

Abstract
Cisplatin is a chemotherapeutic agent that is widely used in the treatment of solid tumors. Ototoxicity is a common side effect of cisplatin therapy and often leads to permanent hearing loss. The sensory organs of the avian ear are able to regenerate hair cells after aminoglycoside ototoxicity. This regenerative response is mediated by supporting cells, which serve as precursors to replacement hair cells. Given the antimitotic properties of cisplatin, we examined whether the avian ear was also capable of regeneration after cisplatin ototoxicity. Using cell and organ cultures of the chick cochlea and utricle, we found that cisplatin treatment caused apoptosis of both auditory and vestibular hair cells. Hair cell death in the cochlea occurred in a unique pattern, progressing from the low-frequency (distal) region toward the high-frequency (proximal) region. We also found that cisplatin caused a dose-dependent reduction in the proliferation of cultured supporting cells as well as increased apoptosis in those cells. As a result, we observed no recovery of hair cells after ototoxic injury caused by cisplatin. Finally, we explored the potential for nonmitotic hair cell recovery via activation of Notch pathway signaling. Treatment with the γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester failed to promote the direct transdifferentiation of supporting cells into hair cells in cisplatin-treated utricles. Taken together, our data show that cisplatin treatment causes maintained changes to inner ear supporting cells and severely impairs the ability of the avian ear to regenerate either via proliferation or by direct transdifferentiation.
Introduction
Cisplatin is a chemotherapeutic agent that is highly effective in the treatment of ovarian, testicular, bladder, and head and neck tumors. Like many antineoplastic drugs, cisplatin can cause a number of deleterious side effects, such as peripheral neuropathy and nephrotoxicity. Among the most serious complications of cisplatin treatment is ototoxicity, which occurs in ∼30% of treated individuals and can lead to permanent hearing loss (Nagy et al., 1999). Numerous studies have demonstrated that cisplatin leads to the death of hair cells (HCs), which are the sensory receptor cells of the cochlea and vestibular organs (Rybak, 2007). Following systemic injection, cisplatin accumulates in the inner ear fluids and is then taken up by otic epithelial cells. Platinated DNA has been observed in both hair cells and supporting cells following cisplatin treatment, leading to the formation of DNA platinum adducts (van Ruijven et al., 2005). Additional studies have shown that cisplatin exposure leads to increases in reactive oxygen species within hair cells (Clerici and Yang, 1996), activation of ERK1/2 (So et al., 2007), release of certain immune cytokines (Kim et al., 2008), and expression of the vanilloid receptor TRPV1 (Mukherjea et al., 2008). Nevertheless, present knowledge of the cellular signaling events that are involved in cisplatin ototoxicity is still rudimentary. In addition, it is not clear whether hair cells are the primary target of cisplatin or whether other cell types within the sensory epithelia of the ear are also affected. Previous anatomical studies, for example, have suggested that cisplatin treatment induces structural changes in cochlear supporting cells (Ramirez-Camacho et al., 2004).
The present study examined the effects of cisplatin on the chick cochlea and utricle. The avian ear—unlike its mammalian counterpart—is able to regenerate sensory hair cells after noise exposure or aminoglycoside ototoxicity. This regenerative response is mediated by epithelial supporting cells, which reenter the cell cycle after hair cell injury and serve as precursors to replacement hair cells (Corwin and Oberholtzer, 1997; Stone and Cotanche, 2007; Brigande and Heller, 2009). Given the antiproliferative effects of cisplatin, we sought to determine whether the avian ear retained its regenerative ability after cisplatin injury. We found that cisplatin treatment inhibited cell division and increased cell death in cultures of inner ear supporting cells in a dose-dependent manner. In organotypic cultures of the chick cochlea and utricle, we found that cisplatin caused apoptotic hair cell death, but we observed very limited supporting cell proliferation and no hair cell regeneration. Further experiments examined the ability of the γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) to promote transdifferentiation of supporting cells into hair cells after cisplatin ototoxicity. We found that, although numerous supporting cells survived cisplatin exposure, treatment with DAPT did not yield new hair cells. Our results indicate that cisplatin treatment not only blocks proliferation in avian supporting cells, but it also inhibits the ability of those cells to transdifferentiate into replacement hair cells. We conclude that cisplatin injury causes long-term changes in supporting cells and abolishes the regenerative ability of the avian inner ear.
