

Abstract
Small conductance Ca2+-activated K+ type 2 (SK2) channels are expressed in the postsynaptic density of CA1 neurons where they are activated by synaptically evoked Ca2+ influx to limit the size of EPSPs and spine Ca2+ transients. At Schaffer collateral synapses, the induction of long-term potentiation (LTP) increases the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor (AMPAR)-mediated contribution to synaptic transmission and decreases the synaptic SK2 channel contribution through protein kinase A-dependent channel endocytosis. Using a combination of electrophysiology and immunoelectron microscopy in mice, the relationship between the dynamics of spine SK2 channels and AMPARs was investigated. Unlike AMPARs, synaptic SK2 channels under basal conditions do not rapidly recycle. Furthermore, SK2 channels occupy a distinct population of endosomes separate from AMPARs. However, blocking vesicular exocytosis or the delivery of synaptic GluA1-containing AMPARs during the induction of LTP blocks SK2 channel endocytosis. By ∼2 h after the induction of LTP, synaptic SK2 channel expression and function are restored. Thus, LTP-dependent endocytosis of SK2 is coupled to LTP-dependent AMPA exocytosis, and the ∼2 h window after the induction of LTP during which synaptic SK2 activity is absent may be important for consolidating the later phases of LTP.
Introduction
The expression of long-term potentiation (LTP) at Schaffer collateral (SC) to CA1 synapses is thought to take place primarily in the postsynaptic cell (for review, see Kerchner and Nicoll, 2008). The increased synaptic strength is largely accomplished by the rapid increase of synaptic GluA1-containing α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors (AMPARs) (Shi et al., 1999; Park et al., 2004; Derkach et al., 2007; Petrini et al., 2009). One model for the activity-dependent insertion of AMPARs into the postsynaptic density (PSD) is that this process occurs in two steps: exocytosis of AMPARs to the plasma membrane, either within or outside of the spine, followed by a lateral translocation into the PSD (Yudowski et al., 2007; Yang et al., 2008; Makino and Malinow, 2009). The PDZ ligand at the C terminus of GluA1 is required for synaptic stabilization, presumably through interactions with PDZ motifs on scaffolding or accessory proteins (Hayashi et al., 2000; Boehm et al., 2006). AMPARs also undergo rapid and constitutive recycling between the PSD and the spine cytosol, distinct from the activity-dependent trafficking during LTP, and this occurs through dynamin-dependent, clathrin-mediated endocytosis into early and recycling endosomes (Carroll et al., 1999; Lüscher et al., 1999; Ehlers, 2000; Passafaro et al., 2001) (but see Adesnik et al., 2005).
We have previously shown that in dendritic spines of CA1 neurons, SK2 channels reside in the PSD, where they are activated by synaptically driven Ca2+ influx. SK2 channel activity provides a repolarizing conductance that diminishes EPSPs and attenuates the spine Ca2+ transient (Ngo-Anh et al., 2005). Upon the induction of LTP, SK2 channels undergo protein kinase A (PKA)-dependent endocytosis (Ren et al., 2006) from the PSD of potentiated spines (Lin et al., 2008). As a result, synaptic strength during LTP is increased by the combination of AMPAR trafficking into the synapse and concomitant SK2 channel trafficking out of the synapse.
To further understand the relationships between the dynamics of spine SK2 channels and AMPARs, we investigated the trafficking of spine SK2 channels under basal conditions and after the induction of LTP. The results show that under basal conditions SK2 channels do not undergo rapid, dynamin-dependent recycling and are present within the spine in a population of endosomes distinct from AMPARs. However, their endocytosis following the induction of LTP does occur through dynamin-dependent endocytosis. Blocking the synaptic delivery of GluA1-containing AMPARs blocks not only the LTP-dependent increase of synaptic AMPARs, but also the endocytosis of SK2 channels. Importantly, following their activity-dependent endocytosis, SK2 channels functionally repopulate the PSD with a delay of ∼2 h after the induction of LTP. The absence of synaptic SK2 channel activity after the induction of LTP may provide a window during which the EPSP is boosted relative to its eventual potentiated state, and that may be important for consolidating the later phases of LTP or subsequent changes in the induction threshold for further plasticity.
Materials and Methods
Slice preparation.
All procedures were performed in accordance with the guidelines of Oregon Health and Science University (Portland, OR) and University of Castilla-La Mancha (Albacete, Spain). Hippocampal slices were prepared from 3- to 6-week-old C57BL/6J male or female mice. Animals were anesthetized with isoflurane and decapitated. The cerebral hemispheres were quickly removed and placed into cold artificial CSF and equilibrated with carbogen (95% O2/5% CO2). Hippocampi and cortex were removed, placed onto an agar block, and transferred into a slicing chamber containing sucrose-artificial CSF (ACSF) (in mm): 70 sucrose, 80 NaCl, 2.5 KCl, 21.4 NaHCO3, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 1.3 ascorbic acid, 20 glucose, equilibrated with carbogen. Transverse hippocampal slices (300–350 μm) were cut with Leica VT1000s or VT1200s (Leica Instruments) and transferred into a holding chamber containing regular ACSF (in mm): 125 NaCl, 2.5 KCl, 21.5 NaHCO3, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, 15 glucose, equilibrated with carbogen. Slices were incubated at 34°C for 30 min and then at room temperature for ≥1 h before recordings were performed.
Electrophysiology.
