

Abstract
Axonal and nerve terminal action potentials often display a depolarizing after potential (DAP). However, the underlying mechanism that generates the DAP, and its impact on firing patterns, are poorly understood at axon terminals. Here, we find that at calyx of Held nerve terminals in the rat auditory brainstem the DAP is blocked by low doses of externally applied TTX or by the internal dialysis of low doses of lidocaine analog QX-314. The DAP is thus generated by a voltage-dependent Na+ conductance present after the action potential spike. Voltage-clamp recordings from the calyx terminal revealed the expression of a resurgent Na+ current (INaR), the amplitude of which increased during early postnatal development. The calyx of Held also expresses a persistent Na+ current (INaP), but measurements of calyx INaP together with computer modeling indicate that the fast deactivation time constant of INaP minimizes its contribution to the DAP. INaP is thus neither sufficient nor necessary to generate the calyx DAP, whereas INaR by itself can generate a prominent DAP. Dialysis of a small peptide fragment of the auxiliary β4 Na+ channel subunit into immature calyces (postnatal day 5–6) induced an increase in INaR and a larger DAP amplitude, and enhanced the spike-firing precision and reliability of the calyx terminal. Our results thus suggest that an increase of INaR during postnatal synaptic maturation is a critical feature that promotes precise and resilient high-frequency firing.
Introduction
To localize sounds, mammals use sub-millisecond differences in the timing of binaural signals (Carr et al., 2001). Preservation of the exact timing of action potential spikes along the ascending auditory pathway is thus thought to be crucial for the rapid and precise pinpointing of sound sources. The calyx of Held synapse is a pivotal element in a brainstem circuit that computes sound localization (Kandler and Friauf, 1993; Grothe, 2003). Fast Na+ channel inactivation kinetics (Leão et al., 2005), together with high levels of expression of high-threshold (HT) Kv3 potassium channels (Wang and Kaczmarek, 1998; Nakamura and Takahashi, 2007), promote short action potential (AP) waveforms at the calyx terminal, making it well suited to preserve the timing of sensory signals (Dodson and Forsythe, 2004; Kaczmarek et al., 2005) Moreover, a fast and narrow AP waveform allows the mature calyx terminal to fire short spike trains at frequencies up to1 kHz without failures (Wu and Kelly, 1993; Taschenberger and von Gersdorff, 2000). Sodium currents in the auditory system may thus have been under evolutionary pressure for fast inactivation and rapid recovery from inactivation (Koishi et al., 2004).
After the action potential spike, recordings from the calyx of Held exhibit a depolarizing after potential (DAP) characterized by a fast upstroke and a slower decay phase (Borst et al., 1995). Previous studies indicate that passive discharge currents regulate the decay phase of the DAP (Borst et al., 1995), whereas active Na+ and/or K+ currents influence the amplitude of the DAP (Dodson et al., 2003; Paradiso and Wu, 2009). However, the specific mechanism that generates the calyx DAP has not been established. Moreover, a prominent DAP has also been observed in the action potential waveforms of other CNS nerve terminals (Geiger and Jonas, 2000; Debanne, 2004) and peripheral axons (Barrett and Barrett, 1982; David et al., 1995; Lin, 2008), where the DAP represents a period of subthreshold superexcitability for axons (Bowe et al., 1987). The DAP is therefore of great interest for neuropathies like multiple sclerosis that affect axonal excitability (Baker, 2005).
Pituitary nerve terminals (Ahern et al., 2000) and the calyx of Held (Huang and Trussell, 2008) express a persistent Na+ current (INaP), and modeling studies have suggested that INaP may generate the DAP in some mammalian axons (Burke et al., 2009; McIntyre et al., 2002). In many neurons, non-inactivating INaP currents are active within the subthreshold region and facilitate bursts of neuronal discharges (Crill, 1996; Kay et al., 1998; Bean, 2007). By contrast, resurgent sodium current (INaR) is activated during membrane repolarization, following the action potential depolarization that inactivates the transient Na+ current (INaT). INaR is generated by Na+ channels transiently dwelling in an open state during return to closed states, and its presence promotes rapid recovery from Na+ channel inactivation (Raman and Bean, 2001). INaR thus facilitates neuronal repetitive firing (Raman and Bean, 1997; Afshari et al., 2004). Here, we propose that a developmentally upregulated INaR improves the calyx of Held's ability to fire without failures at high stimulation frequencies.
Materials and Methods
Brainstem slice preparation.
All animal procedures were approved by the institutional animal use committee and followed National Institutes of Health guidelines. Transverse brainstem slices (200 μm thick) were prepared from Sprague Dawley rat pups on postnatal day 5 (P5)–P17. After rapid decapitation, the brainstem of rat pups was quickly removed from the skull and immersed in ice-cold, low-calcium artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 3 MgCl2, 0.1 CaCl2, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 0.4 ascorbic acid, 3 myo-inositol, and 2 Na-pyruvate, pH 7.3–7.4 when bubbled with carbogen (95% O2, 5% CO2; osmolarity of 310–320 mOsm). After cutting with a vibratome slicer (VT1000, Leica), the slices were transferred to an incubation chamber containing normal aCSF bubbled with carbogen and maintained at 35°C for 30–45 min and thereafter at room temperature (RT). The normal aCSF was the same as the slicing aCSF, but with 1 mm MgCl2 and 2 mm CaCl2.
Electrophysiology.
Whole-cell patch-clamp recordings were performed in normal aCSF at RT (22−24°C). Slices were perfused at 2 ml/min and visualized using an infrared differential interference contrast microscope (Axioskop FS1, Zeiss) and a 40× water-immersion objective coupled to a 2× premagnification (Optovar, Zeiss) and a CCD camera (C73, Sony) with contrast enhancement controller (Hamamatsu). Presynaptic action potential recordings used a pipette solution containing the following (in mm): 130 K-gluconate, 20 KCl, 5 Na2-phosphocreatine, 10 HEPES, 4 Mg-ATP, 0.2 or 5.0 EGTA, and 0.3 GTP, pH adjusted to 7.3 with KOH. Presynaptic Na current recordings used a pipette solution containing the following (in mm): 130 Cs-gluconate, 20 CsCl, 10 TEA-Cl, 5 Na2-phosphocreatine, 10 HEPES, 4 Mg-ATP, 5 EGTA, and 0.3 GTP, pH adjusted to 7.3 with KOH. The external solution contained 10 mm TEA-Cl and 0.2 mm CdCl2 to block presynaptic K+ and Ca2+ channels. In some experiments, Alexa 555 (500 μm, Invitrogen) was included in the pipette solution for subsequent morphological analysis by confocal microscopy. Pipettes were pulled from borosilicate glass (World Precision Instruments) with a Sutter P-97 electrode puller (Sutter Instruments) to open tip resistances of 3–4 MΩ. Recordings were continued only if the initial uncompensated series resistance (Rs) was <20 MΩ. For voltage-clamp recordings, Rs was compensated 40–80%, and the average value of Rs after compensation was 7.8 ± 0.57 MΩ (n = 19).