Materials and Methods
Animals.
Chicken eggs (Charles River Laboratories) were incubated until hatching, and chicks were housed in heated brooders for approximately 7–14 d posthatch (P7–14). All protocols were approved by Washington University Institutional Animal Research Committee (Saint Louis, MO).
Cultures of isolated sensory epithelia.
Cultures of sensory epithelia from the chick utricle were prepared following previously described methods (Warchol, 1995, 1999, 2002). Briefly, chicks were euthanized via CO2 asphyxiation and decapitated. Following removal of the skin and mandible, heads were placed in 70% EtOH for 5–10 min to kill surface pathogens. The temporal bones were then exposed and utricles were quickly removed and placed into chilled Medium 199 (M199) with Hanks' salts and HEPES buffer. Otoconia were removed with fine forceps, and the utricles were then incubated for 1 h in thermolysin (500 μg/ml in M199) at 37°C. Following thermolysin treatment, a 30 gauge needle was used to remove the sensory epithelium from the underlying basement membrane and stromal tissue. Isolated sheets of sensory epithelia were cut into small fragments, incubated for 15 min in trypsin (0.05%) at 37°C, and triturated 10× in M199 with 10% fetal bovine serum (FBS). Dissociated cells—consisting mostly of supporting cells—were plated onto laminin-coated culture wells (MatTek). Each well contained cells from approximately one utricular sensory epithelium suspended in 50 μl of M199 with Earle's salts, 2200 mg/L sodium bicarbonate, 0.69 mm l-glutamine, 25 mm HEPES, supplemented with 10% FBS. Cultures were initially incubated overnight at 37°C in a 5% CO2/95% air environment to permit attachment of the cells onto the laminin substrate. The next day, an additional 50 μl of medium was added to the cultures for a final volume of 100 μl. Epithelial cultures were fed fresh medium at 2 day intervals and used in experiments after 7–8 d in vitro.
Organotypic cultures of the chick utricle.
Organotypic cultures of utricles were prepared following previously published methods (Matsui et al., 2002). Chicks were euthanized and utricles were isolated as described above. Single utricles were placed in 1 cm MatTek wells that contained 100 μl of M199 with Earle's salts and 1% FBS. Cultures were incubated at 37°C in a 5% CO2/95% air environment and fed fresh medium every 48 h.
Organotypic cultures of the chick cochlea.
Cochlear cultures were prepared following previously described methods (Warchol and Corwin, 1996). Chicks were euthanized and cochleae were explanted and placed in chilled M199 (with Hanks' salts). The tegmentum vasculosum and lagena were removed, and the remaining sensory organs were transferred to 1 cm MatTek wells that contained 100 μl of M199 (with Earle's salts) and 1% FBS. Cochleae were incubated at 37°C in a 5% CO2/95% air environment and fed fresh medium every 48 h.
Ototoxic injury.
Crystalline cis-platinum(II) diammine dichloride (cisplatin; Sigma-Aldrich) was diluted in PBS and stored at −20°C. Dissociated cultures were treated for 24 h with cisplatin (0.2, 2, or 20 μm). Organotypic cultures of utricles and cochleae were treated for 24 h with 10–20 μm cisplatin. Following cisplatin treatment, utricles and cochleae were thoroughly rinsed in fresh medium and maintained in vitro for 1–7 d. In all experiments, equal numbers of untreated utricles or cochleae were cultured in parallel and served as negative controls. In addition, some cultured utricles received ototoxic injury via treatment for 24 h with 1 mm streptomycin sulfate (Sigma-Aldrich); these specimens served as positive controls for the studies of supporting cell proliferation and hair cell regeneration.
Labeling of proliferating cells and inhibition of γ-secretase.