For synaptically evoked recordings, CA1 pyramidal cells were visualized with infrared/differential interference contrast optics (Leica DM LFS) and a charge-coupled device (CCD) camera. Whole-cell, patch-clamp recordings were obtained from CA1 pyramidal cells using an Axopatch 200B amplifier (Molecular Devices), digitized using an ITC-16 analog-to-digital converter (Heka Instruments), and transferred to a computer using Pulse software (Heka Instruments). Patch pipettes (open pipette resistance, 2–4 MΩ) were filled with a solution containing the following (in mm): 130 K-gluconate, 8 NaCl, 1 MgCl2, 10 HEPES, 4 ATP, 0.3 GTP, and 10 phosphocreatine, pH 7.26. Series resistance was not electronically compensated, and recordings with series resistance that changed >20% during the experiment were discarded. Electrophysiological records were filtered at 5 kHz and sampled at 20 kHz. The input and series resistance was determined from a ∼30 pA (500 ms) hyperpolarizing current injection pulse interspersed between events. All recordings were from cells with a resting membrane potential between −70 and −50 mV and a stable input resistance. For EPSP measurements in current-clamp mode, a bias current was applied to maintain the membrane potential at −60 mV. All electrophysiological recordings were performed at room temperature. Control experiments verified that the apamin-induced enhancement of EPSPs is independent of initial EPSP size (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Synaptic stimulation.
EPSPs or EPSCs were recorded in whole-cell mode. Capillary glass pipettes (tip diameter, ∼5 μm) filled with ACSF and connected to an Iso-Flex stimulus isolation unit (A.M.P.I.) were used to stimulate presynaptic axons in stratum radiatum as described in Results. Pathway independence was verified by the lack of paired pulse facilitation to synaptic stimulation delivered in each electrode with 50 ms intervals. SR95531 (2 μm) and CGP55845 (1 μm) were present to reduce GABAA and GABAB contributions, respectively. To prevent epileptic discharges in the presence of GABAergic blockers, the CA3 region was microdissected out of slices used for field and EPSC recordings.
Theta burst pairing.
The theta burst pairing (TBP) protocol was delivered within 15 min after whole-cell formation to prevent the washout of LTP induction. Back propagating action potentials (b-APs) were initiated in whole-cell, current-clamp mode by somatic current injection (1 ms, 1–2 nA). The standard TBP protocol consists of EPSPs paired with a single b-AP timed so that the b-AP (∼15 ms delay) occurred at the peak of the EPSP as measured in the soma. A single burst consists of five EPSP-b-AP pairs delivered at 100 Hz and 10 bursts delivered at 5 Hz per sweep. Three TBP sweeps were delivered at 10 s intervals. Subthreshold EPSPs were elicited by 100 μs current injections that were ∼1/3 of the stimulus required for evoking an action potential.
Data analysis.
Data were analyzed using IGOR (WaveMetrics). The slope of the rising phase of the EPSP was measured between 10 and 20% of the rising phase. Binning data at 1 min intervals generated summary plots. Data are expressed as mean ± SEM. Paired two sample t tests were used to determine significance of data in the same pathway, and ANOVA with Dunnett's post hoc test was used to determine significance between groups of data; p < 0.05 was considered significant.
Chemicals and solutions.
d-AP5, CNQX, CGP55845, and SR95531 and peptides P4, S4, Pep1-TGL, and Pep1-AGL were obtained from Tocris Bioscience. Scramble-TGL (ALSPGSMGTGL) was custom-made from New England Peptide. Apamin was from Calbiochem. TeTx was from List Biological Laboratories. All other chemicals were obtained from Sigma-Aldrich unless specified. All perfusing solutions were modified from regular ACSF unless otherwise noted. NMDA component of EPSP was isolated by perfusing ACSF with 10 μm CNQX and 0.5 mm MgCl2. Chemical LTP was induced by 5 min bath application of potentiation solution (in mm): 124 NaCl, 5 KCl, 1.25 KH2PO4, 0.1 MgCl2, 24 NaHCO3, 2 CaCl2, 10 glucose, 25 tetraethylammonium, equilibrated with carbogen.
Immunoelectron microscopy.
Antibodies and concentrations used were as follows: anti-SK2 raised in guinea pig (1–2 mg/ml); anti-GluR1–4 raised in rabbit (combined GluA1, GluA2/3, and GluA4; 10 mg/ml; Millipore), anti-NR1 raised in rabbit (1–2 mg/ml; Millipore), and anti-EEA1 (early endosomal antigen 1) and anti-Rab4 mouse monoclonal antibodies (10 μg/ml; BD Transduction Laboratories). Ultrastructural analyses were performed with a JEOL 1010 transmission electron microscope (Synaptic Structure Laboratory, School of Medicine, University of Castilla-La Mancha, Albacete, Spain). Electron photomicrographs were captured with a CCD camera (Mega View III; Soft Imaging System). Digitized electron images were modified for color, brightness, and contrast with Adobe Photoshop, version 7.0. Labeled structures were classified based on unambiguous morphological information in each section. Axon terminals were identified by the presence of synapses and small round and/or large granular vesicles. Synapses were identified as parallel membranes separated by widened clefts that were associated with membrane specializations. Synapses displaying a prominent density on the postsynaptic side were characterized as asymmetrical. Dendritic spines were identified as small protrusions exhibiting membrane continuity with the dendritic shaft. Postembedding immunohistochemistry was performed according to our previously published methods (Lin et al., 2008). Ultrastructural analyses were performed in a JEOL 1010 transmission electron microscope. For colocalization analysis, three independent tissue samples were used and only spines with at least one immunoparticle for each marker were included. EEA1 or Rab4 immunoparticles that had associated SK2 or AMPAR immunoparticles were counted. Averages ± SEM were calculated and used to derive 95% confidence intervals. For time course experiments, three independent tissue samples were used and 20 spines from each sample (60 immunopositive spines) with at least one immunoparticle in each spine were examined per time point. Tangential location of gold particles was measured from the midline of the PSD (Lin et al., 2008). For a given distance from the PSD, the number of particles was counted for each sample to determine the percentage of total particles within 15 nm of the PSD. Data were averaged over the three samples to determine average ± SEM for each time point.
