7.5), and 1× no-salt wash buffer (50 mm Tris, pH 7.5). The surface fraction was eluted from the beads with 2× sample buffer and proteins separated on an 8% gel using SDS-PAGE. Samples were transferred to nitrocellulose membranes and probed for antibodies against NR1 (1:1000; 556308; BD Biosciences Pharmingen), NR2A (1:1000; AB1555; Millipore; 1:500; MAB5216; Millipore; 1:500; A6473; Invitrogen), NR2B (1:1000; AGC-003; Alomone Labs; 1:500; 06-600; Millipore), GABAARα1 (1:1000; 06-868; Millipore), GABAARα2 (1:500; AB5984; Millipore Bioscience Research Reagents), GluR 2/3 (1:1000; 07-598; Millipore), PSD-95 (1:1000; 610496; BD Biosciences Pharmingen), and actin (1:2000; A2066; Sigma-Aldrich). Actin and GABAARs were used as loading controls for total and surface fractions, respectively. Blots were incubated with HRP-conjugated goat anti-mouse or goat anti-rabbit secondary antibodies (1:3000; Cell Signaling), and signals were visualized using chemiluminescence (SuperSignal Chemiluminescent Substrate; Thermo Fisher Scientific). All quantified films were in the linear range of exposure and were digitally scanned, and signals were quantified using NIH ImageJ.
Whole-cell electrophysiological recordings of synaptic NMDA and AMPA receptor-mediated currents.
Whole-cell voltage-clamp recordings were performed as previously described (Elmariah et al., 2004, 2005) from 14 to 21 div pyramidal neurons treated for 24 h with patient CSF containing anti-NR1 antibodies, control CSF, or left untreated. Briefly, neurons were incubated in an extracellular physiological solution without Mg2+ (in mm: 119 NaCl, 5 KCl, 2 CaCl2, 30 glucose, 10 HEPES, pH 7.4). Voltage-clamp recordings were made at RT (22–25°C) using glass microelectrodes (resistance, 4–6 MΩ) filled with a cesium substituted intracellular solution (in mm: 100 cesium gluconate, 0.2 EGTA, 5 MgCl2, 2 ATP, 0.3 GTP, 40 HEPES, pH 7.2). Pipette voltage offset was neutralized before the formation of a gigaohm seal. Membrane resistance, series resistance, and membrane capacitance were determined from current transients elicited by a 5 mV depolarizing step from a holding potential of −80 mV, using the whole-cell application of PatchMaster software (HEKA Elektronik). Criteria for cell inclusion in the data set included a series resistance ≤30 MΩ and stability throughout the recording period. Currents were amplified, low-pass filtered at 2.5 kHz, and sampled at 5 Hz using PatchMaster software. Spontaneous miniature EPSCs (mEPSCs) were recorded at −70 mV in the presence of TTX (1 μm) and picrotoxin (10 μm). APV (50 μm) and CNQX (10 μm) were bath applied to block NMDAR- and AMPAR-mediated currents, respectively. mEPSC events were detected and analyzed using MiniAnalysis (Synaptosoft), which uses a threshold-based event-detection algorithm. NMDAR and AMPAR components of mEPSCs were separated temporally by their distinct kinetics (Hestrin et al., 1990; Watt et al., 2000; Yang et al., 2003). The amplitude of the NMDAR-mediated current was determined in a window between 15 and 25 ms after the peak of the AMPAR-mediated component, which has a fast, <1 ms rise time. All values are presented as mean ± SEM.
Fab fragment preparation and treatment.
Fab fragments were prepared from serum IgG using a kit according to the manufacturer's directions (Fab preparation kit; Pierce Protein Research Products; Thermo Fisher Scientific). Briefly, serum IgG was digested for 2–4 h at 37°C with 1% (w/w) papain, pH 7.0, with 0.01 m cysteine, resulting in cleavage into Fab and Fc fragments. Fab fragments were isolated by chromatography, and concentration was determined by absorption at 280 nm, and then used to treat neurons at a concentration of 4 μg/ml. Control experiments showed that incubating neurons with patient Fab fragments for 30 min resulted in surface staining of NR1 clusters (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Alzet minipump placement, IgG infusion, and analysis of effects on NMDA receptors.
Seven- to 8-week-old female Lewis rats were anesthetized, and a cannula was placed into the left hippocampus using predetermined coordinates (−3.2 mm posterior to bregma, 2 mm lateral, and 3 mm deep to the dura mater). The cannula was secured to a head probe mounted to the skull, and attached with sterile tubing to an Alzet minipump (Alzet brain infusion kit 3; pump model 2002) implanted subcutaneously on the back. Patient or control CSF was then delivered at a rate of 0.5 μl/h for 2 weeks. Rats were then killed, and brain tissue was harvested, immersion fixed in 4% paraformaldehyde in PBS, pH 7.4, for 15 min, cryoprotected in 30% sucrose in PBS, pH 7.4, overnight at 4°C, and snap frozen in isopentane cooled in dry ice. Frozen 10 μm sections from infused hippocampus (where the track of the cannula was visible) and contralateral matched area of the noninfused hippocampus were immunostained in parallel to determine the presence of human IgG and the levels of NR1 using the primary and secondary antibodies described above. The degree of cell death was assayed with terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL). Sections were imaged and thresholded with the same parameters, and confocally imaged and analyzed as described above.
