

Abstract
In the G-protein-coupled receptor phototransduction cascade, visual Arrestin 1 (Arr1) binds to and deactivates phosphorylated light-activated opsins, a process that is critical for effective recovery and normal vision. In this report, we discovered a novel synaptic interaction between Arr1 and N-ethylmaleimide-sensitive factor (NSF) that is enhanced in a dark environment when mouse photoreceptors are depolarized and the rate of exocytosis is elevated. In the photoreceptor synapse, NSF functions to sustain a higher rate of exocytosis, in addition to the compensatory endocytosis to retrieve and to recycle vesicle membrane and synaptic proteins. Not only does Arr1 bind to the junction of NSF N-terminal and its first ATPase domains in an ATP-dependent manner in vitro, but Arr1 also enhances both NSF ATPase and NSF disassembly activities. In in vivo experiments in mouse retinas with the Arr1 gene knocked out, the expression levels of NSF and other synapse-enriched components, including vGLUT1 (vesicular glutamate transporter 1), EAAT5 (excitatory amino acid transporter 5), and VAMP2 (vesicle-associated membrane protein 2), are markedly reduced, which leads to a substantial decrease in the exocytosis rate with FM1-43. Thus, we propose that the Arr1 and NSF interaction is important for modulating normal synaptic function in mouse photoreceptors. This study demonstrates a vital alternative function for Arr1 in the photoreceptor synapse and provides key insights into the potential molecular mechanisms of inherited retinal diseases, such as Oguchi disease and Arr1-associated retinitis pigmentosa.
Introduction
Arrestins play established roles in the regulation of G-protein-coupled receptor (GPCR) signaling by specifically binding to agonist-activated, phosphorylated receptors, terminating additional G-protein activation. There are four distinct members in the arrestin superfamily expressed in vertebrates (Craft and Whitmore, 1995). Arrestin 1 (Arr1) is expressed in both rod and cone photoreceptors, whereas Arrestin 4 (Arr4) (cone or X-arrestin) is expressed in cone photoreceptors (Nikonov et al., 2008). Both visual arrestins are also highly expressed in pinealocytes (Craft et al., 1994), whereas β-arrestin 1 and β-arrestin 2 are ubiquitously expressed and mediate the desensitization of β-adrenergic receptors and many other GPCRs (Lefkowitz et al., 1992). Moreover, β-arrestin 1 also directs receptors to coated pits for internalization and switches signaling to alternative pathways (Gagnon et al., 1998; Orsini and Benovic, 1998; Lefkowitz and Shenoy, 2005).
Previous work, which includes gene knock-outs of mouse Arr1, demonstrated its function in modulating the phototransduction shutoff and recovery in the rod and cone photoreceptor outer segment by binding to light-activated, phosphorylated opsins and quenching the GPCR phototransduction cascade (Wilden et al., 1986; Xu et al., 1997; Nikonov et al., 2008). Furthermore, mutations in the human gene encoding ARR1 lead to either an inherited recessive form of stationary night blindness known as Oguchi disease, which is associated with abnormal electroretinograms (ERGs) of reduced a-wave amplitudes and absent scotopic b-waves (Fuchs et al., 1995; Nakazawa et al., 1997), or retinitis pigmentosa (Nakamachi et al., 1998), which leads to retinal degeneration.
Subcellular localization of signaling molecules is vital for their biological function. Constitutively shuttling of signaling proteins and the redistribution of their interactive partners between subcellular compartments are important for the modulation of their activity. With light exposure, Arr1 is translocated to the outer segments in rods and cones and also contributes to light adaptation, whereas in dark-adapted photoreceptors, high concentrations of Arr1 localizes to the inner segment, perinuclear region, and synaptic terminals (Broekhuyse and Winkens, 1985; Philp et al., 1987; Whelan and McGinnis, 1988; Arshavsky, 2003; Brown et al., 2010). Based on the photoreceptor synaptic localization in the dark of Arr1 and alternative functions of the β-arrestins in neuronal synapses, we examined potential binding partners of Arr1.
In this study, we report a protein–protein interaction between Arr1 and N-ethylmaleimide-sensitive factor (NSF), an ATPase with diverse cellular activities, which is critical for soluble NSF attachment protein receptor (SNARE) complex disassembly in membrane trafficking events, including neurotransmitter release. Arr1 coimmunoprecipitates with NSF and deletion analysis maps the relevant binding site to the junction of two functional domains of NSF. We also provide evidence supporting the interaction of Arr1 with NSF to modulate its ATPase activity and to drive disassembly of the SNARE complex. Finally, we observe that synaptic vesicle recycling in photoreceptors is dramatically decreased and analysis of the photopic ERG b-wave also demonstrate a light adaptation defect in Arr1−/− mice (Brown et al., 2010). These cumulative findings demonstrate that normal photoreceptor synaptic function involves the ability of Arr1 to regulate and to enhance the dark-associated activity of NSF such that photoreceptor synaptic vesicle demand is met efficiently in these specialized sensory cells.
Materials and Methods
Animals.
Mice were dark-reared in the University of Southern California (USC) Vivarium following the appropriate established guidelines of the National Institutes of Health and adapted by the Institutional Animal Care and Use Committee of the University of Southern California. Breeding pairs of the Arrestin 1 (Arr1−/−) knock-out mice (Xu et al., 1997; Burns et al., 2006) and transgenic mouse cone arrestin (mCAR-H) backcrossed to the Arr1−/− (mCAR-Harr1−/−) (Chan et al., 2007) were generously provided by Dr. Jeannie Chen (USC). Colony control (C57BL/6J and SVJ129 [wild-type (WT)] mixed genetic background) mice were created by breeding heterozygote littermates of the Arr1+/−Arr4+/− knock-out mice. Their offspring were verified by PCR genotype analysis for both Arr1+/+ and Arr4+/+ and used as breeders (WT) (Nikonov et al., 2008). All mice were born and maintained in a dark environment. Both male and female mice were used for these experiments.
Antibody pull-down assays and coimmunoprecipitation.
To verify the potential physiological interaction of Arr1 with NSF, antibody pull-down assays and coimmunoprecipitation experiments were performed as previously described (Zhu et al., 2003). The reactions were performed in both light and dark (infrared or dim red light) conditions. Briefly, retinas from WT mice were homogenized in lysis buffer [50 mm Tris-HCl, pH 8.0, 1% Triton X-100, 150 mm NaCl with 1× proteinase inhibitor mixture (Roche)], sonicated on ice for 30 s, incubated at 4°C with gentle shaking for 30 min, and centrifuged at 13,000 × g for 15 min. Equal volumes of supernatant were precleared for 1 h incubation with 50 μl of a 50% slurry of protein G-agarose (KPL). After centrifugation, the Arr1 in the supernatant was immunoprecipitated using 10 μl of mouse monoclonal antibody (MAb) D9F2 specific for Arr1 (amino acids 361-369/PEDPDTAKE). The tubes were incubated at 4°C overnight and then incubated with 50 μl of protein G-agarose at 4°C for 2 h. The affinity-purified anti-rabbit mouse cone arrestin (mCar-LUMIj) polyclonal antibody (PAb) (Zhu et al., 2002) was used as a nonspecific antibody control. The pellets were washed five times with the lysis buffer containing no proteinase inhibitor. The immunoprecipitated proteins were eluted after boiling for 5 min in SDS-PAGE sample buffer and subjected to 10% SDS-PAGE followed by transfer to polyvinylidene difluoride (PVDF) membrane. Immunoblot analysis was performed using a PAb specific for NSF (1:10,000; Millipore; 07-364), and NSF was visualized using the enhanced luminol-based chemiluminescent (ECL) system (GE Healthcare). To confirm equal loading of Arr1, the PVDF membrane was stripped and reprobed to visualize Arr1 using an affinity-purified anti-rabbit PAb C10C10 (1:10,000; amino acids 293-301/RERRGIALD), which was characterized previously (Brown et al., 2010). This antibody is specific for Arr1 and is similar to the results with MAb D9F2.