Results
SK2 channels do not undergo rapid recycling
To examine dynamin-dependent endocytosis, whole-cell recordings of CA1 neurons were performed from hippocampal brain slices. EPSCs were measured in whole-cell, voltage-clamp configuration at a holding potential of −70 mV and were evoked every 30 s by synaptic stimulation of SC afferent fibers in the stratum radiatum. Under these recording conditions, the EPSC is mediated primarily by current flow through AMPARs (Kauer et al., 1988; Isaac et al., 1995; Liao et al., 1995). To disrupt dynamin-dependent endocytosis, a dynamin inhibitory peptide (P4; 100 μg/ml) was included in the patch pipette. P4 interferes with the binding of amphiphysin to dynamin, an interaction that is important for endocytosis (Wigge and McMahon, 1998; Nong et al., 2003). Following whole-cell formation, dialysis of P4 steadily increased the EPSC, and after 22–25 min the EPSC had increased by 72 ± 11.3% of baseline (p < 0.05, n = 8) (Fig. 1 a,c). As a control, interleaved experiments were performed in which cells were dialyzed with S4, a scrambled form of P4 (100 μg/ml); S4 had no significant effect on the EPSC (4 ± 6.8% of baseline, p > 0.05, n = 9) and did not differ from control recordings (14 ± 2.9% of baseline, n = 8) (Fig. 1 a,c). These results are similar to those in previous reports showing that a related dynamin inhibitory peptide, D15, increased the AMPAR-mediated EPSC (Carroll et al., 1999; Lüscher et al., 1999), and this increase reflects the constitutive surface delivery of AMPARs.

SK2 channels do not undergo rapid dynamin-dependent recycling. a , Time course of the EPSC amplitude normalized to baseline (mean ± SEM) from control (circles, n = 8), cells loaded with P4 (filled triangles, n = 8), or cells loaded with S4 (open triangles, n = 9). b , Continued time course after switching to current clamp and perfusing with 0.5 mm Mg2+ and CNQX (10 μm) to block AMPARs of the EPSPNMDA amplitude normalized to the 10 min period before the addition of apamin application. c , Representative average of 10 EPSCs, mean ± SEM (shaded area), taken from the indicated shaded time points in a for baseline (dashed) and after loading (solid) for control, P4, or S4. d , Representative average of 10 EPSPsNMDA, mean ± SEM (shaded area), taken from the indicated shaded time points in b for baseline before (gray) and after (black) apamin application for control-, P4-, or S4-loaded cells. WCF, Whole-cell formation. Vertical and horizontal scale bars are 20 pA and 20 ms ( c ) and 0.5 mV and 50 ms ( d ), respectively.
To determine whether synaptic SK2 channels also undergo rapid dynamin-dependent recycling, the experiments described above were continued and, after 25 min of voltage-clamp recordings, the amplifier was switched to current-clamp mode to record EPSPs. To isolate the effect of P4 on SK2 channels from the effect of P4 on AMPARs, it was necessary to block AMPARs. Therefore, the bath was switched to a solution containing 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm) to block AMPARs and reduced external Mg2+ (0.5 mm) to examine the isolated NMDAR-mediated EPSP (Kauer et al., 1988; Isaac et al., 1995; Liao et al., 1995). After 25 min to ensure complete exchange, 10 min of stable EPSP were recorded before the addition of apamin (100 nm). Blocking SK2 channels with apamin increased the EPSP by the same extent for cells dialyzed with P4 compared with cells dialyzed with S4 or control cells without peptide (P4: 102 ± 9% of baseline, n = 8; S4: 84 ± 4% of baseline, n = 9; control: 92 ± 6% of baseline, n = 8) (Fig. 1 b,d). To ensure that synaptic NMDAR activity was stable over this time period, NMDAR-mediated EPSCs were recorded at −70 mV in cells dialyzed either with P4 or S4 in the presence of CNQX (10 μm), apamin, and reduced external Mg2+ (0.5 mm). Under these conditions, there was no significant change in the EPSCNMDA during 30 min of continuous recording (P4: 6 ± 7%, n = 10; S4: 9 ± 2%, n = 12; control: 4 ± 3%, n = 12; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these results suggest that under basal conditions synaptic SK2 channels do not undergo rapid recycling via dynamin-dependent endocytosis.
Spine SK2 channels and AMPARs occupy distinct endosomes
Under basal conditions, AMPARs have been detected in spine endosomes of cultured hippocampal neurons using antibodies to early endosomal antigen 1, EEA1, and the small G protein, Rab4 (Ehlers, 2000). To determine whether AMPARs or SK2 channels are present in spine endosomes expressing these same markers in acutely prepared hippocampal slices, double label postembedding immunogold electron microscopy (iEM) was performed using antibodies for EEA1 or Rab4 and SK2 or AMPARs (Lin et al., 2008). Immunoparticles for SK2 or AMPARs were detected in the PSD as well as in vesicles that also labeled for EEA1 or Rab4 (supplemental Figs. 3, 4, available at www.jneurosci.org as supplemental material). SK2 channels and AMPARs contribute oppositely to spine membrane potential, raising the possibility that there are mechanisms for separate trafficking of spine SK2 channels and AMPARs. One mechanism would be to segregate SK2 channels and AMPARs into separate populations of vesicles that could respond differently to synaptic signals. Therefore, triple label iEM was performed using antibodies for SK2, AMPARs, and EEA1 (Fig. 2 a,b). The results showed that in 92 EEA1-positive spines representing 115 EEA1-positive vesicles that also contained either an SK2 or an AMPAR immunoparticle, there was colocalization of immunoparticles for AMPAR and EEA1 in 67 vesicles, and SK2 and EEA1 immunoparticles in 48 vesicles. However, in no case were both SK2 and AMPARs colocalized with the same immunolabeled EEA1 vesicle. Similar results were obtained from triple labeling for SK2, AMPARs, and Rab4 (Fig. 2 c,d). From 75 Rab4-positive spines representing 75 Rab4-positive vesicles that also contained either an SK2 or an AMPAR immunoparticle, there was colocalization of AMPARs and Rab4 immunoparticles in 53 vesicles and SK2 and Rab4 immunoparticles in 22 vesicles, but triple colocalization was not observed. These results show that, under basal conditions, if AMPARs and SK2 channels are ever colocalized in EEA1 or Rab4 endosomes, colocalization occurs at a rate so low that it is maximally 3–4% of the time, suggesting that endosomes containing AMPARs or SK2 channels comprise distinct membrane compartments that are not readily distinguished by either EEA1 or Rab4.