For current-clamp recordings, we first established a cell-attached voltage-clamp configuration to set the pipette capacitance compensation (C-fast), and we then broke into the whole-cell configuration and set membrane potential at −80 mV. Once a stable whole-cell voltage-clamp configuration was achieved, we switched to the fast current-clamp configuration of the EPC-9 patch-clamp amplifier (HEKA Electronik) (see also Taschenberger and von Gersdorff, 2000). Presynaptic resting membrane potentials were adjusted by small amounts of current injection (<−50 pA) to have a resting value near −80 mV (Borst et al., 1995). Recordings were not corrected for a calculated liquid junction potential of ∼11 mV. The membrane time constant (τm) was measured from membrane voltage responses to −50 pA current injections (e.g., see Fig. 2b), and data points were fit by a single exponential function using IgorPro software (Wavemetrics). Presynaptic spikes were elicited with a bipolar platinum/iridium electrode (Frederick Haer Company) placed near the midline spanning the afferent fiber tract of the medial nucleus of the trapezoid body (MNTB). An Iso-Flex stimulator delivered 100 μs pulses (<10 V constant voltage) and was driven by a Master 8 pulse generator (A.M.P.I.) triggered by a Macintosh computer (Apple). Data were filtered at 2.9 kHz and acquired at a 5–10 μs sampling rate, using an EPC-9 amplifier controlled by Pulse 8.4 HEKA software.
Confocal fluorescence microscopy.
Slices used for patch-clamp recordings were subsequently fixed in 4% paraformaldehyde for 1 h and permeabilized with 0.5% Triton X-100 for 30 min. Calyxes were filled with Alexa 555 (500 μm, Invitrogen) via the patch pipette (see Fig. 7a; and supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Slices were mounted onto Superfrost slides in photobleaching-protective medium. Stained slices were viewed with a 60× oil-immersion objective using a confocal laser-scanning microscope (FluoView 300, Olympus; laser lines at 488 nm for green and 633 nm for red).
Analysis.
Data were analyzed off-line using IgorPro software (Wavemetrics). Differences were considered statistically significant when p values were <0.05 by Student's t test or by one-way ANOVA. Statistical analyses were performed using Prism 4.0 (GraphPad). Means ± SE are reported, unless otherwise noted. Significance is reported as *p < 0.05, **p < 0.01, and ***p < 0.001, using Student's t test or one-way ANOVA test.
Drugs and peptides.
The β4peptide and the scrambled β4peptide, both 95% pure, were synthesized by Gynmed. All other chemicals were obtained from Sigma-Aldrich except for TTX, which was from Tocris Bioscience.
Computational modeling.
Model Na+ currents and action potentials were performed by numerical integration of Hodgkin–Huxley-like equations and the cable equation using the NEURON programming environment (see supplemental materials, available at www.jneurosci.org as supplemental material). The model calyx consisted of two cylindrical compartments: the calyx nerve terminal (capacitance, 12.6 pF) and the axon (diameter, 3 μm; length, 100 μm; capacitance, 9.4 pF) (Leão et al., 2005). Conductances included were passive leak conductance, HT and low-threshold (LT) K+ conductances, and transient, persistent, and resurgent Na+ currents (Raman and Bean, 2001; Leão et al., 2005; Huang and Trussell, 2008). Auditory neuron HT and LT potassium current models were as previously published (Manis and Marx, 1991; Wang et al., 1998). For the parameters used and further details of the model, see supplemental materials (available at www.jneurosci.org as supplemental material).
Results
Presynaptic DAP and AP timing
Direct recordings from the rat calyx of Held revealed a prominent DAP following a single action potential spike (Fig. 1a) (see also Borst et al., 1995). This presynaptic DAP could be triggered by afferent fiber stimulation in the brainstem midline (Fig. 1a) or by a brief suprathreshold depolarizing current injection in a calyx terminal with a short axon stump (axon length in these cases was measured after filling the calyx with Alexa 555 and varied from 20 to 200 μm, n = 5 calyces) (Fig. 1b; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). A long axon, extending back to the midline, was thus not necessary for generating the DAP, suggesting that the DAP was generated near the calyx nerve terminal.

The presynaptic DAP at the calyx of Held. a, Afferent fiber stimulation evokes a single presynaptic AP spike followed by a depolarizing DAP in the calyx of Held (P13). Inset, Same trace on an expanded time and voltage scale showing the rise of the DAP and the fAHP. b, A brief suprathreshold depolarizing current injection (500 pA for 2 ms) evokes an AP followed by a DAP in a calyx terminal with a severed axon. c, A pair of APs in response to afferent fiber stimulation with 4 ms interval (black trace) and then combined with a short hyperpolarizing current injection (−100 pA) to the calyx to remove the DAP (red traces, arrowhead). Note the delay in the second AP (arrow) or failure to fire in the absence of the DAP.
The presynaptic DAP showed a clear rising phase that depolarized the calyx from the downward peak of the fast after-hyperpolarization (fAHP) to near −60 mV (Fig. 1a, inset). However, the membrane potential at the peak of the DAP rarely surpassed −60 mV and thus was always safely below spike threshold. The DAP amplitude measured from the resting potential to the peak of the DAP depended on the resting potential and was larger at more hyperpolarized potentials (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). However, the DAP amplitude measured from the peak of fAHP to the peak of DAP had a fairly invariant amplitude of 7.7 ± 0.2 mV (n = 22 calyces) at adjusted resting membrane potentials ranging from −90 to −64 mV (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The 10 to 90% rise time of the DAP upstroke was dependent on the adjusted resting membrane potential (1.0 ± 0.1 ms at −90 mV, 1.7 ± 0.3 ms at −80 mV, 2.2 ± 0.2 ms at −70 mV, and 2.7 ± 0.3 ms at −65 mV; n = 3, 3, 6, and 5, respectively, for P13; p < 0.01, one-way ANOVA).
The presynaptic DAP of a first AP influenced the exact timing of a second AP spike. For a pair of APs with a 4 ms interval (corresponding to 250 Hz activity, well within the physiological range of the calyx of Held), a hyperpolarizing current injection of ∼−100 pA removed the DAP and increased the latency of the second AP (latency, measured from the stimulus artifact to the peak of the AP, increased from 0.8 ± 0.06 to 0.9 ± 0.08 ms, n = 5, p < 0.05) (Fig. 1c). In addition, suppressing the DAP also reduced reliability as the failure rate for the second AP was increased. The increase in AP failures may be mediated by the back-propagation of the hyperpolarization down the axon (Paradiso and Wu, 2009). However, we also performed the same experiment on calyces with severed axons of different lengths using a brief depolarizing current injection. Similar to the afferent fiber stimulation case, the complete elimination of the DAP via a step-like hyperpolarizing current injection significantly increased the second AP latency (n = 5; data not shown). These results thus indicate the DAP is generated near the calyx terminal, likely at the axonal heminode that contains a large density of Na+ channels (Leão et al., 2005), and also that the DAP influences the exact timing and reliability of subsequent APs.
Presynaptic DAP: developmental changes
To study the role of DAP during synaptic maturation, we characterized its kinetics at three postnatal ages: shortly after formation of the calyx terminal at P5–P6 (Kandler and Friauf, 1993; Rodríguez-Contreras et al., 2008); just before the onset of hearing at P9–P10; and at P15–P16, when the synapse assumes an adult-like morphology and rat pups respond to sound stimulation (Kandler and Friauf, 1993; Sonntag et al., 2009). Measured from the downward peak of the fAHP to the peak of the DAP (Fig. 2a, inset), the amplitude of DAP rise was 2.4 ± 0.4 mV at P5–P6, 6.7 ± 0.7 mV at P9–P10, and 13.8 ± 0.6 mV at P15–P16 (n = 8, 9, and 4, respectively; p < 0.001, one-way ANOVA). For the same ages listed above, the single exponential decay time constants (τdecay) of DAP were 79 ± 3.6, 30 ± 3.1, and 15 ± 2.4 ms, respectively (n = 8, 9, and 4, respectively; p < 0.001). This decrease in τdecay closely paralleled a decrease in τms, which were 50.5 ± 3.7, 22.4 ± 1.5, and 11.4 ± 1.6 ms, respectively, for the same age groups (n = 7, 5, and 5, respectively; p < 0.001) (Fig. 2b,c). The faster membrane time constant in more mature calyces was due in part to reduced input resistance, which was 375 ± 25.1, 322 ± 29.2, and 191 ± 16.2 MΩ, respectively, at P5–P6, P9–P10, and P15–P16 (n = 7, 5, and 5, respectively; p < 0.001). The 10 to 90% rise time of the DAP was also shorter in more mature rats (1.8 ± 0.13 ms in P15–P16 vs 3.0 ± 0.15 ms in P5–P6 calyces; resting potential, −80 mV; n = 4; p < 0.01). The DAP thus has a significantly faster rise and decay phase for more mature calyces.