Isolation of proteins for mass spectrometry analysis.
The identification of potential interacting partners for Arr1 was performed by liquid chromatography–tandem mass spectrometry at the USC School of Pharmacy Proteomics Core Facility. Proteins were prepared and analyzed by mass spectrometry using a method similar to that previously described (Gallaher et al., 2006). Briefly, the Arr1-interacting proteins were separated by electrophoresis on 10% SDS-PAGE, and the proteins were visualized by Coomassie blue stain. Excised bands were destained and dehydrated, and then digested with trypsin at 37°C overnight. The supernatant was collected and analyzed by liquid chromatography–tandem mass spectrometry. Protein identification was performed with the tandem mass spectrometry search software Mascot 1.9 (Matrix Science) with confirmatory or complementary analyses with TurboSequest as implemented in the Bioworks Browser 3.2, build 41 (Thermo Fisher Scientific).
Plasmid construction.
Mouse cDNA encoding NSF was amplified by PCR from Image clone 4506351(Open Biosystems) with oligonucleotide primers, sense, +NSF-f76, 5′-ATGGCGGGCCGGACTATGCA-3′, and antisense, −NSF-r2310, 5′-TCAATCAAAGTCCAGGGGAC-3′ (NM_008740 mouse NSF complete coding sequence) was subcloned into the PtrcHis-TOPO vector (Invitrogen). Mouse Arr1, soluble NSF attachment protein 25 (SNAP-25), α-SNAP, and VAMP-2 cDNAs were amplified with PCR technology with specific 5′-sense and 3′-antisense primers and subcloned into the PtrcHis-TOPO vector after PCR amplification from cDNA from total mRNA isolated from mouse retina. The cDNA encoding the mouse NSF and Syntaxin 4 were subcloned as fragments into the multiple cloning site using restriction endonucleases for EcoRI–XhoI into the pGEX-4T1 vector (GE Healthcare). The sequences of all constructs were confirmed by DNA sequencing. Recombinant fusion proteins were purified on columns using the histidine (His) tags with the Bio-Rad Profinia purification system as recommended by the manufacturer.
Glutathione S-transferase pull-down and in vitro binding assay.
To define the functional domains in NSF that interact with Arr1, His6-tagged, NSF-truncated segments of varying lengths (amino acid residues 1-744, 251-744, 197-744, and 1-205∧478-744) and glutathione S-transferase (GST)-tagged NSF1-250 and NSF1-197 were constructed. GST-Arr1 proteins (3 μg) were immobilized on glutathione-agarose beads in 25 mm HEPES-KOH, pH 7.4, 200 mm KCl, 1% Triton X-100, 10% glycerol, and 1 mm DTT (buffer A), and then incubated with His6-NSF1-744, 251-744, 197-744, or 1-205∧478-744 at 4°C for 1 h. GST-NSF1-250 or GST-NSF1-197 proteins (3 μg) were also immobilized on glutathione-agarose beads in buffer A and then incubated with His6-Arr1 at 4°C for 1 h. After six washes in buffer A plus 2 mm ATP, 8 mm MgCl2 (buffer B), bound proteins were eluted with 20 mm glutathione and detected by immunoblot analysis.
To evaluate the influence of the ATPase state of NSF on its direct interaction with Arr1, GST-tagged Arr1 (amino acids 1-403) (3 μg), or truncated Arr1 (amino acids 1-191), Arr1 (amino acids 1-370) were immobilized on glutathione-agarose beads in buffer A. Beads were washed twice with buffer B, or 2 mm ATP-γ-S and 8 mm MgCl2 in the presence of 1% BSA, and incubated with 3 μg of His6-tagged NSF at 4°C for 1 h. After four washes in buffer B without BSA, bound proteins were eluted with 20 mm glutathione and detected by immunoblot analysis as described above. To determine the effect of the Arr1 binding to NSF-ATPase activity, the same procedure was performed in the presence of 8 mm MgCl2, 10 mm EDTA, and 2 mm ATP or ATP-γ-S. Densitometric analysis was conducted using the ImageQuant TL software (GE Healthcare).
Quantitative real-time PCR technology (reverse transcription-PCR).
Total RNA was prepared from dark-adapted (DA) (24 h) and light-adapted (LA) (1 h) frozen retinas using Trizol reagent (Invitrogen). The cDNA made from 0.5 μg of total retina RNA was prepared using a reverse transcription system from Invitrogen with oligo-dT20. Each quantitative reverse transcription (RT)-PCR was set up in a final volume of 25 μl containing 12.5 μl of SYBR Green from Superarray. Reactions were done in triplicate on 96-well plates and quantified (LightCycler 480 Real-Time PCR System; Roche). Data analysis was performed using the LightCycler software, version LCS480 1.2.0. The housekeeping gene, mouse glyceraldehyde-3-phosphate dehydrogenase (mGAPDH), was used as the reference to normalize the expression levels of the NSF, vesicle-associated membrane protein 2 (VAMP2), excitatory amino acid transporter 5 (EAAT5), and vesicular glutamate transporter 1 (vGLUT1) transcripts. Values for RT-PCR for retinas from light-adapted WT mice were set to 1. Quantitative RT-PCR primer pair sequences, sense/forward (+/f) and antisense/reverse (−/r), were as follows (gene name, accession number; nucleotide numbers): +mGAPDH-f148-5′-ACCCCTTCATTGACCTCAACTACATGG-3′ (NM_008084; 148-174); −mGAPDH-r303-5′-ATTTGATGTTAGTGGGGTCTCGCTCCT-3′ (NM_008084; 277-303); +qNSF-f2158-5′-GCTCAGCAAGTCAAAGGGAA-3′ (NM_008740; 2158-2177); −qNSF-r2248-5′-GGTACTCAGGATCCATCTGC-3′ (NM_008740; 2229-2248); +qVAMP2-f213-5′-GTGGATGAGGTGGTGGACAT-3′ (NM_009497; 213-232); −qVAMP2-r348-5′-GCTTGGCTGCACTTGTTTCAA-3′ (NM_009497; 328-348); +vGLUT1-f1532-5′-GTGCAATGACCAAGCACAAG-3′ (NM_182993; 1532-1551); −vGLUT1-r1601-5′-TAGTGCACCAGGGAGGCTAT-3′ (NM_182993; 1582-1601); +EAAT5-f1250-5′-GCTCTGCTCATTGCGTTG-3′ (NM_146255; 1250-1267); −EAAT5-r1317-5′-AGCAGGCACTTGAAGGTGAT-3′ (NM_146255; 1298-1317).