Additionally, protein extracts from 20 μm sections of the infused and contralateral hippocampus were separated electrophoretically, transferred to nitrocellulose, and incubated with anti-NR1 antibody (Millipore Bioscience Research Reagents), and the amount of NR1 protein was quantified as described above, using tubulin as a loading control.
Immunostaining, imaging, and image analysis of human tissue.
Hippocampal sections of human tissue were immunostained in parallel as described above. Control and patients' tissue sections were imaged with a Zeiss Axioskop 2 plus (software, AxioVision 4.5) with identical optical settings and exposure times. For analysis of high-magnification regions, 7–10 images were collected from the CA1 region of the hippocampus. These images were inverted, and a cumulative histogram of pixel intensity was calculated for each image. The average cumulative histogram of pixel intensity was generated for each sample, and the cumulative probability of pixel intensity for each sample was determined, plotted, and compared using a paired Komolgorov–Smirnov test (see below).
Statistical analysis.
Titer dependence was assessed with a linear regression analysis. In experiments involving two conditions, the data were analyzed with a two-tailed unpaired Student t test. In experiments involving three or more conditions, the normality of the data was analyzed with the D'Agostino and Pearson omnibus normality test, before using a one-way ANOVA test followed by Bonferroni's multiple-comparison test. Differences in distributions of NR1 intensity were assessed with a paired Komolgorov–Smirnov test. All values are presented as mean ± SEM.
Results
Patients' antibodies reduce surface NMDA receptor clusters and protein in a titer-dependent fashion
Hippocampal neurons were cultured for 1 d with CSF or purified IgG containing anti-NR1 antibodies from patients with anti-NMDAR encephalitis (supplemental Table 1, available at www.jneurosci.org as supplemental material), followed by immunohistochemical and Western blot analyses of surface and total NR1 protein. Patients' antibodies significantly decreased NR1 or NMDAR surface and total cluster density in a titer-dependent fashion, compared with CSF or IgG from control patients (Fig. 1 a,c). Similar findings were obtained after treating the neurons for 3 or 7 d with patients' antibodies (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).

Patients' antibodies reduce surface NMDA receptor clusters and protein in a titer-dependent fashion. a , Hippocampal neurons immunostained for surface and total NMDAR clusters and presynaptic Bassoon. Surface NMDARs are defined as the colocalization between nonpermeabilized patient CSF staining (extracellular NR1 epitope) and commercial NMDAR staining (intracellular epitope; left). Patient CSF treatment for 1 d reduces surface and total NMDAR cluster density without affecting Bassoon cluster density. Scale bar, 10 μm. b , Western blot analyses of surface biotinylated and total lysate NMDAR protein. Patient IgG treatment for 1 d reduces surface as well as total NMDAR protein; GABAAα1 and actin are loading controls. Control NR1 levels have been overexposed in this image to visualize patient treated NR1 bands. c , Surface (left) and total (right) NMDAR cluster density after treatment with CSF from several patients with different antibody titer, showing a titer-dependent decrease in NMDA cluster density (linear regression analysis; surface R 2 = 0.59, p < 0.001; total R 2 = 0.44, p < 0.008). All values are mean ± SEM (n = 18 cells, 3 independent experiments; 12 patient, 2 control samples) (supplemental Table 1, available at www.jneurosci.org as supplemental material). d , Surface (left) and total (right) NMDAR protein after treatment with IgG from several patients with different antibody titer (n = 3–5 Western blots; 10 patient, 2 control samples), showing a titer-dependent decrease in NMDA protein (linear regression analysis; surface R 2 = 0.53, p < 0.0001; lysate R 2 = 0.30, p < 0.002). e , Surface (left) and total (right) NMDAR cluster density after treatment with CSF from the same patient at two time points with different antibody titer; a higher antibody titer decreases surface and total NMDAR clusters to a greater extent. Asterisk indicates significant difference (n = 18 cells, 3 independent experiments; 2 samples from 2 patients; Student's t test, p < 0.02). f , Surface (left) and total (right) NMDAR protein after treatment with IgG from two patients at two time points with different antibody titer; a higher antibody titer decreased surface and total NMDAR protein to a greater extent than IgG isolated from the same patient when a lower antibody titer was present. Asterisk indicates significant difference (n = 3, Western blots; 2 samples from 2 patients; Student's t test p < 0.05).