Cultures were given bromodeoxyuridine (BrdU; 3 μg/ ml) for either 4 h (dissociated sensory epithelia) or 3–7 d (cultured cochleae and utricles). In other experiments, cultured utricles received DAPT (10 or 25 μm in 0.1% DMSO; Sigma-Aldrich) for 7 d to promote the transdifferentiation of supporting cells into hair cells (Daudet et al., 2009). Control cultures in these experiments received 0.1% DMSO.
Immunohistochemistry.
All cultures were fixed for 20 min with 4% paraformaldehyde in 0.1M phosphate buffer. Specimens were then thoroughly rinsed with PBS and nonspecific antibody binding was blocked by incubation for 2 h in PBS with 5% normal horse serum and 0.2% Triton X-100. Hair cells were identified with the HCS-1 antibody (1:500; a mouse monoclonal that labels nonmammalian hair cells) (Cyr et al., 2006) provided by Dr. Jeffery Corwin (University of Virginia, Charlottesville, VA) or with anti-calretinin (1:500, Calbiochem). Supporting cell nuclei were labeled with a goat polyclonal antibody against Sox2 (1:250; Santa Cruz Biotechnology) (Oesterle et al., 2008). Cells undergoing apoptosis were identified by immunoreactivity for activated caspase-3 (rabbit polyclonal, 1:400; Cell Signaling Technology). Macrophages in the chick utricle were labeled with the KUO01 antibody (1:100, mouse monoclonal; Southern Biotechnology) (Mast et al., 1998). Immunoreactivity for BrdU was determined using previously published protocols (Matsui et al., 2002; Warchol, 2002). All specimens were maintained in primary antibodies overnight at 4°C. They were then thoroughly rinsed in PBS and incubated with Alexa Fluor 488 donkey anti-mouse, Alexa Fluor 546 donkey anti-rabbit, or Alexa Fluor 546 donkey anti-goat secondary antibodies (1:500; all Invitrogen) for 2 h at room temperature.
Actin and nucleic acid staining.
Some specimens were stained with Alexa Fluor 546- or Alexa Fluor 488-phalloidin (5 units/ ml; Invitrogen), to visualize F-actin. Nuclei in all specimens were stained with 4′,6-diamidino-2-phenyindole (DAPI; 2.7 μm; Sigma-Aldrich). Specimens were then mounted on glass slides in 90% glycerol/10% PBS (cochleae and utricles, or coverslipped directly in MatTek dishes (isolated utricular epithelia).
Imaging and quantification of cultures.
Specimens were visualized using either Nikon Eclipse 2000 or Zeiss Axiovert 135 inverted microscopes (both equipped with epifluorescent illumination). Confocal images were obtained with a Bio-Rad Radiance 2000 MP system using a Nikon Eclipse inverted microscope. Conventional epifluorescence images were captured with cooled CCD digital cameras (Q-Imaging or Photometrics) and stored as TIFF files. Confocal image stacks were assembled with Volocity software (PerkinElmer) and processed with Adobe Photoshop. Cell counts were made from the stored images. In cultures of isolated epithelia, DAPI- and BrdU-labeled cells were counted from four randomly selected regions per culture of approximately equal cell density per culture (∼200 cells/field for BrdU; ∼100 cells/field for pyknotic nuclei). Proliferation indices (BrdU-labeled nuclei/total nuclei) and apoptosis indices (pyknotic nuclei/total nuclei) were then calculated. Quantification of BrdU-labeled cells and hair cells in whole-mount utricles was carried out from four or five randomly selected extrastriolar and striolar 138,195 μm2 regions per specimen, and then normalized to 10,000 μm2. Quantification of hair cell stereocilia in cochlear whole mounts was conducted from seven discrete regions that began 250 μm from the distal tip and were separated by 250 μm intervals. Cells that labeled positively for BrdU or activated caspase-3 were counted from sampled regions located 500, 1000, and 1500 μm from the distal tip of the sensory epithelium. Resident tissue macrophages in streptomycin- and cisplatin-treated utricles were labeled with the KUL01 antibody (Mast et al., 1998). Confocal microscopy was then used to image macrophages in the sensory epithelia and in underlying stromal tissue. Quantification of macrophages was made directly from confocal images and resulting values were normalized to 10,000 μm2.