Immunoblot analysis.
Protein extracts from dark-adapted (24 h) and light-adapted (1 h) WT or Arr1−/− mouse retina were prepared using NP-40 lysis buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% NP-40, and 1 mm EDTA). The protein concentrations were determined using the BCA (bicinchoninic acid) protein assay kit (Pierce). Twenty micrograms of extracts were separated on 12% SDS-PAGE and transferred to PVDF membrane. After 1 h blocking with 5% nonfat milk, the membranes were incubated in different primary antibody [anti-NSF PAb (1:2500; Abcam), anti-EAAT5 PAb (1:5000; a gift from Dr. David Pow, University of Queensland, Brisbane, Queensland, Australia), anti-vGLUT1 MAb (1:2000; Millipore), anti-VAMP2 MAb (1:5000; Synaptic System), anti-SNAP-25 PAb (1:3000; Abcam)] at 4°C overnight. After washing, the blots were then incubated with appropriate anti-horseradish peroxidase-conjugated secondary antibodies (1:10,000; Bio-Rad) at room temperature for 1 h. The proteins on the membranes were detected using the enhanced chemiluminescence system (ECL) (GE Healthcare). The blots were also probed with mouse GAPDH as an internal loading control. Densitometric analysis was conducted using the ImageQuant TL software (GE Healthcare). Each experiment was repeated at least three times with independent retinal samples from different groups of mice.
Immunohistochemistry.
To further verify the potential physiological relevance of the in vitro interaction between Arr1 and NSF, we performed indirect fluorescent dual immunohistochemical localization as described previously (Zhu et al., 2003). Briefly, the eyes were enucleated under infrared or light conditions, the cornea was removed, and the eyes were immediately immersed in 4% (w/v) paraformaldehyde (PFA) in 0.1 m PBS for 2 h at room temperature. Eyes were rinsed in PBS, pH 7.4, and cryoprotected in 30% sucrose–PBS solution at 4°C overnight, and then embedded in OCT (ornithine carbamyl transferase) (Tissue-Tek). Sections (7 μm) of the retina were cut through the optic nerve with a cryostat, and retina sections were washed in 0.1 m PBS, blocked in blocking buffer (1% BSA, 1% NGS, 1% Triton X-100 in 1× PBS), and incubated with anti-mouse MAb D9F2 (1:20,000) for Arr1 and anti-rabbit PAb (1:2500) for NSF at 4°C overnight. To visualize binding of the primary antibodies, sections were incubated in secondary antibody conjugated to Alexa Fluor 488 or 568, respectively (1:500; Invitrogen), and TO-PRO-3 (1:2500; Invitrogen) nuclear staining for 1 h at room temperature. Samples stained without either or both of the primary antibodies were included as controls to ensure the dual-staining pattern results were reliable (data not shown). The sections were visualized and photographed with a Zeiss confocal laser-scanning microscope (Carl Zeiss).
Cell culture and transfection.
COS-7 cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and antibiotics and maintained at 37°C in a humidified atmosphere with 5% CO2 as previously described (Zhu et al., 2003). The plasmids of interest were transfected into COS-7 cells using Fugene Transfection Reagent according to the manufacturer's instruction (Roche). COS-7 cells were cotransfected with plasmids encoding empty vector, pcDNA4/HisMax-Arr1, pcDNA3.1/GFP-NSF, or pcDNA4/HisMax-Arr1 plus pcDNA3.1/GFP-NSF for 48 h. Cell were washed in PBS and fixed in freshly prepared 4% PFA. Fixed cells were permeabilized and blocked with 0.1% Triton X-100 and 2.5% normal goat serum in PBS for 30 min at room temperature and then incubated with primary and secondary antibodies diluted in the same buffer for 1 h at room temperature. After three washes with PBS, the cells were mounted and viewed under a Zeiss confocal laser-scanning microscope.
NSF ATPase activity assay.
The basal ATPase activity of NSF was measured by a colorimetric assay (Huynh et al., 2004). Recombinant NSF (0.2 μg/μl) was pretreated with 10 mm N-ethylmaleimide (NEM), a NSF inhibitor, as a negative control or with increasing concentrations of recombinant Arr1 protein for 10 min at 37°C. ATPase reaction buffer (25 mm Tris-HCl at pH 9.0, 100 mm KCl, 0.65 mm β-mercaptoethanol, 2 mm MgCl2 and 10% glycerol, 10 mm ATP) was added to the mixture. The release of inorganic phosphate was measured by adding BIOMOL Green (BIOMOL) and the absorbance at 620 nm was determined on a Benchmark Plus microplate reader (Bio-Rad). Corrections were made for minor ATPase contaminants, nonenzymatic hydrolysis of ATP, and preexisting phosphate in protein samples by subtracting the NEM-treated control values from those obtained at 37°C.
NSF disassembly activity assay.
The disassembly activity of NSF was measured by a coprecipitation assay as described previously (Matsushita et al., 2003). Recombinant His6-NSF (0.1 μg/μl) was pretreated with buffer or with increasing concentration of recombinant Arr1 protein for 10 min at 37°C. Immobilized GST-Syntaxin 4 (0.1 μg/μl) on glutathione-agarose beads was incubated with recombinant His6-α-SNAP (0.1 μg/μl) and SNARE polypeptide (0.1 μg/μl each of VAMP-2 and SNAP-25) at 4°C for 1 h. The beads were then washed three times with binding/wash buffer (4 mm HEPES, pH 7.4, 0.1 m NaCl, 1 mm EDTA, 3.5 mm CaCl2, 3.5 mm MgCl2, and 0.5% Triton X-100). The mixture of NSF with increasing concentration of Arr1 was added to the beads and then incubated in binding/wash buffer with 2.5 mm ATP/5 mm MgCl2 for 30 min at 4°C with rotation. The beads were washed with binding/wash buffer six times, mixed with SDS-PAGE sample buffer at 60°C for 3 min, resolved on 12% SDS-PAGE, transferred to PVDF, and then analyzed with specific antibodies on immunoblots.
Evaluation of exocytosis rate using FM1-43 staining.
The procedure for activity-dependent staining with FM1-43 was performed as described previously (Miller et al., 2001; Caicedo et al., 2005). After 1 h of light adaptation, WT and Arr1−/− mice were used. Each retina with its cornea was removed for individual eyecup preparations and immersed in mammalian Ringer's solution. The eyecups were superfused with depolarizing Ringer's solution containing 10 μm fixable FM1-43 (Invitrogen) with or without 25 mm K+ for 30 min. After incubation, preparations were washed three times with Ringer's solution containing 5 mm Co2+ for 5 min to minimize dye release through calcium-dependent exocytosis and to eliminate background staining. Eyecups were fixed in 4% PFA for 1 h, cryoprotected in 30% sucrose, and cut on a cryostat. FM1-43 staining was visualized and photographed with a Zeiss confocal laser-scanning microscope.
Electroretinography.
Photopic, cone-driven ERG responses were recorded from mice in the presence of a steady white background light to suppress the rod response, as previously described (Brown et al., 2010). In this current study, the background light was turned on, and after 1 min of light adaptation, a single maximum intensity flash was delivered and averaged every 2 min until 15 min of recording. The amplitudes from at least eight mice in each group were averaged and two-way ANOVA with Bonferroni's post tests were performed on the data at each time point during recording.