The DAP kinetics during postnatal development. a, A presynaptic AP spike and DAP in the calyx of Held from three developmental stages: P6 (red), P10 (blue), and P16 (black). The AP spike was elicited by afferent fiber stimulation. Resting potential was −80 mV (dashed line). Inset, Same traces on an expanded time and voltage scale. Note the changes in rise and decay times of the DAP at different developments ages. b, Membrane potential changes induced by a −50 pA hyperpolarizing current injection in calyces of different ages indicate that input resistance decreases with age. The P15 trace is also shown scaled to the P6 amplitude to facilitate comparison of decay times (gray trace). c, Summary of the single exponential τdecay (left) of the DAP and the τm (right) for P5–P6, P9–P10, and P15–P16 calyces (p < 0.001, one-way ANOVA). d, A single AP at P10 calyx in the absence (control; black trace) or in the presence of TEA (1 mm; red trace); 1 mm TEA inhibited the fAHP in the calyx terminals. e, Action potential trains in response to 100 Hz afferent fiber stimulation in P5 (red) and P15 calyces (black). The P5 AP train adapted (black arrow). Right, Expanded figure of boxed area of AP trains superimposed. The P5 calyx train of APs showed a large depolarized plateau (large arrowhead) due to the summation of the slow DAPs, and its AP train had spike failures (small vertical arrowheads), whereas the P15 calyx AP train had no failures.
What is the mechanism that governs the fAHP peak? We first note that the fAHP becomes progressively larger (more hyperpolarized) with increasing postnatal age (−69 ± 0.7, −74 ± 0.4, and −78 ± 0.5 mV; n = 8, 9, and 8, respectively, for P5–P6, P9–P10, and P15–P16; p < 0.001) (Fig. 2a). Previous studies indicate that TEA-sensitive Kv3 channels undergo a developmental increase that contributes to shorter AP waveforms and more reliable spiking at high frequencies (Wang and Kaczmarek, 1998; Nakamura and Takahashi, 2007). To test the involvement of Kv3 channels in the generation of the fAHP, we applied 1 mm TEA and found the peak fAHP for P14 calyces was significantly decreased by 7.1 ± 0.3 mV (n = 3) from −75 to −67.7 mV (Fig. 2d), indicating that Kv3 currents are a major determinant of the fAHP peak (Wang and Kaczmarek, 1998).
How does the DAP affect AP firing during a high-frequency train of spikes? We examined this by recording AP trains in P5–P6 and P15–P16 calyces (Fig. 2e). At P5–P6, a long AP train elicited by afferent fiber stimulation at 100 Hz had a large depolarizing plateau, and AP spikes became progressively smaller in amplitude during the train (Fig. 2e, arrow). Action potentials eventually failed in the latter part of the train. However, at P15–P16 calyx AP trains had almost no depolarizing plateau and little spike attenuation during the 100 Hz train (Fig. 2e). Superposition of the two spike trains at higher time resolution shows that the slow P5–P6 DAP summates to produce a large depolarizing plateau (Fig. 2e). This plateau will cause a progressive inactivation of Na+ channels, which explains the attenuation of the spike amplitudes and the eventual failures in spiking (Klyachko et al., 2001; Leão and von Gersdorff, 2002). These developmental changes in the depolarizing plateau may be caused by an increased expression of Kv2-type channels in more mature calyces (Johnston et al., 2008). The fast decay of the DAP in more mature P15–P16 calyces is thus an important developmental change that helps to avoid DAP summation during a train and the subsequent failures in spiking seen in immature calyces.
Presynaptic Na+ currents generate the DAP
We next examined the conductances that underlie the DAP in calyx terminals. We found that a low concentration of TTX (20 nm) significantly decreased the amplitude of DAP from 9.0 ± 1.1 to 1.0 ± 0.7 mV with only a slight reduction of AP spike amplitude by 10% (n = 5) (Fig. 3a,f). For a pair of APs, TTX (20 nm) also had a stronger effect on the second AP latency than the first AP (Fig. 3a,c), indicating that the TTX-sensitive DAP is critical to the timing of subsequent AP. To further test that a Na+ current is the main conductance generating the DAP, we blocked Na+ channels with 1 μm TTX, which completely inhibits Na+ spikes, and then injected a large current (1 to 1.5 nA for 1.5 ms) to depolarize the calyx terminal up to ∼+30 mV, thus mimicking the peak potential reached during an AP. After the membrane potential reached +30 mV, we observed a fast repolarization of the membrane potential, but no DAP was generated (n = 3; P13 calyx) (Fig. 3b, black trace). This rapid decay is likely due to activation of high-threshold Kv3 currents (Dodson and Forsythe, 2004; Kaczmarek et al., 2005). The smaller current injections (0.5 nA), which depolarized the calyx to a peak value <0 mV, resulted in a much slower decay of membrane potential back to the resting potential (Fig. 3b, red trace). In this case, decay was fitted by a double exponential function consisting of a fast component (1.2 ± 0.2 ms) and a slow component (15 ± 0.7 ms; 17% of amplitude; n = 3). The fast component may be due to activation of low-threshold Kv1.1/1.2 channels (Dodson et al., 2003), and the slow component is probably due to the passive discharge of the membrane potential, since this slow τdecay is comparable to the τm (11.4 ms at P15–P16) (Fig. 2a–c). Together, these results indicate that a TTX-sensitive Na+ current, and not depolarization per se, generates the DAP. We conclude that the calyx of Held AP waveform is generated by a primary Na+ influx event during the upstroke of the spike and a secondary Na+ influx event that causes the DAP. This secondary Na+ influx may also have important consequences for energy consumption in axons and nerve terminals (Alle et al., 2009; Carter and Bean, 2009).