A significant titer-dependent decrease in surface and total NR1 protein was also observed with Western blot analyses (Fig. 1 b,d). Moreover, Western blot analyses of the effect of patients' antibodies on NR2 subunits (which assemble with NR1 to form NMDARs) showed that patients' antibodies significantly decreased surface and total NR2A and NR2B proteins in a titer-dependent fashion (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
To determine whether the effects of patients' antibodies correlate with the change of titers during the course of the disease, hippocampal neurons were cultured with CSF samples obtained at two different time points of the disease of two patients. The initial CSF was obtained at the time of symptom presentation and the second sample during symptom improvement in one patient and during symptom worsening in the other. The CSF obtained at symptom presentation had a higher NR1 antibody titer than the CSF obtained during symptom improvement of the first patient; in contrast, the CSF obtained during symptom worsening had a higher antibody titer than the CSF obtained at symptom presentation of the second patient. In both cases, the CSF with higher NR1 antibody titer decreased NMDAR surface and total cluster density (or total NMDAR protein measured by Western blot) (Fig. 1 e,f) to a greater extent than the CSF with the lower titer. Together, these results show that NR1 antibodies from patients with anti-NMDAR encephalitis decrease NMDAR surface cluster density and protein in a titer-dependent manner and that the effects of the antibodies vary with the change of titers during the course of the disease.
Patients' antibodies reversibly reduce synaptic NMDA receptor clusters without affecting the number of synapses and other synaptic components
Because patient antibodies decreased overall NMDAR surface cluster density and protein, we determined whether the antibodies also affected NMDAR synaptic localization, the number of synapses, and other synaptic components. Hippocampal neurons were cultured with CSF or purified IgG for 3 or 7 d, followed by immunostaining or Western blot analysis of NR1 and synaptic components such as presynaptic VGlut, postsynaptic PSD-95, AMPA receptor subunits GluR1 and GluR2, and GABA receptors.
Although the overall structural integrity of excitatory neurons and synapses was not affected (see below), patients' antibodies dramatically reduced the synaptic localization of NMDAR clusters in a titer-dependent fashion compared with controls (Fig. 2 a,c; supplemental Fig. 5, available at www.jneurosci.org as supplemental material), consistent with the overall decrease in surface NMDAR cluster density (Fig. 1). To determine whether the antibody-mediated decrease in NMDAR synaptic localization is reversible, patient antibodies were removed from the culture medium after 3 d of treatment and neurons were cultured for 4 additional days. The density of synaptically localized NMDAR clusters returned to baseline levels 4 d after patient antibodies were removed (Fig. 2 a,c). These results show that patients' antibodies cause a specific loss of NMDARs from excitatory synapses and that this loss is reversed after antibody removal.

Patients' antibodies reversibly reduce synaptic NMDA receptor clusters without affecting the number of synapses. a , Hippocampal neurons immunostained for total NMDAR clusters, a presynaptic glutamatergic terminal protein, VGlut, and a postsynaptic protein localized to glutamatergic synapses, PSD-95. Treatment with patient CSF for 3 d reduces the density of synaptic NMDAR clusters without affecting the number of excitatory synapses. After removal of patient CSF, the proportion of NMDAR clusters localized to synapses returns to baseline. Scale bar, 5 μm. b , Colocalization of presynaptic and postsynaptic proteins at excitatory synapses (n = 36 cells, 3 independent experiments; 2 patient, 1 control samples). c , NMDAR cluster density at excitatory synapses (synaptic NR1). The asterisk indicates significant difference (one-way ANOVA test followed by Bonferroni's multiple comparison test, p < 0.001). Error bars indicate SEM.