Statistical analysis.
Analysis of statistical significance was determined using two-tailed Student's t test and one-way ANOVA followed by Bonferroni's multiple-comparison test. Data are presented as means ± SEM. In all cases, p < 0.05 denotes statistical significance.
Results
NSF is an interacting partner for Arr1 in the photoreceptor synapse
To investigate potential physiologically relevant photoreceptor synaptic partners of Arr1, we performed a pull-down immunoprecipitation assay from mouse colony control WT mouse retina homogenates with an Arr1-specific MAb D9F2, followed by SDS-PAGE separation and analysis. Among the major bands identified by mass spectrometry, the major band identified was NSF (NP_032766) (for details, see supplemental Fig. 1a, available at www.jneurosci.org as supplemental material). Protein database search using Mascot analysis (Matrix Science) revealed ∼22% tryptic peptide coverage with a total Mascot score of 610 of a protein corresponding to NSF (supplemental Fig. 1b, available at www.jneurosci.org as supplemental material). NSF is a well characterized ATPase that is an essential component of various membrane fusions including the exocytosis of synaptic vesicles (Kawasaki et al., 1998; Tolar and Pallanck, 1998; Singh et al., 2004).
To verify the interaction between Arr1 and NSF, we performed additional coimmunoprecipitation assays from retinal homogenates from WT and Arr1 knock-out (Arr1−/−) mice. Immunoblot analysis with a rabbit anti-NSF PAb identified a single band in WT and Arr1−/− retinal homogenates (Fig. 1a), whereas the anti-Arr1 MAb identified a 48 kDa band corresponding to Arr1 only in the WT, but not in the Arr1−/− retinal homogenates (Fig. 1b). A protein of ∼76 kDa, corresponding to NSF, was coimmunoprecipitated with Arr1 MAb from the WT but not from the Arr1−/− retina homogenates (Fig. 1c), confirming the specific protein–protein interaction of Arr1 with NSF. The interaction between Arr1 and NSF is greater in DA retinas compared with LA retinas (Fig. 1d,e). We also observed that NSF was not coimmunoprecipitated with anti-rabbit cone arrestin Pab mCAR-LUMIj in WT retinal homogenates (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material).

NSF coimmunoprecipitated with Arr1 in mouse retina. Arr1 was immunoprecipitated from the homogenates of 10 mouse retinas using the Arr1 monoclonal antibody (MAb D9F2). NSF and Arr1 proteins are shown in the initial retinal homogenates (a, b). NSF coimmunoprecipitated with Arr1 in the WT retinal homogenates (c). The retinal homogenates from Arr1−/− mice were used as a negative control (c). Immunoprecipitated proteins were isolated from mouse retinas under either light- or dark-adapted conditions, analyzed by SDS-PAGE, transferred to PVDF membranes, and detected by ECL using anti-rabbit NSF (d) or anti-mouse Arr1 (e) antibodies. IP, Immunoprecipitation; IB, immunoblot analysis.
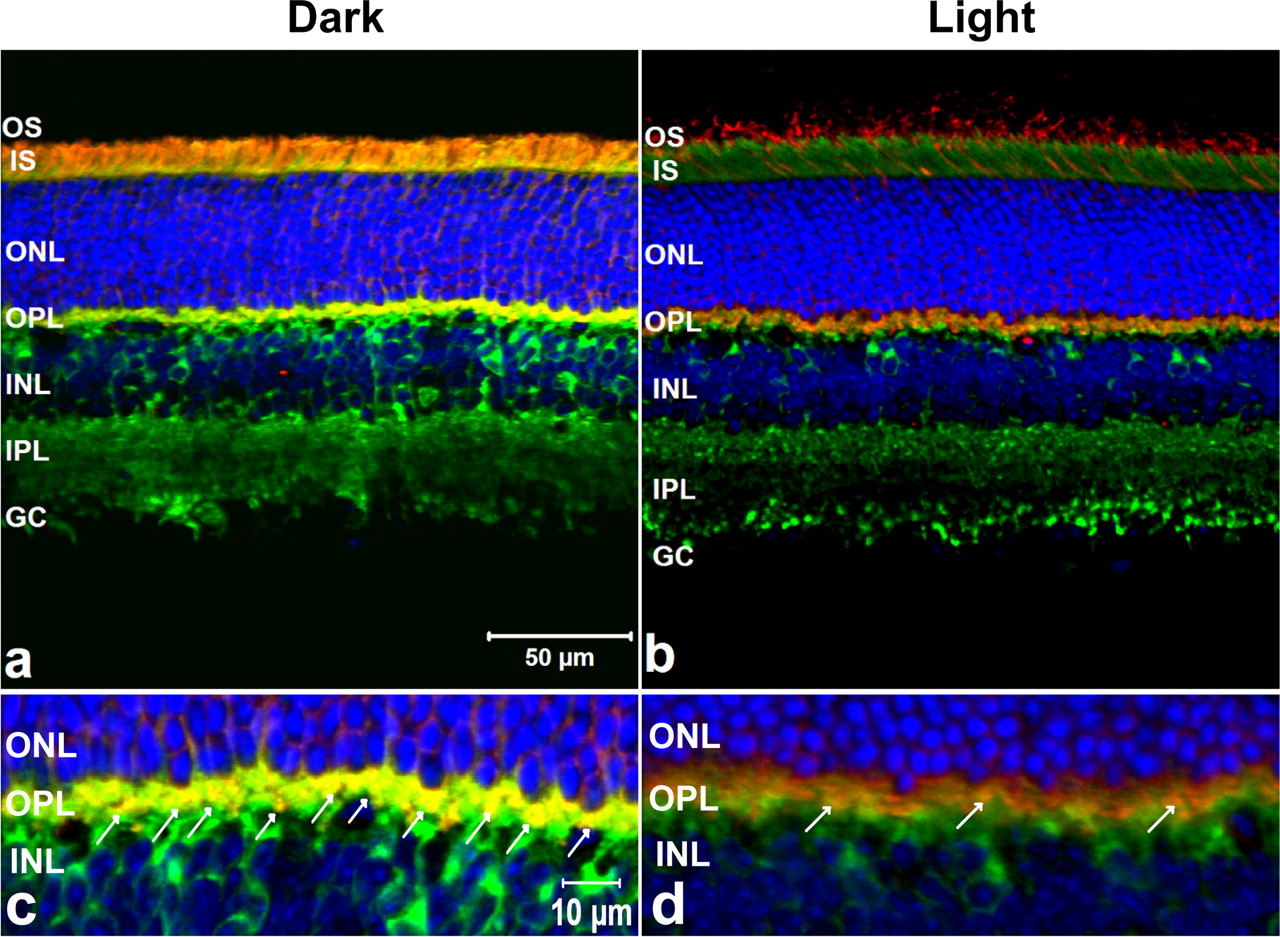
To be a relevant interacting partner for Arr1, NSF must colocalize with Arr1 in the appropriate subcellular compartments within the retina. In this study, retinas from WT mice were either light- or dark-adapted; their retinas were prepared for immunohistochemistry and sections were examined with a confocal microscope. The retinal sections were incubated with Arr1 MAb and NSF PAb that were used in the coimmunoprecipitation experiments, followed with the appropriate secondary antibodies, Alexa Fluor 568 goat anti-mouse IgG antibody and Alexa Fluor 488 goat anti-rabbit IgG antibody, respectively. In the DA retinas, the majority of immunoreactive Arr1 was localized to the inner segments, perinuclear region with a small fraction in the synaptic terminals (Fig. 2a). In the LA retinas, the majority of Arr1 translocated to the outer segments (Fig. 2b). NSF showed some perinuclear labeling in the inner nuclear layer and intensive staining in the outer plexiform layer (OPL) and inner plexiform layer (IPL) in retinas from both LA and DA mice. Dual immunohistochemical labels showed that Arr1 and NSF extensively colocalized in the photoreceptor terminals (OPL) (Fig. 2c). In the DA retinas, Arr1 and NSF colocalized in the junction between the outer segment and inner segment; however, in the LA retinas, Arr1-immunoreactive label was limited and colocalized with NSF only in the OPL (Fig. 2d). These observations further confirm the potential physiological interactions between Arr1 and NSF in vivo. Their intense dual expression pattern at the specialized ribbon synapse of photoreceptors in the dark condition suggests that Arr1 and NSF may be critical partners for modulating neurotransmitter transmission. Alternatively, Arr1 may be acting at the synapse as a cochaperone for NSF in the SNARE complex to regulate exocytosis.