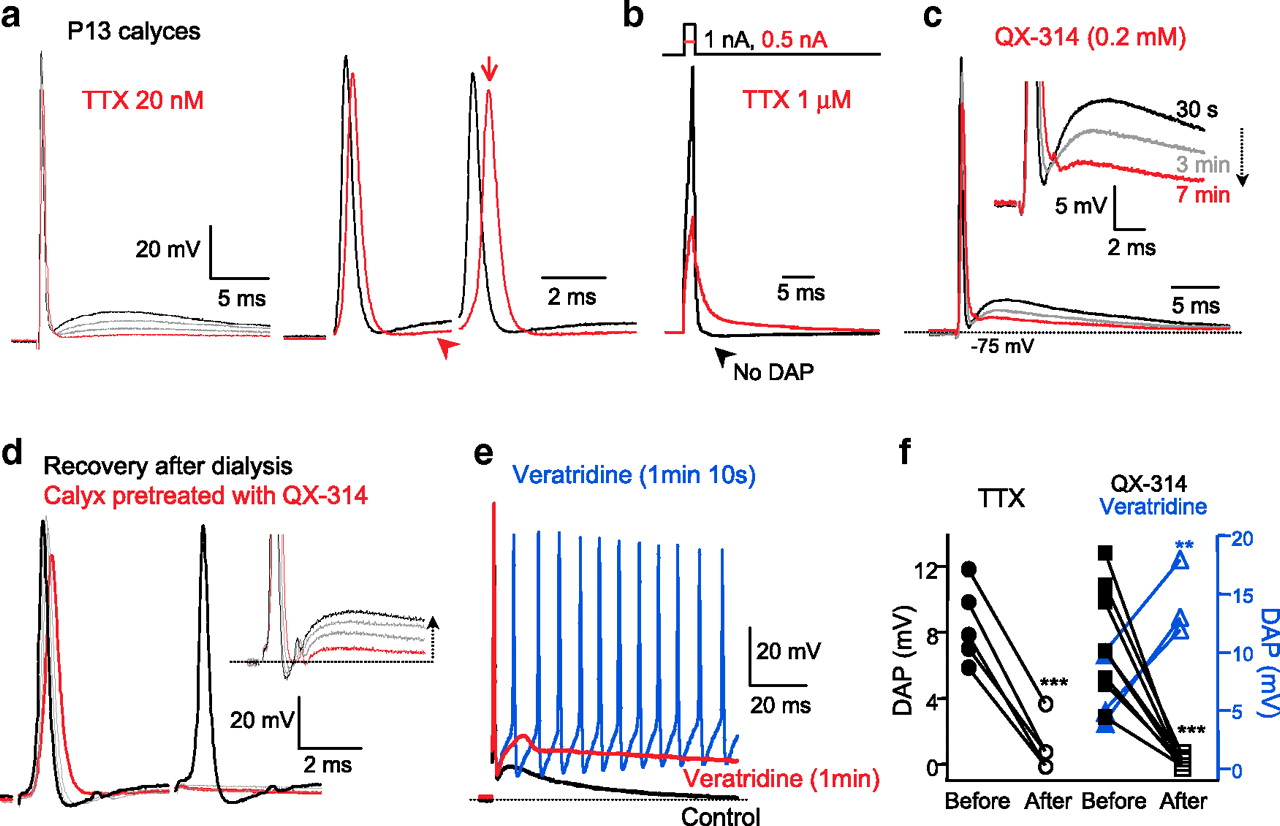
Presynaptic Na+ current generates the rising phase of the DAP. a, Spikes evoked by afferent fiber stimulation in the absence (control; black trace) and presence of external TTX (20 nm, gray traces during onset of application and red traces end of application). For a pair of APs, with 4 ms stimulus interval, removing the DAP (arrowhead) delayed the latency to the peak of the second AP (red arrow). b, After the complete inhibition of voltage-dependent Na+ currents with 1 μm TTX, brief current injections [1.5 ms duration, 0.5 nA (red trace) or 1 nA (black trace)] depolarized the calyx to 0 mV (red) or +30 mV (black). Note the absence of a DAP after the larger spike, whereas the smaller spike has a monotonically decaying DAP with no hump. c, P13 calyx AP recorded with a pipette solution containing a low concentration of QX-314 (0.2 mm), a Na+ channel blocker. The black trace was obtained within a few seconds after break-in, and the red trace was recorded after 7 min of whole-cell recording. After 3 min of QX-314 dialysis, the rising phase of the DAP was significantly inhibited, although spike amplitude was little changed (gray trace). d, The calyx of Held was dialyzed for 5 min with QX-314 (0.3 mm), and the stunted rise of the DAP was recorded (red trace). The recording pipette was then removed, and the same calyx was patched again using normal internal pipette solution without QX-314. The normal rise and amplitude of the AP spike and DAP recovered after 7 min of dialysis, which removed QX-314 from the calyx through the patch pipette. Note the broader AP spike with QX-314 (inset). The removal of QX-314 also recovered the second AP, which failed to fire in the presence of QX-314. e, Veratridine (an activator of Na+ channels; 1 μm) increased significantly the amplitude of the DAP after 1 min (red trace) and elicited repetitive spontaneous spiking in response to a single afferent fiber shock (blue trace; recorded 10 s after red trace). f, Summary of DAP amplitude in the presence of TTX (20–50 nm, n = 5), QX-314 (0.2–0.3 mm, n = 7), and veratridine (1 μm, blue triangles, n = 3). Data were analyzed with paired t test (**p < 0.01; ***p < 0.001).
To test whether local Na+ spike after currents close to the calyx and its axonal heminode play a role in DAP generation, we recorded using a low concentration of the permanently charged lidocaine analog QX-314 in the pipette solution (0.2–0.3 mm). Shortly after whole-cell break-in, calyx spikes were followed by a DAP with a fast rising phase (Fig. 3c, black trace). During dialysis of QX-314, the amplitude of the DAP decreased markedly. After 5 min of QX-314 dialysis, the DAP, measured from the fAHP to the DAP peak, decreased from 8.0 ± 1.1 to 0.2 ± 0.1 mV (n = 8) (Fig. 3c,f), while the amplitude of the AP was only slightly decreased to 94 ± 6% (n = 8). In addition, the internal application of QX-314 significantly hyperpolarized the calyx of Held resting membrane potential by −4 mV (from −80 ± 0.6 to −84 ± 0.7 mV; n = 4; p < 0.01), suggesting a tonic Na+ channel activation at rest (Huang and Trussell, 2008). Moreover, this inhibition of the DAP with QX-314 was reversible. Patching the same calyx terminal a second time using a pipette without QX-314 restored the original DAP amplitude, and the adjusted resting membrane potential moved back to the original −80 mV. In addition, a second spike, which had failed to fire with QX-314 pretreatment, was revived after washing out of QX-314 (n = 3) (Fig. 3d). Na+ currents close to the calyx terminal thus generate the DAP.
By contrast, application of the Na+ channel activator veratridine (1 μm) significantly increased the DAP amplitude from 6 ± 1.8 to 14 ± 1.3 mV and slowed the decay of the DAP (Fig. 3e,f) (n = 3; p < 0.01). The progressive increase in the DAP amplitude with veratridine eventually resulted in repetitive spontaneous spiking in response to a single afferent fiber stimulation (Fig. 3e, blue trace). Veratridine increases the open time of Na+ channels, slows their inactivation rate (Garber and Miller, 1987), and shifts the activation curve to the left (Ulbricht, 1998). This suggests that the decay rate of the DAP may be influenced by the inactivation rate of the underlying Na+ channels that generate the DAP. Furthermore, an abnormally large DAP amplitude produces aberrant spiking and results in hyperexcitability at the calyx of Held (Dodson and Forsythe, 2004).
INaR and INaP at the calyx of Held
To identify the specific type of Na+ current responsible for the DAP, we first performed voltage-clamp recordings of calyx Na+ currents. A step-depolarization from the holding potential of −90 to +30 mV generated a large and rapidly inactivating INaT (Leão et al., 2005). Upon repolarization to potentials ranging from −20 to −70 mV, we observed a voltage- and time-dependent current with biophysical properties similar to the INaR present in Purkinje cells, cerebellar granule cells, vestibular neurons, and MNTB principal cells (Fig. 4a,b) (Raman and Bean, 1997; Leão et al., 2006; Magistretti et al., 2006; Gittis and du Lac, 2008). The current–voltage (I-V) relationship for INaR exhibited a peak of −408 ± 63 pA at ∼−30 mV in P9–P12 calyces (n = 16; no liquid-junction potential correction) (Fig. 4c). The decay kinetics of INaR was voltage dependent, with a single exponential τdecay ranging from 5 to 40 ms (Fig. 4b,d). In addition, we also observed a INaP that remained after the decay of INaR (Fig. 4a) (Huang and Trussell, 2008). We also obtained outside-out patches from the calyx terminal that contained well clamped INaTs (n = 3; data not shown) (Leão et al., 2005), but these did not show INaR or INaP currents (see also Scott et al., 2010).