SK2 and AMPARs reside in distinct populations of endosomes. a – d , Electron micrographs of the stratum radiatum of the CA1 region of mouse hippocampus showing immunoreactivity for SK2 (15 nm particles), AMPARs (GluR1–4; 10 nm particles), and EEA1 (20 nm particles; a , b ) or Rab4 (20 nm particles; c , d ) as detected using triple-labeling postembedding immunogold methods. SK2 and AMPARs were found at postsynaptic densities of excitatory synapses and at intracellular sites (arrowheads) associated with intracellular membranes in dendritic spines (s) of pyramidal cells establishing synapses with axon terminals (b), presumably Schaffer collaterals. SK2 or AMPA was colocalized with EEA1 or Rab4 but were never colocalized with EEA1 or Rab4 to the same intracellular membranes. Scale bar, 0.2 μm.
LTP-dependent endocytosis of SK2 channels is dynamin-dependent
When SC-CA1 synapses experience a TBP protocol that induces LTP, synaptic SK2 channels are rapidly internalized from the PSD in a process that requires PKA (Lin et al., 2008). To determine whether SK2 channel endocytosis upon the induction of LTP is dynamin dependent, EPSPs evoked from two independent SC pathways were recorded in whole-cell, current-clamp mode with either P4 or S4 (100 μg/ml) included in the patch pipette. Two stimulating electrodes were placed on either side of the apical dendrite in stratum radiatum ∼100 μm from the soma and ∼20 μm lateral to the main dendritic shaft. Pathway independence was verified by a paired-pulse protocol (see Materials and Methods). Baseline EPSPs, measured as the maximal slope, were measured for no longer than 15 min in pathways 1 and 2, with stimulations alternating every 20 s. To induce LTP, a TBP protocol that paired presynaptic stimulation with b-APs (see Materials and Methods) was applied to one pathway while synapses in the other pathway experienced only b-APs without paired presynaptic stimulation (Lin et al., 2008).
In cells dialyzed with P4, this TBP protocol induced post-tetanic potentiation followed by a robust LTP in the paired, stimulated pathway. At 30 min after TBP, the relative EPSP slope of the stimulated pathway increased by 241 ± 8% (Fig. 3 a; n = 8). The EPSP in the control pathway also increased by 89 ± 10% consistent with P4 effects on constitutive AMPAR recycling (Fig. 1), but this increase was clearly and significantly less than the LTP seen in the stimulated pathway (p < 0.05, n = 8). After LTP, synaptic stimulation of the potentiated pathway often evoked action potentials. Therefore the stimulus strength in the potentiated pathway was reduced below action potential threshold to match the sizes of EPSPs evoked from either pathway, and a new baseline was established for ≥10 min. Apamin was then applied to assess the contribution of SK2 channel activity to the mixed AMPA/NMDA EPSPs. Apamin application similarly increased the amplitude of EPSPs (p > 0.05) independent of the pathway used to evoke them (potentiated pathway: 53 ± 8% of baseline, p < 0.05; control pathway: 72 ± 6% of baseline, p < 0.05, n = 8) (Fig. 3 b).

SK2 channel endocytosis after LTP is dynamin dependent. a , Top, Time course of the normalized EPSP slope (mean ± SEM) in cells loaded with P4 from control (closed symbols, n = 8) and TBP-stimulated pathway (open symbols). The TBP protocol was delivered at time 0 (arrow). In most cells, the amount of LTP was sufficient to induce action potentials in the evoked pathway only. Therefore, before addition of apamin the stimulus strength in the evoked pathway was reduced below action potential threshold and a new baseline was established. Middle, LTP pathway; bottom, control pathway. Representative average of 10 EPSPs, mean ± SEM (shaded area), were taken from the indicated shaded time points in the top panel for baseline (1) and after TBP induction of LTP (2). b , Continued time course of the EPSP amplitude normalized to the 10 min period before the addition of apamin from control (closed) and stimulated pathways (open) before and after apamin application. Middle, LTP pathway; bottom, control pathway. Representative average of 10 EPSPs, mean ± SEM (shaded area), was taken from the indicated shaded time points in top panel before (3) and after (4) apamin application Vertical and horizontal scale bars are 2 mV and 50 ms ( a ) and 1 mV and 50 ms ( b ), respectively.
In cells dialyzed with S4, TBP also induced a robust LTP only in the paired pathway (158 ± 9.8 vs 18 ± 7.4% of baseline in the control pathway). However, after re-establishing the baseline, apamin increased the EPSP only in the control pathway, as expected from prior results without peptide dialysis (69 ± 6 vs 9 ± 3% of baseline in stimulated pathway; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Together, these results show that SK2 channel endocytosis upon the induction of LTP is dynamin-dependent.