Immunohistochemical fluorescent labeling of NSF and Arr1 in the WT mouse retinas. Adult WT mouse retina frozen sections were triple labeled fluorescently with the anti-mouse Arr1 MAb D9F2 (red), anti-rabbit NSF PAb (green), and appropriate secondary antibodies and TO-PRO-3 for the nuclei (blue). The immunoreactive staining pattern of NSF is mainly in the OPL and IPL in DA (a) or LA (b) retinas. The Arr1 MAb-immunoreactive label is predominantly in the inner segment, perinuclear area, and a fraction in the photoreceptor terminal in DA retinas, whereas the Arr1 MAb immunoreactivity is translocated to the outer segment in LA retinas. The Arr1 MAb immunoreactivity is extensively dual localized with NSF immunological staining in the OPL in DA retinas (c; arrows) and only limited dual staining in the OPL in LA retina (d; arrows). OS, Outer segment; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; RGC, retinal ganglion cell layer. Scale bars: a, b, 50 μm; c, d, 10 μm.
The Arr1–NSF complex formation is enhanced by ATP
Each NSF monomer has three functional domains (Whiteheart et al., 1994; Nagiec et al., 1995): an N-terminal domain (N, amino acids 1-205) that is required for the binding to α-SNAP and SNARE proteins; and two homologous ATP-binding domains, D1 (amino acids 206-477), in which the ATP hydrolytic activity is associated with NSF-driven SNARE complex disassembly, and D2 (amino acids 478-744), which is responsible for hexamerization. To define the potential functional domain in NSF that interacts with Arr1, His6-tagged or GST-tagged NSF-truncated segments of varying lengths were constructed and a GST pull-down assay was performed. Our results indicate that only the NSF fragments that included amino acids 197-250, which is in the junction of the N and D1 domains (Fig. 3i), interact with Arr1.

Functional analysis of the interaction between Arr1 and NSF. i, Mapping the binding site of Arr1 on NSF. NSF has three functional domains: N domain is for SNARE protein complex binding, D1 domain is for fusion complex disassembly, and D2 domain is for NSF homo-hexamer formation. To define the region in NSF that interacts with Arr1, His6-tagged truncated segments of NSF with varying lengths (amino acid residues 1-744, 197-744, 1-477, and 1-205∧478-744) and GST-tagged NSF (amino acids 1-250) and NSF (amino acids 197-250) were constructed, and GST pull-down assay was performed. Bound proteins were detected by immunoblot analysis with anti-mouse His-tag MAb and anti-mouse Arr1 MAb D9F2. Arr1 only bound NSF fragments that included the amino acids 197-250, which was located in the junction of the N and D1 domains. ii, Defining the binding site of NSF on Arr1. GST pull-down assay demonstrated a direct interaction between the N-terminal domain of Arr1 and NSF. GST alone or GST-Arr1 (amino acids 1-191), GST-Arr1 (amino acids 1-370), and GST-Arr1 (amino acids 1-403) coupled to beads were incubated with purified His6-tagged NSF in the binding buffer containing 2 mm ATP plus 8 mm MgCl2 or 2 mm ATP-γ-S plus 8 mm MgCl2. Bound NSF was detected by immunoblot analysis using anti-rabbit NSF antibody. iii, Association of Arr1 with NSF in COS-7 cells. COS-7 cells coexpressing pcDNA4-HisMax-Arr1 and pcDNA3.1-GFP-NSF (amino acids 1-744) or NSF (amino acids 197-250) were processed for immunofluorescence and analyzed by confocal microscopy for the extent of colocalization of pcDNA4-HisMax-Arr1 [b, e, h, red (anti-mouse His-tag MAb); c, f, i, merge] with pcDNA3.1-GFP-NSF (amino acids 1-744) [a, green (GFP); c, merge], NSF (amino acids 197-250) [d, green (GFP); f, merge], and empty vector [g, green (GFP); i, merge].
We further mapped the regions of Arr1 that interact with NSF using in vitro GST pull-down assays. GST-Arr1 (amino acids 1-191), GST-Arr1 (amino acids 1-370), GST-Arr1 (amino acids 1-403), and GST control proteins were immobilized on glutathione-agarose beads and incubated with recombinant His6-NSF. His6-NSF bound specifically to each of the GST-Arr1 recombinant proteins but not the GST control (Fig. 3ii). These data demonstrate that the N-terminal domain (amino acids 1-191) of Arr1 interacts with NSF. Others have shown the association of NSF with other binding partners exhibited an ATP dependence (Banerjee et al., 1996; Nishimune et al., 1998). In the presence of ATP-γ-s/MgCl2 (nonhydrolyzable ATP) or ATP/EDTA (Fig. 4), the interaction of each GST-Arr1 recombinant construct and NSF showed enhanced binding compared with the ATP/MgCl2 (hydrolyzable ATP). These results also confirm that the Arr1 binding to NSF is enhanced by ATP.

Arr1–NSF complex formation is enhanced by ATP. GST-hybrid Arr1 proteins (3 mg) were immobilized on glutathione-agarose beads and then incubated with His6-tagged NSF protein (3 mg) and buffers containing the following: lane 1, 2 mm ATP/8 mm MgCl2/without EDTA; lane 2, 2 mm ATP/8 mm MgCl2/with EDTA; lane 3, only 8 mm MgCl2; lane 4, 2 mm ATP-γ-S/8 mm MgCl2. Bound proteins were eluted with 20 mm glutathione and detected by immunoblot analysis. The amount of NSF bound was normalized to the amount of Arr1 immunoprecipitated. Results represent the means ± SEM for three independent experiments. **p < 0.01; ***p < 0.001.