INaR in the calyx of Held. a, INaT and INaR were elicited in a P10 calyx terminal by a 10 ms step depolarization to +30 mV, followed by step repolarizations to −70, −60, −50 (gray), −40 (green), −30 (blue), and −20 mV (red) as indicated in the voltage-clamp protocol (top). Note that after the decay of INaR, an INaP remains activated. b, Individual current traces for repolarizations to −70 and −30 mV are shown together with single exponential decay fits. The INaT peaks have been truncated. Note the fast deactivating tail currents (arrowheads) before the full activation of INaR. c, The current–voltage dependence (I-V curve) for peak INaR for P9–P12 calyces. The peak INaR was measured from the end of INaT to the peak of INaR. d, The τdecay of INaR as a function of test membrane potentials. INaR decayed more slowly at more depolarized potentials.
Tetrodotoxin at high concentration (1 μm) blocked all three currents described above (INaT, INaR, and INaP), confirming their identity as Na+ currents (Fig. 5a,b). In contrast, internal QX-314 (0.3 mm) blocked the persistent and resurgent currents but spared 60% of the transient current after 10 min of dialysis of the calyx (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material). Bath application of veratridine (5 μm) increased the peak amplitude of INaR from −273 ± 44 pA to −400 ± 62 pA (n = 4) and slowed the inactivation decay of INaR, whereas the peak of INaT was not significantly changed (−6.0 ± 0.5 nA versus −5.6 ± 0.8 nA; n = 4; p > 0.1; paired t test) (supplemental Fig. 3b, available at www.jneurosci.org as supplemental material). Low concentrations of TTX (50 nm) also blocked the INaR and INaP of more mature P15–P16 calyces (n = 3) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Together, these results indicate that the calyx INaR and/or INaP, both of which are blocked by low doses of TTX and QX-314, and are enhanced by veratridine, underlie the upstroke phase of the DAP.

The calyx INaR increases during postnatal development. a, INaT, INaP, and INaR were evoked at a P5 calyx by a short depolarization to +30 mV followed by a step repolarization to −30 mV. TTX (1 μm) completely inhibited INaT, INaP, and INaR in the calyx of Held. The bottom trace is the TTX-sensitive current obtained by subtraction. The large INaT peak has been truncated. b, INaT, INaP, and INaR at a P10 calyx. TTX (1 μm) completely inhibited INaP, INaR, and INaT. c, I-V curve for peak INaR at P5–P6 (black) and P10–P12 calyces (red). All sodium currents were leak subtracted (p/5) and TTX subtracted. There is a significant increase in INaR between the two age groups at the peak of INaP near −30 mV. d, I-V curve for the INaP for P5–P6 calyces (black) and P10–P12 calyces (red). There is a slight increase in INaP between the two different age groups at the peak of INaP near −10 mV, but this was not statistically significant (p = 0.38, Student's t test). Membrane potentials were not corrected for a 10 mV liquid junction potential. Persistent sodium current thus starts to activate at about −80 mV and may contribute to the resting membrane potential.
Developmental increase of calyx INaR
The rising phase of the DAP became significantly faster from P5 to P15 (Fig. 2a), a period when Na+ currents undergo developmental changes at the calyx of Held (Leão et al., 2005). To help distinguish the individual contributions of INaR and INaP in generating the DAP, we examined changes in INaR and INaP amplitudes during the first two postnatal weeks. We found that INaR increased in amplitude during the developmental period (Fig. 5a,b). The I-V curve of INaR showed an inverted-bell shape at P5–P6 and P10–P12, and a peak value at −30 mV (Fig. 5c). Immature calyces (P5–P6) exhibited significantly smaller inward peak INaR measured at −30 mV (−86 ± 10.6 pA, n = 3) than P10–P12 calyces (−313 ± 71 pA, n = 5; p < 0.02, Student's t test). An INaP remained as a noninactivating current after full decay of INaR, and it slightly increased from −335 ± 103 pA at P5–P6 (n = 4) to −533 ± 88 pA at P10–P12, but this increase was not significant (INaP measured at −10 mV; n = 5; p = 0.38, Student's t test) (Fig. 5c). INaR and INaP had different voltage dependences, since INaR and INaP displayed two distinct peaks in their I-V curves (at ∼−30 and −10 mV, respectively) (Fig. 5c,d). We thus conclude that INaR and INaP are distinct sodium currents. Furthermore, the faster rise of DAP amplitudes observed with more mature calyces correlates well with the large increase in INaR during development.
Computer modeling of the calyx DAP
The pharmacological compounds available to us are not selective for INaR or INaP. To obtain further insight into the Na+ conductance that generates the DAP, we thus performed computer simulations to test for the specific role of persistent and resurgent Na+ currents. Conductances included in our modeling were passive (leak) conductance; HT and LT potassium conductances; and transient, persistent, and resurgent sodium currents (Fig. 6, black lines) (Dodson and Forsythe, 2004; Kaczmarek et al., 2005). In this manner, we could investigate the effect of eliminating persistent current (Fig. 6, blue lines) or eliminating resurgent current (Fig. 6, red lines) on the DAP. Full details of the model and its parameters are given in Materials and Methods and in supplemental material (available at www.jneurosci.org as supplemental material).

Computer model of the role of INaR and INaP in the generation of the DAP. a, Model Na+ currents in four scenarios: control (black); after removal of persistent current (blue); after removal of resurgent current (red); and after removal of resurgent current with two times higher persistent current (green). b, Model action potentials in the same scenarios as described in a. Note that the DAP is normal without persistent current, but the DAP is absent without resurgent current. The action potential waveform is also broader without resurgent current. c, Effect of four different densities of persistent current on the DAP (relative to that measured in the calyx of Held). Black trace, 2.4 × INaP; blue trace, 3.2 × INaP; red trace, 4.2 × INaP; and green trace, 5.6 × INaP. Top, Persistent Na+ tail currents evoked by steps from 0 to −80 mV. Bottom, Resulting DAP waveforms. The deactivation time constant (τ) was fixed at τ = 0.24 ms. d, Same as in c except without resurgent Na+ current. No DAP hump was generated. e, The deactivation time constant of the persistent Na+ currents from 0 to −80 mV was varied (black, 0.2 ms; blue, 0.3 ms; red, 0.5 ms; green, 0.9 ms; and purple,1.6 ms). f, Model action potentials with only persistent Na+ currents in the same scenarios as described in e. g, h, Representative experimental data traces of deactivation of calyx Na+ currents from −30 to −80 mV in the P16 calyx (g) and P10 calyx (h). Single exponential fits are shown to the data (red). The more mature P16 calyx had a faster τ.