Patients' antibodies did not affect the number of excitatory synapses compared with controls (Fig. 2 a,b). Moreover, patients' antibodies did not affect the density of postsynaptic PSD-95, GluR1, GluR2 receptor clusters, or the surface or total amount of these proteins or the amount of surface GABA receptor protein (Fig. 3), dendritic branching, dendritic spine density, or Bassoon cluster density (Fig. 4). In addition, patients' antibodies did not affect cell survival (Fig. 4 h,i). The effects of patients' antibodies on NMDAR cluster density were not mediated by complement, because purified patient IgG from serum or heat-inactivated patient CSF decreased NMDAR cluster density and localization to a similar extent as non-heat-inactivated patient CSF (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
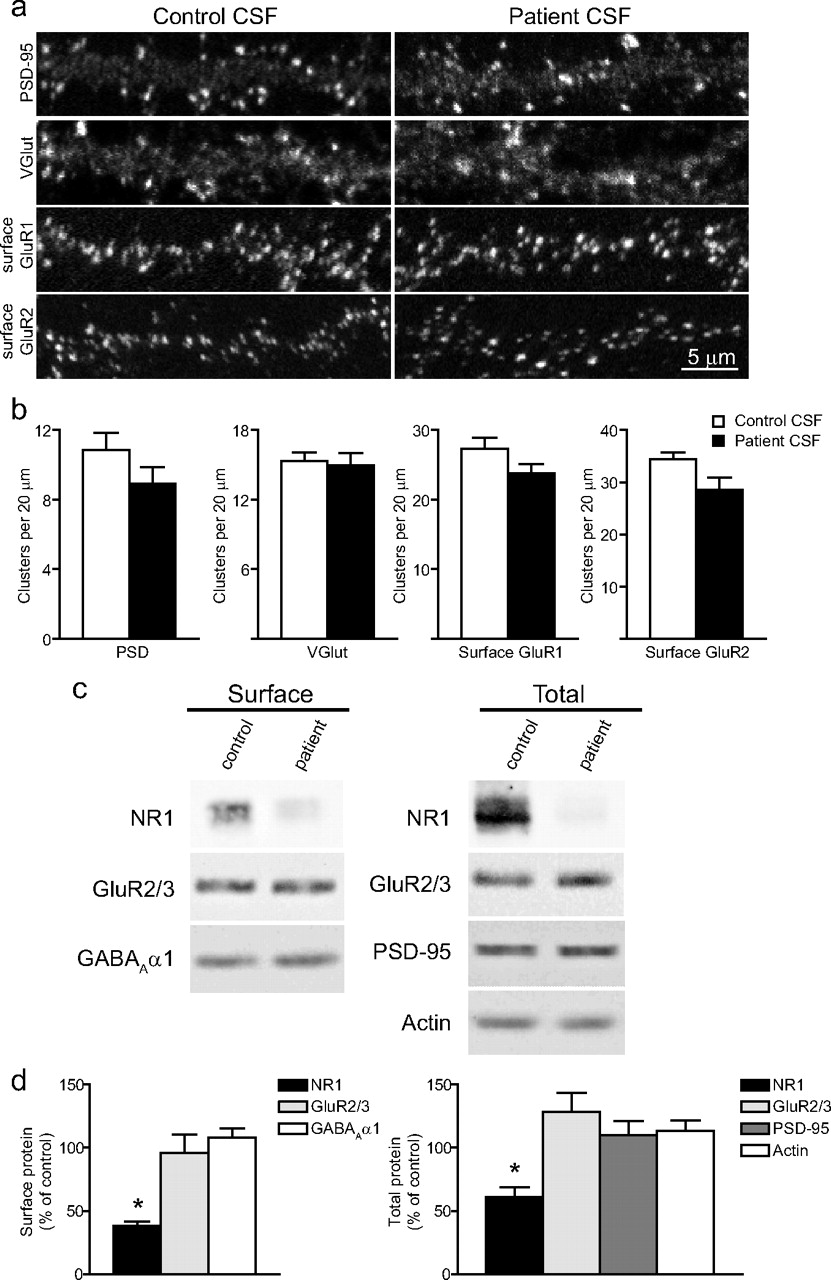
Patients' CSF and IgG treatment does not affect other synaptic components. a , Hippocampal neurons immunostained for an excitatory postsynaptic protein, PSD-95, excitatory presynaptic protein, VGlut, and surface clusters of AMPA receptor subunits GluR1 or GluR2. Scale bar, 5 μm. b , Quantification of excitatory synaptic protein density. Treatment with patient CSF did not affect the density of these excitatory synaptic proteins. All values are shown as mean ± SEM (n = 18 cells, 3 independent experiments; 1 patient, 1 control sample; Student's t test, p > 0.09). c , Western blots of excitatory postsynaptic proteins, AMPA receptor subunits GluR2/3, excitatory postsynaptic protein PSD-95 and GABAA receptors. Treatment with patient IgG did not affect surface or total neurotransmitter receptor or excitatory synapse protein levels. d , Quantification of surface (left) and total (right) protein after treatment with IgG from several patients with different antibody titer. Patient IgG resulted in a decrease in surface and total NMDAR NR1 protein but did not affect the levels of other synaptic proteins. All values are shown as mean ± SEM. The asterisk indicates significant difference (n = 3–5 Western blots; 10 patient, 2 control samples; one-way ANOVA test followed by Bonferroni's multiple-comparison test, p < 0.001).