LTP-dependent SK2 channel endocytosis and delivery of synaptic AMPARs require vesicular exocytosis
SK2 channels and AMPARs provide opposing influences on spine membrane potential and are oppositely regulated upon the induction of LTP (Shi et al., 1999; Hayashi et al., 2000; Lin et al., 2008; Yang et al., 2008). Therefore, we tested whether AMPAR and SK2 channel activity-dependent changes are coupled following the induction of LTP. First, two-pathway experiments were performed in the presence of tetanus toxin (TeTx) light chain. This toxin selectively cleaves some vesicle-associated membrane proteins (VAMPs) (Sikorra et al., 2006), and prevents the Ca2+-evoked dendritic exocytosis of postsynaptic vesicles that contain new AMPARs (Lu et al., 2001). Therefore, TeTx (100 nm in 1 mg/ml BSA), or the boiled toxin as a control, was loaded into the postsynaptic cell. Consistent with previous reports of studies performed on cultured hippocampal neurons (Lu et al., 2001), TeTx largely blocked LTP induced by TBP in acutely prepared hippocampal slices (active TeTx: 38 ± 5% of baseline, n = 10; inactive TeTx: 184 ± 22%, n = 7; p < 0.05) (Fig. 4). After re-establishing baseline EPSPs in both pathways, apamin was applied to assess the contribution of SK2 channel activity to the EPSP in either pathway. The results showed that in cells loaded with TeTx, but not the boiled peptide, EPSPs evoked from either the stimulated or the control pathway were similarly increased by apamin application (TeTx stimulated pathway: 48 ± 6% of baseline, p < 0.05; TeTx control pathway: 58 ± 8% of baseline, p < 0.05, n = 10; boiled peptide stimulated pathway: −7 ± 3% of baseline, p > 0.05; TeTx control pathway: 56 ± 7% of baseline, p < 0.05, n = 7) (Fig. 4). These results suggest a process that couples SK2 channel endocytosis with the exocytosis-dependent delivery of synaptic AMPARs. However, TeTx affects many forms of exocytosis, not just that associated with synaptic delivery of AMPARs. It was therefore necessary to employ a more selective approach to address the requirement for AMPAR trafficking in the endocytosis of SK2 channels.
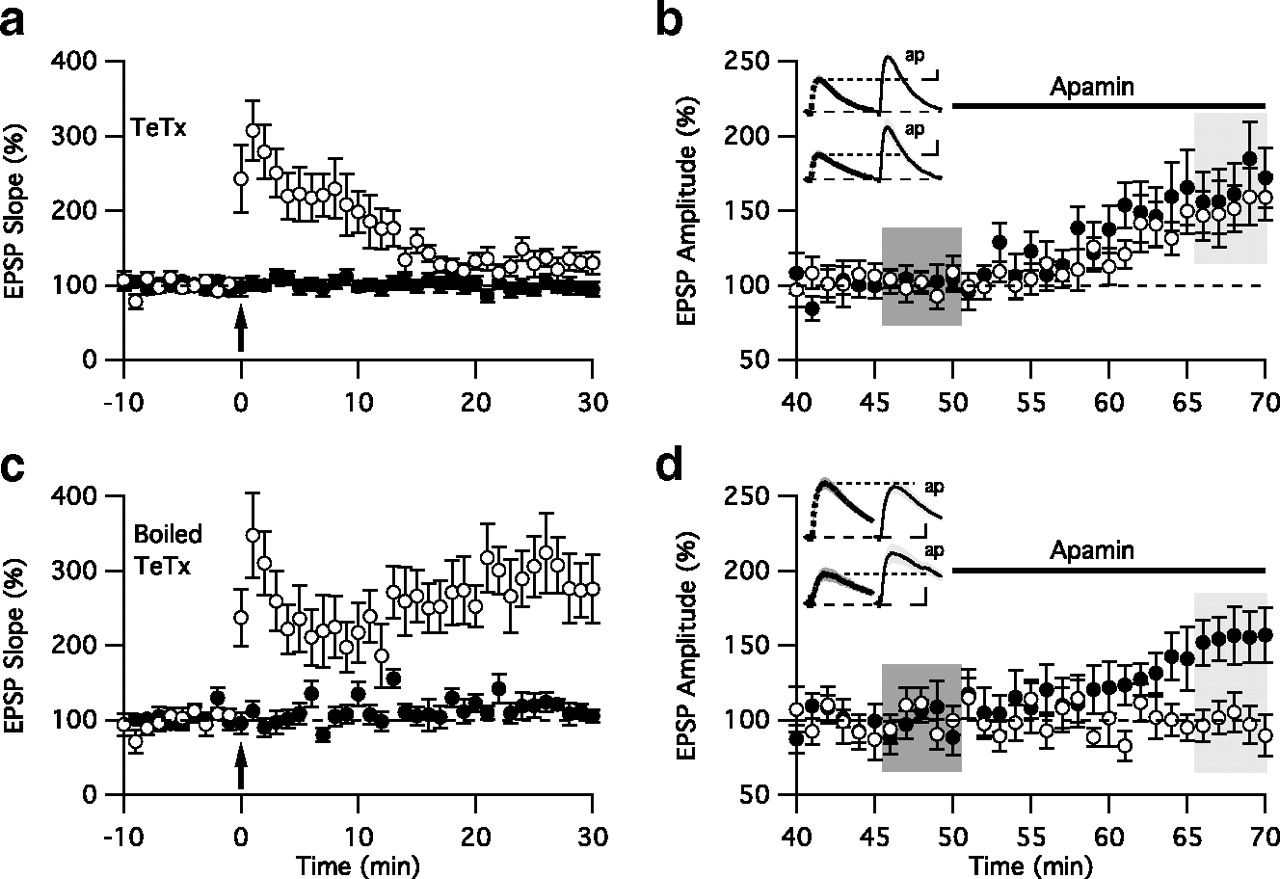
SK2 channel endocytosis requires vesicular exocytosis. a , Time course of the normalized EPSP slope (mean ± SEM) in cells loaded with TeTx (100 nm) from control (closed circles) and TBP-stimulated pathway (open circles). The TBP protocol was delivered at time 0. In most cells, the amount of post-tetanic potentiation was sufficient to induce temporary potentiation in the evoked pathway, but LTP was inhibited 30 min follow TBP stimulation (n = 10). b , Continued time course of the EPSP amplitude normalized to the 10 min period prior to the addition of apamin from control (closed circles) and stimulated pathways (open circles) before and after apamin application. c, d , Similar to panels a and b , except that the cells were loaded with boiled tetanus toxin (100 nm, 95°C for 5 min). LTP was maintained in the TBP-stimulated pathway after 30 min (n = 7). Insets in b and d show representative average of 10 EPSPs, mean ± SM (shaded area), taken from the indicated shaded time points for baseline (left) and after apamin (ap, right) in the TBP pathway (top) and control pathway (bottom). Vertical and horizontal scale bars for the insets are 1 mV and 20 ms ( b ), and 0.5 mV and 20 ms ( d ), respectively.