Model sodium currents (Fig. 6a) were generated by turning off the other conductances and performing linear leak subtraction to remove capacitive currents. The simulated voltage-clamp protocol to elicit mixed resurgent and persistent Na+ current was the same as that used for the recordings shown in Figure 4a: a step depolarization to +30 mV for 5 ms that generated transient Na+ current followed by a repolarizing step from −20 to −70 mV in 10 mV steps. Simulated INaR showed a peak in the bell-shaped I-V relationship near −40 mV, and the decay of INaR also showed the same voltage dependency as the experimental data (simulation results not shown). Importantly, the rate of inactivation of the transient Na+ current was significantly faster with an INaR present in the simulations (Fig. 6a), as expected from the extra effect of the inactivation binding particle used to generate INaR (Raman and Bean, 2001). To generate an I-V relationship for the persistent Na+ current, we used a slow voltage ramp (20 mV/s) from a potential of −90 mV. This I-V relationship showed current activation at −70 mV (no liquid junction potential correction) and a peak near −20 to −10 mV, as we observed experimentally (Fig. 5d; simulation results not shown).
Action potentials were generated in the computer model by current injection (4 nA for 0.2 ms) into the distal end of the axon. Four Na+ current scenarios were studied: normal resurgent and persistent current (black traces), resurgent current without persistent current (blue traces), no resurgent current with normal persistent current (solid red traces), and no resurgent current with two times higher persistent current (broken green traces). Without INaR, we obtained a calyx action potential with no DAP (Fig. 6b). In addition, note that the half-width of the action potential was broader without INaR (as is also the case experimentally with 0.3 mm QX-314) (Fig. 3c,d). Removal of INaP had very little effect on the DAP (blue traces), and even increasing INaP two times over experimentally recorded values did not rescue the DAP in the absence of INaR. These simulations thus capture the main basic features of our previous experimental results, and they show that INaP by itself does not generate the DAP. They thus represent a proof of principle for our main hypothesis that INaR generates the DAP.
We next explored the effect of varying the amplitude of INaP (from 2.4 to 5.6 times the measured values) on the fAHP or DAP (Fig. 6c), while the time constant of deactivation was kept constant at 0.24 ms (close to the value measured in P16 calyces). In the presence of INaR, a 2.4-fold to 3.2-fold higher density of INaP increased the peak amplitude of the DAP (measured relative to the fAHP) only slightly, whereas a 4.2-fold to 5.6-fold increase in INaP was necessary to produce a significantly larger DAP with a prolonged time course (Fig. 6c). However, without INaR we observed that increasing persistent current removed the fAHP but did not produce a rebounding DAP “hump,” as observed experimentally (Fig. 6d). We conclude that persistent current in the calyx of Held is unlikely to generate the DAP, although it may modulate its amplitude and kinetics.
The persistent current used in the simulation shown in Figure 6a–d has fast deactivation kinetics (0.24 ms at −80 mV). However, we reasoned that an INaP with sufficiently slow deactivation kinetics could contribute to the DAP since during membrane repolarization following an AP the increased driving force for Na+ could result in a depolarizing drive similar to that caused by resurgent Na+ current. To explore this, we tested several different models in which the time constant for deactivation at −80 mV was varied from 0.2 to 1.6 ms (Fig. 6e). As the kinetics of persistent current gating were slowed, the fAHP was reduced, and for very slow time constants approaching 1 ms a DAP with a hump was indeed generated (Fig. 6f). However, in the calyx of Held, the experimentally measured persistent Na+ current deactivated with a fast time constant of 0.27 ± 0.07 ms (P16; n = 4) or 0.6 ± 0.1 ms (P10; n = 3) (Fig. 6g–h). We therefore conclude that the INaP in the calyx of Held deactivates too rapidly to generate a significant DAP.
β4 subunit peptides induce INaR in immature calyces
To further test for a causal relationship between INaR and the DAP, we next used a portion of the β4 sodium channel subunit to induce INaR in immature calyces. A leading model for how INaR can arise is through the voltage-dependent binding of a blocking particle (Raman and Bean, 2001), which may originate from the β4 subunit to the NaV1.6 channel (Grieco et al., 2005). According to this model, when the β4 subunit particle binds it both blocks current flow and also prevents the normal “classical” inactivation process from occurring. On repolarization, and dissociation of the β4 subunit particle, the channel conducts again until normal deactivation or closure occurs. Interestingly, very high levels of β4 subunit mRNA are found throughout the adult rat brainstem (Yu et al., 2003), but the level of β4 subunit protein in the developing rat calyx of Held is unknown. If protein levels follow mRNA, an increased expression of β4 subunits during early postnatal development could explain the observed developmental increases in INaR and the DAP amplitude. Unfortunately, due to a lack of reliable antibodies, we have not been able to confirm a developmental increase in protein levels of the β4 subunit in rat MNTB.
We thus tested whether a peptide that mimics the putative action of β4 subunits on Na+ channels could generate INaR in immature P5–P6 calyces, which have little resurgent Na+ current (Fig. 7a,b). In recordings without peptide, the size of INaR in P5 calyces was relatively small and stable during whole-cell recording (control) (Fig. 7c). When 100 μm β4peptide (KKLITFILKKTREK, Grieco et al., 2005) (Fig. 7b, red trace) was included in the pipette solution, INaR increased 10-fold, from 23 ± 8 pA at 2 min to 227 ± 53 pA after 8 min of intracellular dialysis of the peptide (n = 5, p < 0.001) (Fig. 7c); whereas, the increase of INaP with β4peptide was not significant (from 32 ± 11 pA to 53 ± 20 pA after 8 min of dialysis; n = 5, p = 0.11). In the presence of the β4peptide, the I-V relation of INaR was bell shaped and similar to that observed in more mature calyces (n = 8) (Fig. 7d).

Induction of INaR by dialysis of a β4 peptide into P5–P6 calyces of Held. a, An immature calyx terminal (P5) expresses a very small amplitude INaR, which was recorded with repolarizing step pulses from +30 to −20 mV. Inset, The P5 calyx of Held (arrowhead) and attached axon were filled with Alexa-555 via the patch pipette for morphological identification. b, INaR was recorded with control internal solution (black trace) or with an internal solution that contained β4peptide (100 μm; amino acid sequence from the β4 subunit of Na+ channels, red trace) or with a peptide with a scrambled sequence of β4peptide (blue trace). Inset, Expanded traces of the INaR shown from the shadowed box. c, The peak amplitude of INaR at −30 mV progressively increased during whole-cell dialysis of β4peptide (100 μm; colored dots) in five individual P5–P6 calyx recordings. However, no significant change in peak amplitude of INaR was detected with control internal solution (black dots; n = 5). d, Current–voltage relationship of INaR recorded without β4peptide (control, black; n = 5), with β4peptide (red; n = 8), or with scrambled β4peptide (blue; n = 5) for P5–P6 calyx terminals.
To confirm that the effects produced by the β4peptide depended on the specific amino acid sequence, we tested the effect of a scrambled peptide on INaR in which charge was conserved but the clusters of hydrophobic and positively charged residues were disrupted (β4scramble: KIKIRFKTKTLELK) (Grieco et al., 2005) (Fig. 7b,d, blue trace). In the presence of β4scramble, the INaR did not significantly increase during 15 min of dialysis, and its I-V relationship remained similar to control (n = 5) (Fig. 7d). These data indicate that the β4peptide can significantly increase INaR in immature P5–P6 calyces of Held.