Interactions between Arr1 and NSF were examined in COS7 cells that were transiently cotransfected with plasmids encoding either empty vector, pcDNA4-HisMax-Arr1 and pcDNA3.1-GFP-NSF (amino acids 1-744), or pcDNA3.1-GFP-NSF (amino acids 197-250) for 48 h. As shown in Figure 3iii, both GFP-NSF (amino acids 1-744) (Fig. 3iiia) and GFP-NSF (amino acids 197-250) (Fig. 3iiid) showed a cytoplasmic diffuse fluorescent pattern. HisMax-Arr1 was localized to the cytosol and perinuclear region. The GFP empty vector (Fig. 3iiig) showed diffuse staining throughout the cell, including the nucleus. The GFP-NSF (amino acids 1-744) and GFP-NSF (amino acids 197-250) extensively colocalized with HisMax-Arr1 (Fig. 3iiib,e, His-Arr1; c,f, merge), whereas the GFP empty vector had no dual staining with HisMax-Arr1 in the COS7 cells (Fig. 3iiih, HisMax-Arr1; i, merge).
Arr1 enhances NSF ATPase activity and NSF-driven SNARE complex disassembly
To examine the effect of Arr1 binding on NSF ATPase activity, which is critical for NSF function, increasing concentrations of recombinant His6-Arr1 were added to 10 μg of recombinant His6-NSF, and the ATPase activity of the NSF was measured by a colorimetric assay. Arr1 significantly enhanced NSF ATPase activity in a dose-dependent manner (Fig. 5). We next explored the effect of Arr1 on NSF disassembly activity. NSF has been shown to bind stably to SNARE complex molecules using α-SNAP as an adaptor that locks it in the ATP state (Barnard et al., 1997; Müller et al., 1999). Hydrolysis of ATP enables NSF to separate from and disassemble the SNARE complex. Accordingly, we examined the effect of Arr1 on NSF disassembly of purified, recombinant SNARE molecules. The recombinant His6-NSF proteins were pretreated with increasing concentrations of recombinant Arr1. The GST-tagged-Syntaxin 4 proteins were immobilized to glutathione-agarose beads and then incubated with α-SNAP and SNARE polypeptides, including VAMP-2 and SNAP-25 in binding buffer. The mixture of NSF, Arr1, and SNARE complex was precipitated with beads, and the precipitated proteins were separated by SDS-PAGE and analyzed on immunoblots with specific antibodies to NSF, Syntaxin-4, SNAP-25, and VAMP2. These data verified that Arr1 enhanced NSF disassembly activity in a dose-dependent manner (Fig. 6b).

NSF ATPase activity assay. The ATPase activity of NSF was measured with a colorimetric assay. Recombinant NSF (0.2 μg/μl) was pretreated with 10 mm NEM, a NSF inhibitor, as a negative control or with increasing concentrations of recombinant Arr1 protein for 10 min at 37°C. The release of inorganic phosphate was measured by adding BIOMOL Green (BIOMOL) and reading the absorbance at 620 nm on a Benchmark Plus microplate reader (Bio-Rad). Arr1 significantly increases the NSF ATPase activity in a dose-dependent manner. Corrections were made by subtracting the NEM-treated control values. Results of the ATPase assay represent the means ± SEM for four independent experiments. **p < 0.01; ***p < 0.001.

SNARE complex disassembly assay. a, Assembly status of SNARE complexes. GST-Syntaxin 4 was immobilized on glutathione-agarose beads and incubated with His6-tagged VAMP2 and SNAP-25 with (lane 2) or without (lane 1) NSF and α-SNAP for 30 min at 4°C. Proteins precipitated with beads were separated by 12% SDS-PAGE and visualized by Coomassie blue staining. b, Arr1 enhanced NSF disassembly activity in a dose-dependent manner. The NSF disassembly assay was performed by pretreating recombinant His6-tagged NSF with increasing concentration of Arr1 and then mixing with immobilized GST-Syntaxin 4 with His6-tagged-α-SNAP, VAMP2, or SNAP-25. Proteins precipitated with glutathione-agarose beads were transferred to PVDF and incubated with (from top to bottom) anti-rabbit NSF PAb, anti-mouse Syntaxin 4 MAb, anti-rabbit SNAP-25 PAb, and anti-mouse VAMP2 MAb and appropriate secondary antibodies. SNARE complexes of ∼250 kDa were detected with all the antibodies listed above.
Arr1 deletion markedly reduces the expression level of synapse-enriched proteins
Unlike conventional synaptic terminals that release neurotransmitters episodically in response to action potentials, the photoreceptor ribbon synapses are depolarized in the dark, resulting in maintained activation of voltage-gated calcium channels, continual Ca2+ influx, and higher rates of exocytosis required for tonic release of the neurotransmitter glutamate. Increasing light intensity induces a graded hyperpolarization that turns off these events and suppresses glutamate release (Morgans, 2000; von Gersdorff., 2001; Heidelberger et al., 2005). Many presynaptic proteins regulate synaptic vesicle exocytosis and neurotransmitter release. These proteins are differentially distributed among the different synapses of the mouse retina. For example, glutamate transporters, such as vGLUT1, which refills the synaptic vesicles with glutamate, is essential for transmission of visual signaling from photoreceptors to second- and third-order neurons (Johnson et al., 2007). EAAT5, which is expressed in synaptic terminals of photoreceptors and rod bipolar cells, plays a predominant role in reuptake of glutamate from the synaptic cleft to ensure reliable synaptic transmission (Pow and Barnett, 2000; Wersinger et al., 2006). Also, a synaptic vesicle protein, VAMP2, which is the predominant isoform in the mouse retina, is an integral membrane protein associated with forming the fusion core complex required for docking and fusing of synaptic vesicles at the synaptic active zone (Sherry et al., 2003). To investigate whether the presence of Arr1 alters the expression levels of NSF and the synapse-enriched encoded genes such as vGLUT1, EAAT5, and VAMP2, we performed quantitative RT-PCR to compare the mRNA expression level of these genes in WT and Arr1−/− retinas under different lighting conditions. Comparing DA and LA WT retinas, the mRNA levels of NSF, vGLUT1, EAAT5, and VAMP2 were significantly higher in the dark (Fig. 7a). In the light, there was no significant difference in the transcriptional level of NSF and VAMP2 in Arr1−/− retinas compared with WT retinas, but the mRNA expression levels of vGLUT1 and EAAT5 were lower in Arr1−/− retinas than the WT retinas. Difference in expression levels between WT and Arr1−/− retinas is light dependent. In the dark, the mRNA levels of NSF, vGLUT1, VAMP2, and EAAT5 were markedly decreased in the Arr1−/− retinas compared with the WT retinas. The protein expression level of NSF, vGLUT1, VAMP2, and EAAT5 in WT and Arr1−/− retinas under different lighting conditions correlated to the transcription level of these genes (Fig. 7b). For SNAP-25, the protein expression level was lower in the DA Arr1−/− retinas compared with DA WT retinas, and there was no significantly difference between LA WT and Arr1−/− retinas.