Patient CSF treatment does not affect dendritic branching, spines, Bassoon cluster density, or cell survival. a , Hippocampal neurons transfected with the fluorescent protein, Tomato-td, imaged before and after 1 d of treatment with control or patient CSF, and traced with NeuronJ. Control (top) or patient (bottom) CSF treatment did not affect dendritic branching or complexity. Scale bar, 100 μm. b , Quantification of primary dendrite number. c , Quantification of primary dendrite length. All values are shown as mean ± SEM (n = 9 cells, 3 independent experiments; 1 patient, 1 control sample; Student's t test, p > 0.6). d , Sholl analysis of dendrite complexity before (white) and after (black) 1 d of control (left) or patient CSF (middle) treatment. Comparison of the difference before and after control and patient CSF treatment (right). e , Hippocampal neurons transfected with fluorescent protein, Tomato-td, and treated for 1 d with control or patient CSF. Control (top) or patient (bottom) CSF treatment did not affect dendritic protrusion density. Scale bar, 5 μm. f , Quantification of the density of dendritic protrusions (Student's t test, p > 0.3). g , Patient CSF treatment for 1 d does not affect Bassoon cluster density (linear regression analysis; R 2 = 0.005, p = 0.79). All values are mean ± SEM (n = 18 cells, 3 independent experiments; 12 patient, 2 control samples) (supplemental Table 1, available at www.jneurosci.org as supplemental material). h , Quantification of the density of dissociated hippocampal cells in vitro after 1 d treatment of control or patient CSF. i , Quantification of the percentage of TUNEL-positive neurons in vitro (apoptotic cells). These measures were not significantly different between control or patient CSF treatment [n = 30 fields (750 μm2), 4 independent experiments; 1 patient, 1 control sample; Student's t test, p > 0.6].
These results show that patients' antibodies specifically affect NMDAR without any demonstrable effect on AMPA or GABA receptors, other synaptic proteins, the number of excitatory synapses, and neuronal morphology or viability.
Patients' antibodies selectively decrease synaptic NMDAR currents
We next assessed the effects of patient antibodies on NMDAR function using whole-cell patch recordings of mEPSCs, which consist of a fast AMPA receptor-mediated current and a slow NMDAR current. Neurons were treated for 1 d with patient or control CSF, and spontaneous mEPSCs were recorded at −70 mV in a 0 Mg2+ extracellular solution to unmask the synaptic NMDAR-mediated component. TTX was used to block action potentials, CNQX was used to block AMPA receptor-mediated mEPSCs, APV was used to block NMDAR-mediated mEPSCs, and picrotoxin was used to block GABA receptor-mediated miniature IPSCs (Fig. 5 a).

Patients' antibodies selectively decrease synaptic NMDA currents. a , mEPSCs recorded in physiological saline with TTX, picrotoxin, and 0 Mg2+ to isolate synaptic NMDAR-mediated currents (left top trace). APV, an NMDAR antagonist, blocks the slow decay of mEPSCs leaving only AMPA receptor-mediated currents, which account for the fast rise of mEPSCs (left top middle trace). CNQX, an AMPA receptor antagonist, blocks the fast rise of mEPSCs, allowing NMDAR-mediated currents to be isolated (left bottom middle trace). Both AMPA- and NMDAR-mediated synaptic currents are blocked by CNQX plus APV (left bottom trace). Note that, under the same recording conditions, treatment of hippocampal neurons with patient CSF for 1 d dramatically reduces synaptic NMDAR-mediated currents (right traces). b , Representative average mEPSCs from neurons treated for 1 d with control CSF (left) or patient CSF (middle). The difference between the 0 Mg2+ and the 0 Mg2+ plus APV traces, plotted at right, shows the NMDAR current. Neurons treated for 1 d with patient CSF have less NMDAR-mediated synaptic current than neurons treated with control CSF. c , Effect of patient antibodies on NMDA (left) and AMPA (right) receptor-mediated synaptic currents (n = 13 cells, 7 control CSF, 6 patient CSF, 4 independent experiments; 2 patient, 2 control samples). The asterisk indicates significant difference (Student's t test, p < 0.001). Error bars indicate SEM.
In neurons treated for 1 d with CSF from control patients, CNQX blocked large, fast AMPA receptor-mediated currents, revealing small, slower NMDAR-mediated currents that were completely blocked by APV (Fig. 5 a, left). In contrast, in neurons treated for 1 d with patient CSF, CNQX blocked all mEPSCs, and no additional reduction was observed after APV (Fig. 5 a, right). This result shows that patient antibody treatment decreases NMDAR-mediated current.
To quantify the reduction in synaptic NMDAR-mediated currents, currents were examined before and after APV application. In neurons treated for 1 d with CSF from control patients, APV reduced or abolished the late, slow NMDAR-mediated component of the mEPSC (Fig. 5 b, left; c, left). In contrast, in neurons treated for 1 d with patient CSF, APV application did not further reduce the NMDAR-mediated component of the mEPSC (Fig. 5 b, middle; c, left). The difference between the 0 Mg2+ and the 0 Mg2+ plus APV traces shows that neurons treated for 1 d with patient CSF have less NMDAR-mediated synaptic current than neurons treated with control CSF (Fig. 5 b, right; c, left). No difference was observed in the peak AMPA receptor-mediated component of the mEPSC (Fig. 5 c, right). Patient antibody treatment did not affect mEPSC frequency or amplitude (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), suggesting that presynaptic release probability is unaltered. These data are also consistent with structural analyses that showed that patients' antibodies do not affect the number of excitatory synapses or the number of postsynaptic sites containing AMPA receptors. These results show that patients' antibodies specifically decrease synaptic NMDAR-mediated currents and do not affect AMPA receptor-mediated currents, consistent with the specific loss of surface, synaptically localized NMDAR clusters.