β4 subunit peptides increase the DAP amplitude and firing reliability
Next, we investigated the effect of β4peptide dialysis on the AP waveform of immature P5–P6 calyces. AP spikes were evoked once every 10 s by afferent fiber stimulation, and β4peptide (100 μm) was dialyzed into the calyx via the patch pipette. The amplitude of the DAP in the presence of β4peptide was significantly increased from 2.2 ± 0.2 to 4.0 ± 0.5 mV after 10–15 min of dialysis (P5–P6; n = 12, p < 0.01; DAP amplitude was measured from the fAHP) (Fig. 8a1). The potential reached at the peak of the DAP measured from the resting membrane potential was also increased by intracellular β4peptide dialysis from −67 ± 0.4 to −63 ± 0.8 mV (n = 12, p < 0.01) (see supplemental Fig. 5a, available at www.jneurosci.org as supplemental material). The β4peptide depolarized the average calyx adjusted resting membrane potential from −80 ± 0.4 to −76 ± 0.5 mV and significantly shortened the spike latency from 1.6 ± 0.2 to 1.3 ± 0.1 ms (n = 12, p < 0.001) (Fig. 8b). In addition, the 10 to 90% rise time of the spike was reduced from 328 ± 16.9 to 273 ± 14.3 μs (n = 9; p < 0.001) (Fig. 8b). Similar effects on AP spike latency and rise time are also observed in postsynaptic MNTB neurons after the dynamic clamp incorporation of INaR into normal MNTB neurons (Leão et al., 2006).

Dialysis of β4peptide into P5–P6 calyces increases the DAP amplitude and improves reliability of presynaptic firing. a1, Presynaptic AP spikes and DAP evoked by afferent fiber stimulation without β4peptide (black) and with β4peptide (100 μm, red) in the same P5 calyx terminal. The rising phase of the spikes are superimposed. The β4peptide depolarized the calyx and increased the DAP. a2, Current injection (+15 pA) depolarized the calyx from −80 to −75 mV in the control, but this small depolarization had no effect on the amplitude of the DAP. b, Internal dialysis of β4peptide depolarized the calyx and decreased spike latency, which was measured as the time from stimulus artifact (S.A.; arrow) to the peak of the spike. c, Spike trains were elicited by afferent fiber stimulation at 133 Hz (7.5 ms stimulus interval, 50 stimuli) in a P5 calyx without β4peptide (black). The same calyx was then patch-clamped with a pipette containing β4peptide (red). In control, the P5 calyx exhibited spike failures at 133 Hz (arrows), but dialysis of β4peptide allowed the same calyx to fire spikes without any failures at 133 Hz (red). Inset, Spikes in the two shadowed boxes are superimposed and expanded in time scale. d, In control, the P5 calyx could not follow 200 Hz stimulus train (black trace). Depolarization from −80 to −70 mV by current injection (30 pA) had no effect on these failures (blue traces). The same calyx with β4peptide had a reduced rate of failure at 200 Hz (red trace). Inset, Spikes in the shadowed boxes are superimposed. Note the shorter latency with β4peptide (red arrowhead).
The increase in DAP amplitude by the β4peptide was not due to membrane depolarization because depolarizing the calyx by 4 mV via current injection in control calyces lacking β4peptide had no effect on the amplitude of DAP (n = 3) (Fig. 8a2). In three recordings, we also observed the diffusion of the β4peptide using tetramethylrhodamine-tagged β4peptide and found that it diffused along the axon for at least 100–200 μm in the 7–10 min after break-in to whole-cell recording mode (supplemental Fig. 5b, available at www.jneurosci.org as supplemental material). This indicates that the β4peptide reaches the whole extent of the heminode region of the axon where NaV1.6 channels are primarily located (Leão et al., 2005). To test whether the effects of the β4peptide are due to changes in K+ current induced by the peptide, we recorded calyx K+ currents with pipettes containing β4peptide. Insertion of β4peptide into P5 calyces had no significant effect on the amplitude of voltage-dependent K+ currents after 10 min of β4peptide dialysis (K+ currents at 0 mV were 1.9 ± 0.46 nA in control and 1.8 ± 0.59 nA with β4peptide; n = 3, p > 0.05; data not shown). Thus, we conclude that an increase of INaR by β4peptide dialysis enhances the amplitude of the DAP and reduces the AP latency.
We next examined the consequences of increased INaR and DAP for high-frequency firing by recording from P5–P6 calyces before and after β4peptide dialysis. To reduce calyx-to-calyx variability (Sonntag et al., 2009), we first recorded spike trains (100–200 Hz) in control without β4peptide. We then removed the patch pipette and repatched the same calyx with a pipette containing 100 μm β4peptide. P5 calyces fired trains of 50 spikes without failure at 100 Hz but showed several failures at 200 Hz. The success rate (number of spikes per total of 50 stimuli) was smaller in control than after dialysis with β4peptide. At 133 Hz (stimulus interval, 7.5 ms), several control P5 calyces (6 of 10) exhibited spike failures during the late phase of the stimulus train. After dialysis with β4peptide, these same calyces showed no failures at 133 Hz (Fig. 8c, black and red traces, respectively). The success rate at 133 Hz increased from 92 ± 3.5% to 100% with β4peptide (n = 10; p < 0.05).
To control for the possibility that the depolarization of ∼4 mV caused by the peptide could have effects on the calyx firing fidelity, we performed a control experiment without β4peptide. Most P5 calyces could not follow 200 Hz stimulus trains for 250 ms, showing a large number of AP failures (e.g., 25 failures of 50 stimuli) (Fig. 8d). Constant current injection (∼30 pA) depolarized the calyx resting potential from −80 to −70 mV; nevertheless, the calyx still had the same number of AP failures at 200 Hz. In the same calyx, however, the dialysis of 100 μm β4peptide reduced AP failures in the early phase of the train. The success rate increased from 59 ± 3.3% to 71 ± 3.9% with β4peptide at 200 Hz (n = 10; p < 0.01) (Fig. 8d). Together, our results suggest that INaR induced by β4peptide increases peak DAP amplitude, reduces the spike latency, and leads to an increase in firing reliability at high stimulation frequencies.
Discussion
We have uncovered the presence of a presynaptic INaR in a mammalian nerve terminal whose action potential waveform has a prominent DAP. Direct calyx of Held recordings show that the INaR amplitude is upregulated during the first 2 weeks of postnatal development. Introducing a small β4 peptide into an immature calyx mimicked this developmental increase in INaR and expanded the range of reliable firing of the immature terminals. We thus propose that a developmental increase of INaR in auditory calyx of Held nerve terminals promotes reliable high-frequency firing, a critical feature for encoding signals for sound localization (Yin, 2002; Grothe, 2003).
Local Na+ currents mediate the presynaptic DAP
The mechanism that generates the DAP at CNS bouton-type nerve terminals and axons has not been clearly established because their small size makes direct recordings very difficult. At the calyx of Held, a local puff application of TTX on the calyx terminal reduced the spike amplitude but did not affect the DAP amplitude measured from the adjusted resting membrane potential, suggesting that the DAP may originate in the axon (Borst et al., 1995). More recently, a local puff application of riluzole, a nonspecific blocker of Na+ channels, reduced the DAP and AP spike amplitude (Paradiso and Wu, 2009). However, riluzole also blocks Ca2+ and K+ channels (Lamanauskas and Nistri, 2008), so the specific mechanism that generates the DAP remained uncertain. Our QX-314 dialysis experiments show that the rising phase of the DAP is caused by local Na+ current activation because dialysis of 0.3 mm QX-314, a blocker of INaR and INaP (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material), reversibly blocked the rising phase of the DAP (Fig. 3c,d). Moreover, a low concentration of TTX (20 nm) also blocked the DAP, although it had a relatively small effect on AP spike amplitude (Fig. 3a). Importantly, the DAP was observed in calyces with short axon stumps after a brief current injection (Fig. 1b; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). So, a long intact axon was not necessary for generating the DAP. Instead, we propose that local Na+ currents in the calyx pre-terminal heminode region generate the DAP.