LTP-dependent SK2 channel endocytosis requires delivery of synaptic GluA1-containing AMPARs
Delivery of GluA1-containing AMPARs to the synapse during the induction of LTP requires protein–protein interactions mediated by the C-terminal PDZ binding domain (Hayashi et al., 2000; Shi et al., 2001). Pep1-TGL is a synthetic peptide corresponding to the C-terminal region of GluA1, containing the PDZ ligand TGL. Dialyzing postsynaptic neurons with Pep1-TGL has been shown to block the delivery of GluA1-containing AMPARs to the spine membrane, presumably by competing for the binding partners of the GluA1 C terminus (Toyoda et al., 2007; Yang et al., 2008), and thus blocks synaptic potentiation. Pep1-AGL contains a single amino acid substitution that renders the PDZ ligand ineffective. Therefore, CA1 neurons were dialyzed with either Pep1-TGL or Pep1-AGL (150 μm), and TBP was used to induce LTP. In cells dialyzed with Pep1-TGL, LTP was markedly reduced in the pathway that experienced TBP, while LTP was intact in cells loaded with Pep1-AGL (Fig. 5) (TGL: 23 ± 4%, n = 8; AGL: 182 ± 18%, p < 0.05, n = 8). After re-establishing the baseline, apamin increased EPSPs evoked at both potentiated and control pathways in cells dialyzed with Pep1-TGL (Fig. 5) (stimulated: 56 ± 2%; control: 49 ± 2%), but apamin only increased EPSPs evoked from control pathway in cells dialyzed with Pep1-AGL (stimulated: −2 ± 3%; control: 50 ± 8%, p < 0.05). Recent studies have demonstrated that PDZ interactions, including those for GluA1, usually require more than the minimal C-terminal tripeptide PDZ ligand and thereby impart greater specificity (von Ossowski et al., 2006; Tonikian et al., 2008). Therefore, to further support the role of GluA1, CA1 neurons were dialyzed with Scramble-TGL, a peptide with the same amino acid composition as Pep1-TGL but with the sequence scrambled except for the final three residues, TGL. In cells dialyzed with Scramble-TGL, LTP was not different from that evoked in the presence of Pep1-AGL in the pathway that experienced TBP (supplemental Fig. 6, available at www.jneurosci.org as supplemental material; Scramble-TGL: 180 ± 15%, n = 7; Pep1-AGL: 182 ± 18%, n = 8). After re-establishing the baseline, apamin increased EPSPs evoked only in the control pathway (supplemental Fig. 6, available at www.jneurosci.org as supplemental material; stimulated: −3 ± 4%; control: 78 ± 15%, p < 0.05). Together, these results show that blocking the activity-dependent synaptic delivery of GluA1-containing AMPARs blocks LTP and also blocks the activity-dependent endocytosis of synaptic SK2 channels.

SK2 channel endocytosis requires synaptic delivery of AMPARs. a , Time course of the normalized EPSP slope (mean ± SEM) in cells loaded with a peptide, preventing AMPAR insertion into the postsynaptic membrane, pep1-TGL (150 μm) from control (closed circles) and TBP-stimulated pathway (open circles). The TBP protocol was delivered at time 0 (arrow). In most cells evoked pathway showed a brief post-tetanic potentiation, but LTP was not maintained (n = 8). b , Continued time course of the EPSP amplitude normalized to the 10 min period before the addition of apamin from control (closed circles) and stimulated pathways (open circles) before and after apamin application. c , d , Similar to panels a and b , except the cells were loaded with control peptide pep1-AGL (150 μm). LTP was maintained in the TBP-stimulated pathway after 30 min (n = 8). Insets in b and d show representative average of 10 EPSPs, mean ± SEM (shaded area), taken from the indicated shaded time points for baseline (left) and after apamin (ap, right) in the TBP pathway (top) and control pathway (bottom). Vertical and horizontal scale bars for the insets are 0.5 mV and 20 ms ( b ) and 1 mV and 20 ms ( d ), respectively.
SK2 channels repopulate the PSD after LTP
To determine whether synaptic SK2 channels eventually repopulate the synapse after LTP, hippocampal slices were treated with or without a chemical LTP protocol (Roth-Alpermann et al., 2006; Lin et al., 2008), allowing us to periodically monitor the effects of apamin on synaptically evoked EPSPs over a long period of time. A population of slices was treated for 5 min (time 0), and the effect of apamin on synaptically evoked EPSPs was examined by sampling single cells in individual slices at different times after treatment. Similar to TBP pairing-evoked LTP, this procedure causes a rapid loss of SK2 activity (Lin et al., 2008). Whole-cell recordings performed at different times following chemical LTP revealed that synaptic SK2 channel activity was still absent 100 min after chemical LTP treatment, as apamin had no significant effect on EPSPs (chemical LTP: 6 ± 5%, n = 8; control: 57 ± 4%; n = 4). However, synaptic SK2 channel activity had recovered within 200 min after chemical LTP treatment, as apamin increased the EPSP to the same extent as in control slices (chemical LTP: 85 ± 19%, n = 11; control: 51 ± 8%, n = 9). The effect of apamin remained constant for up to 5 h after chemical LTP treatment (Fig. 6 a,b). The electrophysiological results were supported by iEM studies that showed SK2 immunoparticles were detected mostly associated with the PSD at time 0 (74.8 ± 1.2% in PSD) but were largely lost from the PSD (8.6 ± 0.9% in PSD) as soon as 5 min after chemical LTP treatment and were still absent at 60 min (10.9 ± 1.5% in PSD). SK2 immunoparticles had repopulated the PSD 4 h later (62.9 ± 1.3% in PSD) (Fig. 7 a – c). During this time the location of PSD95 did not change (Fig. 7 d,e). Together, these data show that SK2 channels repopulate the synapse following LTP but only after ∼2 h.

