

 Cellular/Molecular
Cellular/Molecular

Tonic and Burst Spiking Open Distinct Calcium Channels
Adam C. Errington, John J. Renger, Victor N. Uebele, and Vincenzo Crunelli
(see pages 14843–14853)
Thalamocortical (TC) neurons exhibit distinct spike modes during sleep and wakefulness. In the latter state, TC neuron membrane potential (Vm) is relatively depolarized, and continual sensory and cortical inputs drive tonic spiking. During sleep, Vm hyperpolarizes and the neurons fire in bursts, generating low-frequency oscillations that impede relay of sensory information through the thalamus. Bursting relies on T-type calcium channels, which are inactivated during wakefulness and deinactivate when neurons hyperpolarize. Small depolarizing steps then evoke sufficient T current to drive Vm above threshold, producing so-called low-threshold spikes. Errington et al. report that T-type channels are located throughout the dendritic arbor of rat TC neurons, so low-threshold spikes led to near-synchronous all-or-none calcium influx throughout the tree, regardless of whether stimulation was delivered to the soma or distal dendrites. In contrast, tonic spiking caused calcium influx only in proximal dendrites. This calcium influx, produced by backpropagating spikes, did not involve activation of T-type channels.
Development/Plasticity/Repair

Loss of Protein Function Might Underlie Cognitive Decline
Robert Tamayev, Luca Giliberto, Wei Li, Cristina d'Abramo, Ottavio Arancio, et al.
(see pages 14915–14924)
Many neurodegenerative diseases, like Alzheimer's, are characterized by aggregation, fibrillization, and deposition of normally soluble proteins. Such amyloidosis often results from abnormal processing of a precursor protein, and the aggregates are widely believed to initiate cognitive decline and degeneration associated with the diseases. But in mice expressing human mutated forms of these proteins, cognitive deficits and amyloid deposition are not always correlated: some mutations cause amyloidosis without cognitive effects, and others produce cognitive deficits without amyloidosis. Tamayev et al. conclude that cognitive decline sometimes results from loss of normal protein function rather than abnormal aggregation. They introduced a mutation linked to familial British dementia into the homologous mouse gene Bri2. Unlike human patients, knock-in mice showed no abnormal amyloidosis, neurodegeneration, or tau pathology. Nonetheless, like humans, knock-in mice had reduced levels of BRI2 protein and exhibited impaired hippocampal-dependent memory. Similar deficits occurred in Bri2 heterozygous-null mice, supporting the hypothesis that cognitive decline was caused by loss of normal function.
Behavioral/Systems/Cognitive

Fast and Slow Adaptation Involve Different Processes
Aysha Keisler and Reza Shadmehr
(see pages 14817–14823)
Motor adaptation has a fast component, in which performance improves rapidly but deteriorates shortly after training, and a slow component, in which improvement and decay take longer. Noting that fast adaptation is strongest when errors are consciously perceived, Keisler and Shadmehr hypothesized that it is akin to declarative learning. If so, performing another declarative task immediately after training should interfere selectively with fast adaptation. To test this, they had subjects move a robotic arm. The robot induced slow adaptation by imposing a unidirectional force over many trials, then induced fast adaptation by imposing force in the opposite direction. During testing, fast adaptation initially masked the effect of slow adaptation, but as fast adaptation decayed, the effect of slow adaptation emerged. If subjects performed a declarative memory task between training and test periods, however, the effect of fast adaptation was eliminated, suggesting that fast and slow adaptation involve different processes, and the former shares resources with declarative memory.
Neurobiology of Disease