DAP is generated predominantly by INaR in the calyx terminals
Is the DAP generated by INaR, INaP, or both types of Na+ currents? Here, we present three separate pieces of evidence that INaR is sufficient to produce a DAP. First, our modeling studies suggest that INaP contributes to the DAP only if its deactivation kinetics is slowed significantly to values near 1.36 ms, whereas we measured an INaP time constant for deactivation of ∼0.25 ms in P15–P16 calyces (Fig. 6g). Similarly, adult cerebellar Purkinje cells have an INaP time constant for deactivation of 0.2 ms (Kay et al., 1998). Second, the rising phase of the DAP became progressively faster and larger during development from P5 to P15 (Fig. 2a). Likewise, peak INaR amplitudes increased significantly from P5 to P12 (Fig. 5c; supplemental Fig. 4, P15, available at www.jneurosci.org as supplemental material), while the developmental increase in INaP was less significant (Fig. 5d). A resurgent current of ∼100 pA at nearly −70 mV (the peak of the fAHP) would depolarize P15–P16 calyces with an input resistance of ∼150 MΩ by ∼15 mV. By contrast, the immature P5–P6 calyx has a higher input resistance of ∼300 MΩ, so a much smaller INaR of ∼20 pA could generate a DAP of ∼6 mV. An increase in INaR may thus be necessary to overcome the low input resistance of mature calyces, whereas an unchecked increase in INaP may generate spontaneous firing (Raman and Bean, 1997). Third, dialysis of the β4 peptide increased the DAP amplitude in immature P5–P6 calyces (Fig. 8a1; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Because β4 peptide increases significantly INaR amplitude (Fig. 7b), but does not significantly change the average INaP amplitude, we suggest that the DAP is caused mainly by INaR activation. Recently, the β4 peptide has been shown to also increase INaP in transfected cells expressing NaV1.1 channels (Aman et al., 2009). However, the calyx of Held heminode expresses a high density of NaV1.6 channels (Leão et al., 2005), and high levels of β4 subunit mRNA are found in adult rat brainstem (Yu et al., 2003). Interestingly, the coexpression of β4 subunits with NaV1.6 channels is not sufficient to reconstitute INaR in transfected cells (Chen et al., 2008). Additional proteins beyond Nav1.6 α and β4 subunits may thus be required for induction of INaR.
DAP kinetics and repetitive firing
The peak amplitude and time course of the DAP influences the fidelity of repetitive firing at high frequencies (Fig. 2e). Since the passive properties of the axon and its myelin sheath contribute to the kinetics of the DAP at peripheral axons (Barrett and Barrett, 1982; Bowe et al., 1987; David et al., 1995; Lin, 2008), the lack of myelin on immature calyx axons at P5–P6 could contribute to the slower decay of DAP at immature calyces, whereas the faster DAP decay observed in P15–P16 calyces may arise in part from a more compact myelin sheath that promotes a faster axonal τm (Fig. 2c). During repetitive firing, DAP summation in the immature calyx causes a large depolarizing plateau that probably leads to a reduction in spike amplitudes, due to an accumulation of Na+ channels in the inactivated state, and eventual spike failures (Fig. 2e, arrow). Thus, the faster DAP decay kinetics and decrease in membrane time constant of more mature calyces help them to fire spikes at higher frequencies without failures.
What is the mechanism for reduced failure rates with the β4 peptide (Fig. 8d)? The β4 peptide increases the DAP amplitude (Fig. 8a1), and the DAP helps to avoid AP failures (Fig. 1c). In addition, we propose that the β4 peptide increases the availability of Na+ channels for opening by reducing the number of channels in the inactivated state. Studies at the nodes of Ranvier and calyx of Held indicate that at a resting membrane potential of −80 mV ∼20% of the Na+ channels are inactivated (Schwarz et al., 1995; Nakamura and Takahashi, 2007). A large surplus of Na+ channels that are available for opening is necessary for the reliable firing of spikes at high frequencies (as shown by studies using low doses of TTX; Madeja, 2000). By blocking the “classical inactivation” pathway for Na+ channels the β4-peptide may effectively increase the number of channels that can be opened during a stimulus train. Larger Na+ currents due to increased availability of channels in the calyx during a stimulus train will thus lead to a reduced number of failures, as well as a reduction in the spike latency and a faster AP rise time. The presence of INaR in calyx terminals thus promotes an acceleration of firing and greater spiking reliability (Fig. 8c,d).
INaR contributes to faster Na+ channel inactivation and recovery
Previous studies have suggested various specializations that are acquired during early development that aid the calyx of Held in its remarkable capacity for high-frequency firing (Schneggenburger and Forsythe, 2006). One is the faster rate of Na+ channel inactivation of the more mature calyx, and another is an increase of Kv3 channel density during development (Leão et al., 2005; Nakamura and Takahashi, 2007). Both will promote a shorter AP waveform (Rudy and McBain, 2001). Indeed, the more mature calyx of Held has a shorter AP waveform and a faster recovery rate from Na+ channel inactivation than other CNS synaptic boutons (Jackson and Zhang, 1995; Engel and Jonas, 2005). The presence of INaR in a neuron promotes faster Na+ channel inactivation and faster recovery from inactivation, and thus facilitates repetitive firing (Raman and Bean, 1997). Our computer simulations also support a role of INaR in maintaining AP half-widths short via fast Na+ channel inactivation (Fig. 6a,b). We thus propose that the faster recovery from inactivation of Na+ channels in the more mature calyx of Held may be due in part to the more copious expression of INaR.
Functional implications of INaR for the auditory system
Sound-evoked signals must be conveyed through the ascending auditory pathways at high speed and low jitter, and with great reliability (Carr et al., 2001; Yin, 2002). A short AP waveform is thus critical for the transfer of high-frequency auditory signals. Our computer simulations show that the presence of a relatively large INaR aids in the production of short APs (Fig. 6b). Moreover, INaR also produces a fast rising DAP that accelerates the firing of a subsequent AP (Fig. 1c), and thus the precise timing of APs during a spike train (Fig. 8c,d). Postsynaptic MNTB neurons also exhibit a DAP (Johnston et al., 2009), and a developmental increase in INaR, which improves their ability to fire at higher frequencies, and INaR expression is altered by deafness (Leão et al., 2006). Of course, speed, precision, and reliability of spiking are also augmented through the myelination of axons, which increases during early postnatal development (Vabnick and Shrager, 1998). Resurgent Na+ currents in nerve terminals and axons may thus be yet another specialization of auditory circuits that promotes fast, precise, and resilient action potential signaling.
Footnotes
-
J.H.K. was supported by the American Heart Association, C.K. was funded by Fundação de Amparo à Pesquisa do Estado de Minas Gerais and Conselho Nacional de Desenvolvimento Científico e Tecnológico (Brazil), and H.v.G. was supported by an RO1 grant from the National Institute on Deafness and Other Communication Disorders. We thank Paul Brehm, Maxim Dobretzov, Ricardo M. Leão, Robert Renden, Geetha Srinivasan, Larry Trussell, and John Williams for discussions.
- Correspondence should be addressed to Dr. Henrique von Gersdorff, The Vollum Institute, Oregon Health and Science University, 3181 SW Sam Jackson Park Road, Portland, Oregon 97239. vongersd{at}ohsu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}