Quantitative RT-PCR and protein expression levels of NSF, vGLUT1, VAMP2, and EAAT5 in the retinas. a, Expression levels of NSF, VAMP2, EAAT5, and vGLUT1 in WT and Arr1−/− mouse retina were measured by quantitative RT-PCR. Each column represents the average of three amplification reactions (mean ± SEM), performed on a cDNA sample reverse transcribed from total RNA prepared from 10 pooled retinas using Trizol reagent and transcribed into cDNA with oligo-dT20 using the Superscript III system (Invitrogen). Values for light-adapted WT retinas were set to 1. The NSF, vGLUT1, VAMP2, and EAAT5 mRNA levels were significantly higher in the dark-adapted (D) WT retinas compared with light-adapted (L) WT retinas. In the light, significantly lower expression levels of vGLUT1 and EAAT5 were observed in the Arr1−/− retinas compared with WT retinas. In the dark, NSF, vGLUT1, VAMP2, and EAAT5 mRNA levels were markedly decreased in the Arr1−/− retinas compared with the WT retinas. *p < 0.05; **p < 0.01; ***p < 0.001. b, Immunoblots analysis of NSF, vGLUT1, EAAT5, VAMP2, and SNAP-25. Mouse mGAPDH was used as internal control. In the bar graph, the expression level of these proteins is expressed as a ratio to GAPDH expression. Values for light-adapted WT retinas were set to 1. Results represent the means ± SEM for three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
Arr1 deletion suppresses the synaptic activity in the photoreceptor synapse in vivo
To further test whether photoreceptor synapses are dysfunctional in vivo in the Arr1−/− retinas, we performed experiments that were based on the widely used fluorescent dye, FM1-43, which can selectively label structures and living cells that are undergoing exocytosis and endocytosis. In WT retinas depolarized with 25 mm KCl, FM1-43 was avidly sequestered into the presynaptic terminals in the OPL and IPL (Fig. 8a), whereas in the depolarized Arr1−/− retinas FM1-43 staining in the OPL and IPL was strongly reduced (Fig. 8b). In the non-depolarized WT (Fig. 8c) and Arr1−/− (Fig. 8d) retinas, the FM1-43 uptake was diminished in both OPL and IPL. These results further illustrate that when Arr1 expression is absent in the retina, the synaptic activity is dramatically reduced in the photoreceptors. We also observed FM1-43 staining in the outer segment (Choi et al., 2005) in WT retinas, and this staining pattern may indicate the continuous renewal process of the outer segment and dynamic communication of the disc and extracellular space (Chen et al., 2002).

Synaptic uptake of FM1-43 revealed a decrease of synaptic activity in the Arr1−/− mouse retina. Enhanced FM1-43 uptake in the OPL (arrows) and IPL was observed in depolarized (25 mm K+) WT retina (a), compared with depolarized Arr1−/− retina (b). FM1-43 intensity was also significantly reduced in the OPL (arrows) and IPL in non-depolarized WT (c) and Arr1−/− (d) retinas. Scale bars, 50 μm.
In this study, unlike Arr1, we observed that Arr4 does not interact with NSF (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), which suggests a divergence of the function of Arr1 in the cone photoreceptor. In cones, we have demonstrated in isolated cone photoreceptors that at least one visual Arrestin is essential for normal photoreceptor recovery with increasing light intensity (Nikonov et al., 2008). When the averaged ERG b-wave amplitudes are recorded from the WT, Arr1−/−, and Arr4−/− every 2 min from 1 to 15 min during light adaptation (Fig. 9a), the photopic b-wave amplitudes for these three genotypes are similar at 1 min; however, only WT and Arr4−/− mice demonstrated equivalent light adapting increases in the b-wave, reaching maximum amplitudes at 9 min. In contrast, Arr1−/− mice showed no b-wave amplitude increase over the 15 min of light adaptation. Similar results were observed with 20 multiple-intensity flashes to further saturate rhodopsin (Brown et al., 2010).

ERG analysis of WT, Arr1−/−, Arr4−/−, and mCAR-Harr1−/− mice. Average photopic b-wave amplitudes (in microvolts) recorded every 2 min during 15 min of light adaptation of the WT, Arr1−/−, Arr4−/− (a), and mCAR-Harr1−/− (b) mice. Two-way ANOVA with Bonferroni's post tests used for statistical comparisons with WT (**p < 0.01; ***p < 0.001).
To further delineate the functional divergence between Arr1 and Arr4 in synaptic regulation from their established function in phototransduction inactivation, we also tested mouse cone arrestin (Arr4) transgenic mice (mCAR-Harr1−/−). These Arr1-null mice have normal levels of Arr4 in their cones plus high rod photoreceptor expression levels, which is driven by a rhodopsin promoter to all the rods. Previously, single-cell recordings of rods from these mCAR-Harr1−/− mice were observed to restore partial recovery of rod function and morphologically reduced levels of light-dependent rod degeneration associated with the loss of Arr1 (Chan et al., 2007). In Figure 9b, the 15 min of light adaptation showed no appreciable increase in amplitudes of b-wave ERG responses in the mCAR-Harr1−/−, similar to the observed phenotype in the Arr1−/−.
Discussion
In this study, we demonstrate the alternative structural and potential functional interactions between visual Arr1 and NSF in the photoreceptor synapse using a combination of biochemical, cellular, and molecular biological techniques.
The crystal structures of Arrestin family members, including Arr1, Arr4, and β-arrestin 1, are remarkably similar (Wilden et al., 1997; Hirsch et al., 1999; Han et al., 2001; Sutton et al., 2005). In a previous in vitro yeast two-hybrid study, β-arrestin 1 was shown to interact in vitro with NSF and it bound to the identical region (amino acids 197-250 of NSF) as observed for Arr1 binding. The β-arrestin 1–NSF complex formation was also ATP-dependent (McDonald et al., 1999). Overexpression of NSF rescued the dominant-negative effect of a β-arrestin 1 phosphorylation mutant (S412D) by restoring normal sequestration of the GPCR, the AR (β2-adrenergic receptor). It is important to note that other members of the Arrestin family do not possess the Ser412 residue and therefore are not subject to the same regulation in receptor internalization as β-arrestin 1. In our work, we show that the Arr1 N-terminal motif (amino acids 1-191) contains a key domain involved in activation–recognition and phosphorylation-specific binding of the Arr1 (Gurevich and Benovic, 1992, 1993; Gurevich et al., 1994) for interaction with NSF, but no evidence documents that this region of β-arrestin 1 interacts with NSF. We also determined that cone Arr4, which is also expressed in cone pedicles with Arr1 (Nikonov et al., 2008), did not interact with NSF (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In the previous studies, the interaction of β-arrestin 1 with NSF directs the clathrin-mediated receptor internalization. In our experiments, Arr1 binds directly to the junction of the NSF N and D1 domains, which are required for SNARE complex binding and ATP hydrolysis, to modulate the process of exocytosis.
Unlike conventional synaptic terminals that release neurotransmitter episodically in response to action potentials, the photoreceptor ribbon synapses, which are specialized structures at active zones in the rod spherules and cone pedicles, are depolarized in the dark, resulting in maintained activation of voltage-gated calcium channels, continual Ca2+ influx, and higher rates of exocytosis required for tonic neurotransmitter (l-glutamate) release (Morgans, 2000; von Gersdorff, 2001; Heidelberger et al., 2005). Increasing light intensity induces a graded hyperpolarization that turns off these events and suppresses neurotransmitter release. This higher rate of exocytosis in the photoreceptor synapses must be balanced by compensatory endocytosis to retrieve vesicle membrane and vesicle proteins incorporated into the plasma membrane during fusion.