Patients' antibodies cross-link and internalize NMDA receptors
We next determined the mechanism by which patients' antibodies decrease surface NMDAR cluster density and protein. The Fc IgG domain was enzymatically removed from patients' antibodies to generate Fab fragments. These Fab fragments, like intact patient IgG, bound to surface NR1 clusters identified with commercial anti-NR1 immunostaining (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Neurons treated for 1 d with patients' Fab fragments had the same NMDAR cluster density and surface protein as neurons treated with control IgG (Fig. 6 a,b). In contrast, neurons treated for 1 d with patients' Fab fragments and anti-Fab secondary antibodies (linking two Fab fragments in a conformation similar to unmodified patients' antibodies) had significantly lower NMDAR cluster density and surface protein compared with neurons treated with control IgG (Fig. 6 a,b). These results show that patients' antibodies mediate the loss of surface NMDARs in part by binding to, capping, and cross-linking NMDARs, resulting in their internalization (Fig. 6 c).

Patients' antibodies bind, cross-link, and internalize NMDA receptors. a , Hippocampal neurons immunostained for surface and total NMDAR clusters. Treatment with patient IgG decreases surface and total NMDAR cluster density (middle left). Treatment with patient Fab fragments does not affect surface or total NMDAR cluster density (middle right), whereas treatment with divalent patient Fab fragments (Fab fragments plus anti-Fab secondary antibodies) decreases surface and total NMDAR cluster density (right). Scale bar, 5 μm. b , Effects of patient IgG, Fab fragments, and divalent Fab fragments on surface and total NMDAR cluster density (n = 30 cells, 4 independent experiments; 2 patient, 2 control samples). The asterisk indicates significant difference (one-way ANOVA test followed by Bonferroni's multiple comparison test, p < 0.001). Error bars indicate SEM. c , Diagram that outlines the effect of each treatment on surface receptor clusters.
Patients' antibodies decrease NMDA receptor cluster density and protein in rodent and human hippocampus in vivo
Our results show that, in vitro, patients' anti-NR1 antibodies lead to a selective loss of surface NMDAR clusters and their function, without loss of other synaptic components or neuron viability. To determine the effects of patients' antibodies in vivo, CSF from patients with high titers of NR1 antibodies, or control CSF from individuals without NR1 antibodies, was infused directly into the hippocampus of adult rats for 2 weeks, followed by immunostaining for human IgG to examine the diffusion and deposition of patients' antibodies, immunostaining and Western blot analysis of NMDARs and other synaptic components to assess the effects of patients antibodies, and analysis of cell death using the TUNEL assay. Patients' antibodies colocalized with NMDAR clusters in vivo as in vitro (supplemental Fig. 7a, available at www.jneurosci.org as supplemental material). Moreover, IgG from infused patient CSF, but not from control CSF, was found bound to rat hippocampus in a predictable pattern that was dependent on NMDAR density (e.g., high density in proximal dendrites of dentate gyrus) (supplemental Fig. 7b, available at www.jneurosci.org as supplemental material). This pattern was similar to the direct immunostaining of bound IgG reported in the autopsy of two patients with anti-NMDAR encephalitis (Dalmau et al., 2007). Moreover, in regions where human IgG was deposited, there was a significant decrease in NMDAR cluster density and intensity of NR1 immunostaining without affecting the number of synapses, the density of other synaptic components (Fig. 7 a–e; supplemental Fig. 7b, available at www.jneurosci.org as supplemental material), or cell death (supplemental Fig. 7c, available at www.jneurosci.org as supplemental material). The magnitude of the effects of each patient's CSF was significantly correlated with the titer of NR1 antibodies infused into rat brains (Fig. 7 b), as in in vitro studies (Fig. 1). Furthermore, the total amount of NR1 protein was reduced in rodent hippocampus infused with patients' CSF compared with the contralateral, uninfused hippocampus (Fig. 7 c).