Synaptic SK2 channel activity is restored following LTP. a , Time course of the apamin sensitivity of EPSP amplitude recorded from slices at different times following chemical LTP (time 0). Closed symbols represent control slices not treated with chemical LTP solution. Immediately following chemical LTP, the EPSP amplitude was not affected by apamin, consistent with previous findings that SK2 channels are removed from the PSD region following chemical LTP. SK2 channel function began to return between 1 and 2 h after chemical LTP. Inset shows the representative average of 10 EPSPs, mean ± SEM (shaded area), taken from the indicated time points (aa: 80 min; ab: 240 min) for baseline (left) and after apamin (ap, right) in the chemical LTP-treated slices. Vertical and horizontal scale bars for the insets are 0.5 mV and 20 ms (top), 2 mV and 20 ms (bottom), respectively. b , Bar graph showing the normalized apamin response from a at < 100 min or > 200 min following chemical LTP or no LTP treatment (Control).

SK2 immunoparticles repopulate the PSD after LTP. a – c , Electron micrographs of the stratum radiatum of the CA1 region of mouse hippocampus showing immunoparticles for SK2 at 5 min ( a ), 60 min ( b ), and 5 h ( c ) after chemical LTP induction as detected using postembedding immunogold methods. At 5 and 60 min after LTP, SK2 immunoparticles were localized to intracellular sites (arrowheads) associated with intracellular membranes in dendritic spines (s) of pyramidal cells establishing synapses with axon terminals (b), presumably Schaffer collaterals, but by 5 h they were mostly associated with the PSD (arrow). d , Time course of the recovery of SK2 channels to within 15 nm of the PSD as determined by the distribution of immunoparticles. e , The localization of PSD95 immunoparticles to the PSD did not change after LTP. Scale bar, 0.2 μm.
Discussion
Synaptic SK2 channels provide a Ca2+-dependent repolarizing postsynaptic current that limits the size of EPSPs and spine Ca2+ transients, even on the scale of unitary events (Ngo-Anh et al., 2005), and, in addition to an increased contribution of AMPARs that drive spine depolarization, the induction of LTP results in the endocytosis of synaptic SK2 channels (Lin et al., 2008). The results presented here show that the trafficking of spine AMPARs and SK2 channels is coordinately regulated. Upon the induction of LTP, the rapid exocytosis of additional GluA1-containing AMPARs triggers the endocytosis of synaptic SK2 channels. These trafficking patterns reflect the segregation of AMPARs and SK2 channels into distinct vesicle populations. After the establishment of LTP at SC-CA1 synapses, there is a slower recovery of synaptic SK2 channel activity. The ∼2 h window when synaptic SK2 activity is absent would increase spine Ca2+ transients, and this may be important for consolidating the later phases of LTP.
Previous studies have examined trafficking of AMPARs under basal conditions and after the induction of LTP. These studies have largely used either cultured neurons or organotypic slice cultures together with biochemical tags or overexpression of modified receptors to follow receptor trafficking. Similar approaches have been used to manipulate proteins that selectively affect endosomal compartments containing AMPARs (Shi et al., 1999; Ehlers, 2000; Hayashi et al., 2000; Park et al., 2004; Gerges et al., 2005). In the present study, we have used a combination of electrophysiology and iEM to examine the movement of native AMPARs and SK2 channels through endosomal compartments with their endogenous constituents in acute hippocampal slices. These present findings are in agreement with many of the previous studies using exogenous protein expression and extend them to include SK2 trafficking.
Results from several previous studies demonstrated that AMPARs undergo a constitutive recycling between the plasma membrane and spine endosomes as seen by the effects of loading cells with a dynamin inhibitory peptide (Carroll et al., 1999; Lüscher et al., 1999) (but see (Adesnik et al., 2005). We also used a dynamin inhibitory peptide, P4, and saw the same effects on AMPA-mediated EPSCs. Consistent with this, in cultured hippocampal neurons AMPARs have been anatomically localized to EEA1- and Rab4-expressing endosomes (Ehlers, 2000; Park et al., 2004), which are regarded as markers for early and recycling endosomes, respectively (Somsel Rodman and Wandinger-Ness, 2000; Sönnichsen et al., 2000). Using acute slice preparations for iEM, we also detected colocalization of AMPARs and EEA1 or Rab4. In contrast to AMPARs, spine SK2 channels do not appear to undergo constitutive dynamin-dependent recycling on a rapid time scale and are not found colocalized in the same endosomal compartment with AMPARs. However, SK2 does colocalize to EEA1- and Rab4-expressing endosomes (Ehlers, 2000) indicating that distinct subclasses of EEA1- and Rab4-expressing endosomes exist and have different rates of recycling (Fig. 1; supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
The conclusion that synaptic SK2 channels do not constitutively cycle in CA1 pyramidal cells is in apparent contrast to observations of spine SK2 channels expressed in principle neurons in the lateral amygdala, where it has been reported that they do undergo constitutive dynamin-dependent endocytosis and recycling (Faber et al., 2008). This may reflect different trafficking in different cell types. In any case, since AMPARs and SK2 channels oppositely influence spine membrane potential, their segregation to nonoverlapping populations of endosomes provides an effective platform to efficiently modulate spine excitability.