Our study demonstrates that Arr1 interacts with NSF to enhance its ATPase activity and to also stimulate its ability to disassemble the SNARE complex. These functions are crucial for NSF in regulation of vesicular transport and synaptic transmission. Moreover, Arr1 deletion markedly reduces the expression level of NSF and synapse-enriched proteins, including vGLUT1, EAAT5, VAMP2, and SNAP-25. SNAP-25 and VAMP2 are the major components for SNARE complex formation (Roth and Burgoyne, 1994; Søgaard et al., 1994). The vGLUT1 regulates the glutamate sequestered into synaptic vesicles in photoreceptor terminals (Johnson et al., 2007), and EAAT5 is involved in synaptic uptake of glutamate into photoreceptor to ensure reliable synaptic transmission (Wersinger et al., 2006). Dynamic exocytosis, vesicle replenishment, and glutamate removal are principally responsible for regulating the kinetics of synaptic transmission in the photoreceptors (Thoreson, 2007).
We observe that a loss of Arr1 gene expression in the retina of Arr1−/− mice is also associated with a reduced level of mRNAs and proteins involved in these processes, and synaptic activities are dramatically suppressed in the photoreceptors of these mice. On the basis of previous studies (Mendez et al., 2003; Smith et al., 2006; Brown et al., 2010) and our current findings, we suggest that Arr1 serves a dual functional role in distinct subcellular compartments in the photoreceptor. Bright light exposure induces Arr1 translocation to the photoreceptor outer segment for termination and recovery of the light response by binding to light-activated, G-protein receptor kinase 1-dependent phosphorylated rhodopsin (Wilden et al., 1986; Xu et al., 1997). In the dark, Arr1 is highly expressed and maintained in all mouse photoreceptor synapses, where it interacts with NSF for modulation of sustained synaptic vesicles release to fulfill the need to accurately encode subtle membrane potential changes leading to specific adaptations of the synaptic machinery used in the photoreceptor synapses.
Mutations in the ARR1 gene lead to Oguchi disease (Fuchs et al., 1995; Nakazawa et al., 1997) and retinitis pigmentosa (Nakamachi et al., 1998). Oguchi disease is a rare recessive form of congenital stationary night blindness (CSNB) characterized by distinctive golden-brown discoloration of the fundus that disappears after prolonged dark adaptation. The typical ERG findings in patients with Oguchi disease show a negative configuration with subnormal to normal a-waves and nearly absent b-waves, similar to those in complete CSNB. After longer dark adaptation (2–4 h), the mixed rod–cone ERG shows increases in both a- and b-waves. This recovery of light sensitivity was thought to be related to the time course of rhodopsin regeneration (Dryja, 2000). From another perspective, our study suggests an alternative explanation with Arr1 involvement. The b-wave reflects the depolarizing response of bipolar cells and the absence of a b-wave suggests that the synaptic transmission between photoreceptors and depolarizing bipolar cells may be defective.
We also observe an abnormal photopic b-wave phenotype associated with loss of light adaptation in the Arr1−/− mice (Fig. 9a) (Brown et al., 2010). Increased light adaptation gradually changes the synaptic signal transfer from photoreceptor synapse to the second-order neuron. We propose that this phenotype reflects a defect of intercellular feedback mechanisms dependent on synaptic transmission downstream of the light-activated, phosphorylated GPCR recovery observed with Arr1 function (Van Epps et al., 2001). Mouse rod photoreceptors only express Arr1, whereas the cone photoreceptors express both Arr1 and Arr4. When recording electrophysiological signals from single mice cones with either one or both visual Arrestins knocked out, at least one visual Arrestin is required for normal cone inactivation (Nikonov et al., 2008). In other studies, Arr4 was shown to partially substitute for Arr1 in Arr1−/− rod photoreceptors in transgenic mCAR-Harr1−/− retina (Chan et al., 2007), which targeted the expression of Arr4 to rod photoreceptors on an Arr1-null background. In Figure 9b, we showed the light adaptation b-wave curve in mCAR-Harr1−/− retina is almost identical with the Arr1−/−. This suggests that, even though Arr4 can partially substitute for the function of Arr1 in rod phototransduction inactivation, it cannot substitute for the function of Arr1 in photoreceptor synaptic regulation since Arr4 does not interact with NSF. Therefore, the ERG b-wave abnormality in patients with Oguchi disease, who lack normal expression of ARR1, may result from the disruption in synaptic transmission in the photoreceptor terminals. Photoreceptors can properly respond to the change in light stimuli depending on precise control of neurotransmitter release. Reliable photoreceptor synaptic transmission requires accurate detection of the changes in glutamate concentration in the synaptic cleft by the postsynaptic cells. We propose that, without Arr1 acting as a modulator for NSF, the photoreceptor synapse cannot maintain the normal graded exocytosis rate to continuously adjust the release of glutamate to optimize the signal transfer to the postsynaptic horizontal and bipolar cells.
In summary, we have demonstrated that the interaction of Arr1 and NSF is ATP-dependent, and the N-terminal domain of Arr1 interacts with the N and D1 functional domains of NSF. The Arr1–NSF interactions are greater in the photoreceptor synaptic terminal in the dark. Furthermore, Arr1 enhances the NSF ATPase activity and increases the NSF disassembly activities, which are critical for NSF functions in sustaining a higher rate of exocytosis in the photoreceptor synapses and the compensatory endocytosis to retrieve vesicle membrane and vesicle proteins for vesicle recycling. Deletion of Arr1 expression also leads to reduced levels of mRNA and protein expression of NSF, vGLUT1, EAAT5, and VAMP2, which have the potential to markedly depress the exocytosis rate in the dark-adapted retinas. These data provide strong in vitro results and for the first time in vivo evidence that, in photoreceptors, demonstrate that the Arr1 and NSF interaction is necessary for the maintenance of normal vision. This study also reveals a novel functional role of Arr1 in photoreceptor synapses and provides additional insights into potential mechanisms of inherited retinal diseases, such as Oguchi disease and ARR1-associated retinitis pigmentosa.
Footnotes
This work was supported in part by National Institutes of Health Grants EY015851 (C.M.C.) and EY03040 [Doheny Eye Institute (DEI)], Research to Prevent Blindness (RPB) (DEI; C.M.C.), Dorie Miller, Tony Gray Foundation, and Mary D. Allen Foundation [Dr. Richard Newton Lolley Memorial Scholarship (S.-P.H.)]. C.M.C. is the Mary D. Allen Chair in Vision Research, DEI, and a RPB Senior Scientific Investigator. We thank members of the Mary D. Allen Laboratory for scientific discussions and critical reading of this manuscript, Lawrence Rife for his technical expertise with ERG analysis, Drs. Larry A. Donoso for Mab D9F2, Jeannie Chen for providing Arr1−/− and mCAR-Harr1−/− mice, David Pow for anti-EAAT5 polyclonal antibody, and C.-P. Ko for expert technical advice in FM1-43 experiments.
- Correspondence should be addressed to Dr. Cheryl M. Craft, Mary D. Allen Chair in Vision Research, Doheny Eye Institute, Department of Ophthalmology, Keck School of Medicine, University of Southern California, 1355 San Pablo Street, DVRC 405, Los Angeles, CA 90033-9224. ccraft{at}usc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}