Patients' antibodies decrease NMDA receptor cluster density and protein in rodent and human hippocampus in vivo. a , Brain sections from rats infused with control CSF (top left) contain many NMDAR clusters in CA1, whereas brain sections from rats infused with patient CSF (top right) contain significantly reduced NMDAR clusters. Presynaptic synapsin immunostaining is similar between groups (bottom left, right). Scale bar, 2 μm. b , Effect of infusion of patient CSF with varying antibody titer on NMDAR cluster density in CA1. Each point represents the mean NMDAR cluster density from three to five images from an infused rat. Patient CSF with higher antibody titers reduce NMDAR cluster density to a greater extent than low-titer samples. Infusion with patients' CSF for 2 weeks results in a titer-dependent decrease in NMDA cluster density (linear regression analysis; R 2 = 0.32, p < 0.03). All values are mean ± SEM (n = 9 animals killed after 14 d of infusion; 5 patient, 4 control samples). c , Western blot analyses of NR1 protein in patients' CSF infused rat hippocampus (top), ipsilateral (I) and contralateral (C) to infusion; tubulin protein is a loading control (below). NR1 protein is reduced in ipsilateral patient CSF infused hippocampus compared with the contralateral, uninfused hippocampus. “F” indicates NR1 protein in the frontal cortex ipsilateral to the hippocampus infused with patients' CSF. d , Synapsin cluster density (Student's t test, p > 0.6). e , Excitatory synapse density (colocalization between synapsin and AMPAR clusters; Student's t test, p > 0.5). f , Hippocampal section from a control patient (left) and from a patient with anti-NMDAR encephalitis (right) immunostained with a commercial anti-NR1 antibody. g , The boxed areas in f are shown at higher magnification. Scale bars: top, 1 mm; bottom, 25 μm. h , Intensity of NR1 immunostaining is dramatically reduced in the hippocampi of anti-NMDAR encephalitis patients (n = 2) compared with hippocampi of control patients (n = 3). The distribution of both patient values for NR1 intensity is significantly different from the distribution of control values (paired Komolgorov–Smirnov test, p < 0.05).
To investigate whether NMDAR cluster density is reduced in the brains of patients with anti-NMDAR antibodies, paraffin-embedded sections of the hippocampus of two patients with anti-NMDAR encephalitis and the hippocampus of three age-matched, anti-NR1-negative, neurologically normal individuals were immunostained with commercial anti-NR1 antibodies. The intensity of NMDAR immunostaining was significantly decreased in patients' hippocampus compared with controls (Fig. 7 f–h). Moreover, deposits of human IgG, but not complement, were identified in some of the regions with reduced NMDAR clusters (data not shown). These data show that patient anti-NMDAR antibodies reduce NMDAR clusters in rodent neurons in vitro and in vivo as well as in the brain of patients with the disorder.
Discussion
Anti-NMDAR encephalitis is a recently described disorder that is associated with antibodies against the NR1 subunit of the NMDAR and results in a well defined set of symptoms. Our previous studies noted that the resulting syndrome resembled the phenotype of animals in which the NMDAR function had been attenuated pharmacologically or genetically, suggesting that patients' antibodies decreased NMDAR levels (Dalmau et al., 2008). We now demonstrate using in vitro and in vivo studies that patients' antibodies decrease the surface density and synaptic localization of NMDAR clusters via antibody-mediated capping and internalization, independent of the presence of complement, and without affecting other synaptic proteins, AMPA receptors, or synapse density. The magnitude of these changes depends on antibody titer, and the effects are reversible when the antibody titer is reduced. Moreover, patients' NR1 antibodies decrease NMDAR- but not AMPA receptor-mediated synaptic currents. Thus, the selective loss of surface clusters abolishes NMDAR-mediated synaptic currents. These findings indicate that NR1 antibodies from patients with anti-NMDAR encephalitis decrease glutamatergic synaptic function without a substantial loss of synapses.
This reversible loss of NMDARs, and the resulting synaptic dysfunction, may underlie the deficits of memory, behavior, and cognition that are hallmarks of anti-NMDAR encephalitis (Sansing et al., 2007; Dalmau et al., 2008; Iizuka et al., 2008). Indeed, a remarkable feature of this disorder is the frequent reversibility of symptoms, even when these are severe and protracted (Iizuka et al., 2008; Ishiura et al., 2008). Previous studies with 100 patients showed a correlation between clinical outcome and antibody titers, which are often higher in CSF than serum because of intrathecal antibody synthesis (Dalmau et al., 2008; Seki et al., 2008). The work we present here demonstrates that the effect of patients' CSF on surface NMDARs correlates with the antibody titers and is coupled to changes in antibody titers and symptom severity during the course of the disease. Analysis of the hippocampus of two patients who died of this disorder showed a substantial decrease of NMDAR levels compared with the hippocampus of three age-matched, neurologically normal individuals. This decrease of NMDARs was comparable with that observed in rats infused with patients' antibodies. Moreover, we previously reported that patients' hippocampus showed deposits of IgG and absence of complement (Dalmau et al., 2007), consistent with the complement-independent antibody effects demonstrated in in vitro studies.