The present results are consistent with a requirement for exocytosis of AMPARs for the expression of LTP (Shi et al., 1999; Hayashi et al., 2000; Lu et al., 2001; Park et al., 2004; Yang et al., 2008; Makino and Malinow, 2009; Petrini et al., 2009). Even though AMPARs and SK2 channels are trafficking in opposite directions with LTP, our results suggest that the activity-dependent endocytosis of SK2 channels is coupled to the synaptic delivery of AMPARs. Blocking activity-dependent vesicular exocytosis with TeTx inhibits not only synaptic potentiation but also SK2 channel endocytosis. Notably, TeTx did not alter basal synaptic transmission, consistent with previous results that found that activity-dependent insertion of AMPARs to the cell surface was TeTx sensitive, while constitutive insertion of AMPARs was not (Lu et al., 2001). These findings suggest that separate populations of endosomes use distinct VAMPs that differ in their TeTx sensitivity to mediate constitutive and activity-dependent AMPAR exocytosis. In addition, blocking interactions mediated by the C-terminal domain of GluA1-containing AMPARs by filling the cells with Pep1-TGL effectively inhibits LTP as well as SK2 channel endocytosis. This strongly suggests that SK2 channel endocytosis is specifically linked to the synaptic delivery of GluA1-containing AMPARs. The mechanism by which AMPAR delivery drives SK2 removal remains unclear but is presumably not due to a direct competition of AMPARs and SK2 at the same PDZ domain, as SK2 lacks the PDZ-ligand motif. We note that this also makes it unlikely that the TGL peptide has an off-target direct effect on SK2.
The specificity of Pep1-TGL for GluA1 was demonstrated in two ways. First, a single amino acid substitution in Pep1-AGL that disrupts the PDZ binding ligand rendered the peptide unable to affect either LTP or SK2 endocytosis. Second, scrambling the upstream GluA1-specific sequence of Pep1 also disrupted the effect of the peptide when the TGL core of the PDZ ligand was left intact. This result suggests that specificity determinants for PDZ binding reside in the extended sequence of Pep1, consistent with previous GluA1 peptide binding studies (von Ossowski et al., 2006) and with detailed specificity mapping of PDZ binding determinants (Tonikian et al., 2008). Alternatively, we cannot rule out the possibility that the upstream domain provides the binding determinants for a non-PDZ-mediated protein–protein interaction that may be required for activity-dependent synaptic delivery of GluA1-containing AMPARs (Schulz et al., 2004).
The results confirming that the C-terminal domain of GluA1 is essential for LTP expression suggest that it is the exocytosis of GluA1-containing AMPARs that mediate the expression of LTP, consistent with several previous reports (Shi et al., 1999; Zamanillo et al., 1999; Hayashi et al., 2000; Toyoda et al., 2007; Yang et al., 2008) (but see Hoffman et al., 2002). However, we note that this does not address whether these are GluA1 homomers or heteromeric AMPARs that contain GluA1 (Plant et al., 2006; Adesnik and Nicoll, 2007). The role of the C-terminal sequence of GluA1 in LTP is poorly understood, with some studies reporting the PDZ sequence as important for GluA1 trafficking during LTP (Shi et al., 2001; Kopec et al., 2007; Yang et al., 2008), and others reporting the opposite (Kim et al., 2005). It is perhaps noteworthy that similarly divergent results have been reported in mutant mice lacking GluA1 entirely, with both GluA1-dependent (Zamanillo et al., 1999; Hoffman et al., 2002; Frey et al., 2009) and GluA1-independent LTP (Hoffman et al., 2002; Frey et al., 2009; Romberg et al., 2009) reported to varying degrees under different experimental conditions. This may reflect an as yet poorly defined heterogeneity in AMPAR trafficking mechanisms, as GluA2 is necessary for retention of AMPARs at some Schaffer collateral synapses but not others (Lu et al., 2009), and distinct sets of synapses also regulate AMPAR expression with either PSD-93 or PSD-95 (Elias et al., 2006).
The molecular mechanisms that couple the LTP-dependent delivery of synaptic AMPARs and SK2 channel endocytosis are not yet known, but we note with interest that this coupling is apparently not reciprocal, as specifically inhibiting SK2 channel endocytosis does not block AMPAR exocytosis during LTP (Fig. 3 b) (Lin et al., 2008)). This suggests that the delivery of synaptic AMPARs precedes SK2 channel endocytosis. It remains to be seen whether the trafficking of AMPARs and SK2 channels are coupled by a discrete biochemical change that results in PKA-dependent SK2 channel endocytosis or is simply a result of competition for limited space within the PSD. To address this issue, it will be important to identify SK2-interacting proteins and determine the types of protein–protein interactions that mediate their synaptic localization and activity-dependent trafficking.
Synaptic SK2 channels are rapidly removed upon the induction of LTP and then repopulate the PSD by ∼2 h later. The temporary absence of synaptic SK2 channel activity and the decreased SK2 contribution to the EPSP will increase both the local depolarization and the spine Ca2+ transient upon excitatory neurotransmission during this window. This boosting of the EPSP provided by the temporary absence of synaptic SK2 channels may reduce the threshold for subsequent induction of plasticity or aid in the consolidation of the later stages of LTP.
Footnotes
-
This work was supported by National Institutes of Health Grants NS038880 and NS065855 (J.P.A.) and MH081860 (J.M.) and Ministerio de Ciencia e Innovación Grant BFU-2006-01896, Consolider-Ingenio Grant CSD2008-00005, and Consejeria de Educacion y Ciencia, Junta de Comunidades de Castilla-La Mancha Grant PAI08-0174-6967 (R.L.).
- Correspondence should be addressed to either John P. Adelman or James Maylie, Vollum Institute, Department of Obstetrics and Gynecology, Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239, adelman{at}ohsu.edu or mayliej{at}ohsu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}