Blocking NGF Reduces Cancer-Induced Sprouting and Pain
Juan M. Jimenez-Andrade, Aaron P. Bloom, James I. Stake, William G. Mantyh, Reid N. Taylor, et al.
(see pages 14649–14656)
Cancer pain usually increases over time, often becoming resistant to analgesic treatment. In most people who die from prostate cancer, tumors have metastasized to bone, leading to ongoing, analgesic-resistant pain. To investigate the causes of this pain, Jimenez-Andrade et al. injected canine prostate tumor cells into mouse femurs. The cells produced tumors like those in humans: small colonies of tumor cells formed throughout the bone, surrounded by new bone, macrophages, stromal cells, and blood vessels. Furthermore, mice showed evidence of ongoing pain, frequently holding the injected leg aloft while stationary. Pain likely resulted from the sprouting of sensory neurons, which was detected within bone marrow. Many sprouted nerve fibers expressed a nerve growth factor (NGF) receptor, and sustained treatment with anti-NGF antibodies starting 10 days after injection of tumor cells reduced both sprouting and the frequency of hindpaw elevation. Such treatment might also slow the progression of pain in humans.
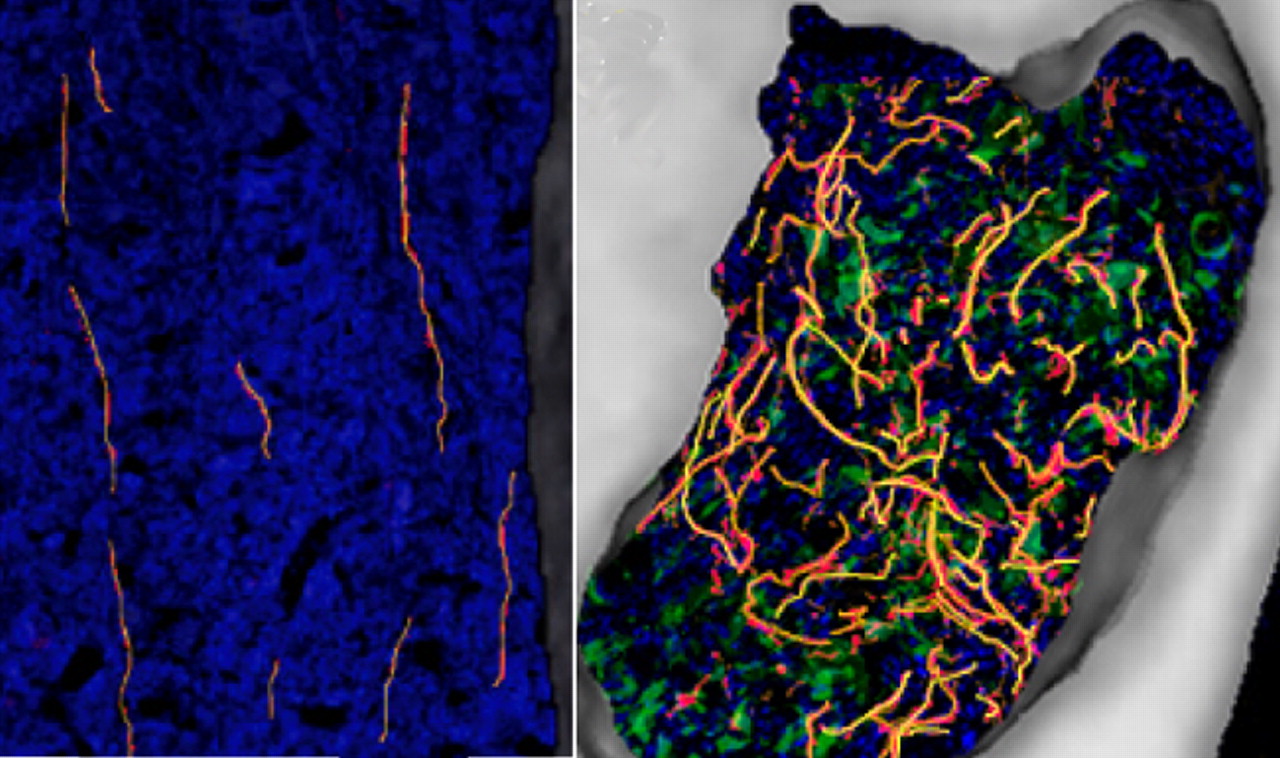
In normal mouse bone (left), sensory nerve fibers (yellow) grow linearly through hematopoietic cells (blue), whereas in bones injected with prostate tumor cells (green, right), the nerves sprout and become dense and highly branched, and colonies are surrounded by new woven bone (white). See the article by Jimenez-Andrade et al. for details.





{kind=link}