In the peripheral nervous system, immune-mediated disruption of synaptic structure and function results in well known disorders of neuromuscular transmission such as myasthenia gravis and the Lambert–Eaton syndrome (Sanders, 2002; Conti-Fine et al., 2006). Anti-NMDAR encephalitis provides a new model of CNS synaptic autoimmunity, antigenically different but mechanistically similar to the Lambert–Eaton syndrome in which autoantibodies, but not monovalent Fab fragments, cross-link and internalize voltage-gated calcium channels, without complement activation (Nagel et al., 1988). Both disorders may occur as paraneoplastic manifestation of a tumor that expresses neuronal proteins (e.g., small-cell lung cancer in Lambert–Eaton syndrome) (Titulaer et al., 2008) or contains ectopic nervous tissue (e.g., teratoma in anti-NMDAR encephalitis) (Dalmau et al., 2007). Moreover, in both disorders the immunological trigger of cases without tumor association is unknown, although a genetic predisposition to autoimmunity has been demonstrated or suggested (Wirtz et al., 2004, 2005; Florance et al., 2009). Although both disorders respond to immunotherapy and when appropriate tumor removal, the response of anti-NMDAR encephalitis is slower and less predictable, particularly in cases with delayed diagnosis or without a detectable tumor (Dalmau et al., 2008; Florance et al., 2009). These patients usually have persistently high CSF antibody titers, despite the effectiveness of plasma exchange or intravenous Ig in reducing serum antibody titers. In these cases, symptoms frequently respond to cyclophosphamide, which crosses the blood–brain barrier, or rituximab, which depletes memory B-cells (Sansing et al., 2007; Dalmau et al., 2008; Ishiura et al., 2008; Florance et al., 2009). As postulated in other disorders, these cells are able to cross the blood–brain barrier and are believed to undergo restimulation, antigen-driven affinity maturation, clonal expansion, and differentiation into antibody-secreting plasma cells (Hauser et al., 2008).
NMDAR dysfunction has been implicated in several other cognitive disorders, including schizophrenia (Olney and Farber, 1995; Gunduz-Bruce, 2009). Studies investigating the effects of phencyclidine and ketamine (noncompetitive antagonists of NMDARs) in human subjects show these drugs induce behaviors similar to the positive and negative symptoms of schizophrenia, along with repetitive orofacial and limb movements, autonomic instability, and seizures (Luby et al., 1959; Bailey, 1978; Castellani et al., 1982; Krystal et al., 1994; Weiner et al., 2000). In rodents, drugs that antagonize NMDAR function induce cataleptic freeze, and locomotor and stereotype behaviors, consistent with schizophrenia-like manifestations (Haggerty et al., 1984; Jentsch and Roth, 1999; Chartoff et al., 2005; Mouri et al., 2007). Furthermore, mice with decreased expression of NR1 have similar behavioral deficits, whereas mice lacking NR1 develop breathing problems and die in the perinatal period (Mohn et al., 1999). Interestingly, most patients with anti-NMDAR encephalitis present with acute schizophrenia-like symptoms and are admitted to psychiatric institutions before they develop catatonia, catalepsy, stereotyped movement disorders, and frequent autonomic instability and hypoventilation. The striking similarity between these phenotypes, the effect of patients' antibodies resulting in a dramatic decrease of surface NMDAR clusters and function, and the reduced levels of NMDARs in autopsied patients, support an antibody-mediated pathogenesis of anti-NMDAR encephalitis. The psychosis and cognitive and behavioral deficits in patients with anti-NMDAR encephalitis most likely result from NMDAR hypofunction, directly and indirectly affecting synapse and circuit structure and function in regions that bind NR1 autoantibodies. Thus, the findings we report here also support the hypothesis that NMDAR hypofunction underlies many manifestations of schizophrenia. Future studies will focus on the circuit-level dysfunction caused by patients' antibodies in hippocampus and other brain regions to begin to connect synaptic and circuit dysfunction with the behavioral abnormalities that are hallmarks of this disorder.
Footnotes
-
This work was supported by grants from the National Institutes of Health, including Grants NS046490 and MH057683 (R.J.B.-G.); Grants CA89054 and CA107192 (J.D.); Grant NS45986 and a Foederer Award (from Children's Hospital of Pennsylvania) (D.R.L.); National Research Service Award (NRSA) Grant NS056549 (E.G.H.); and NRSA Grant MH083395 (A.J.G.). We thank Drs. Marc Dichter, Myrna Rosenfeld, and Steven Scherer for helpful discussions, and Marion O. Scott and Margaret Maronski for technical assistance. E.G.H. and X.P. designed and performed all experiments, except as indicated below, analyzed the data, and wrote the paper; A.J.G. performed the Western blot experiments and analysis; M.L. generated the Fab fragments and performed the human tissue staining; L.Z. designed and performed the rat infusion experiments; R.T. performed the time-lapse imaging experiments; T.D.P. contributed to electrophysiological experiments; D.R.L. contributed to experimental design and interpretation; and J.D. and R.J.B.-G. contributed to all aspects of this project and wrote this paper.
- Correspondence should be addressed to Dr. Rita J. Balice-Gordon, Department of Neuroscience, University of Pennsylvania School of Medicine, 215 Stemmler Hall, Philadelphia, PA 19104-6074. rbaliceg{at}mail.med.upenn.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}