Statistics.
Data were analyzed using an unpaired Student's t test or ANOVA with multiple comparisons (Tukey's test; SPSS). An α of 0.05 was chosen for significance. All data are expressed as either percentage or mean ± SD.
Results
Cisplatin blocks proliferation and causes apoptosis in dissociated supporting cells
Initial experiments characterized the effects of cisplatin on dissociated cultures prepared from isolated sensory epithelia of the chick utricle. Previous studies have shown that such cultures are comprised primarily of supporting cells that proliferate at high levels (Warchol, 2002). Cultures were treated for 24 h with 0.2, 2.0, or 20.0 μm cisplatin and received BrdU for the final 4 h in vitro (to label proliferating cells). After fixation, cell nuclei were also stained with DAPI. Numerous DAPI-labeled pyknotic nuclei were present in cisplatin-treated cultures, but pyknotic nuclei were rare in untreated cultures (Fig. 1A,B). Also, the relative numbers of pyknotic nuclei appeared greater in regions of lower cell density (i.e., where supporting cell proliferation is the highest) (Warchol, 2002), while areas of higher cell density contained proportionally fewer apoptotic cells (data not shown). Apoptosis indices (number of pyknotic nuclei/total nuclei) were calculated from regions of the cultures that contained ∼100 nuclei/100,000 μm2; within these regions, we observed a dose-dependent increase in cell death in response to cisplatin treatment (Fig. 1C). In addition, quantification of BrdU labeling revealed a dose-dependent decrease in cell division following exposure to cisplatin (Fig. 1D). Treatment with 20 μm cisplatin reduced proliferation levels by almost 30-fold, compared with control cultures.

Cisplatin causes increased apoptosis and decreased proliferation in cultures of dissociated supporting cells. A, B, Dissociated cultures of epithelial cells from the chick utricle were either untreated (A) or treated (B) for 24 h with 20 μm cisplatin. The cultures also received a 4 h pulse of BrdU immediately before fixation. Increased numbers of pyknotic nuclei (arrows) and decreased numbers of BrdU-labeled nuclei (green) were observed in cisplatin-treated cultures. C, D, Quantification from these specimens revealed that cisplatin caused a dose-dependent increase in apoptosis (C) and decrease in proliferation (D). Scale bar: (in B) A, B, 100 μm. *p < 0.05 [F(3,52) = 39.6], compared to 0 μm (control) group.
Cisplatin kills hair cells in organotypic cultures of the cochlea and utricle
We next examined the effects of cisplatin on the survival of vestibular hair cells. Chick utricles were cultured for 24 h in medium that contained 20 μm cisplatin. They were then thoroughly rinsed and maintained for an additional 24 h in cisplatin-free medium. After fixation, surviving hair cells were immunolabeled with the HCS-1 antibody (Cyr et al., 2006) and apoptotic cells were labeled by immunoreactivity for activated caspase-3. Extensive apoptosis was observed in cisplatin-treated cultures but not in untreated controls (Fig. 2A,B). Hair cell loss was assessed by quantifying the numbers of HCS-1-labeled cells in 10,000 μm2 regions that were located in both the striolar and extrastriolar portions of cisplatin-treated and control utricles. One day after cisplatin exposure, hair cell densities were reduced by 36% in the extrastriolar region and by 43% in the striolar regions, compared with untreated controls (p < 0.005) (Fig. 2C,D).

Cisplatin causes apoptosis of utricular hair cells. A, B, Organotypic cultures of chick utricles were either untreated (A) or treated (B) with 20 μm cisplatin for 24 h (n = 5/6). They were then rinsed and maintained in culture for an additional 24 h in drug-free medium. After fixation, specimens were immunolabeled for HCS-1 (green, hair cells) and activated caspase-3 (red, apoptotic cells). C, D, Quantification of hair cells in both the extrastriolar (C) and striolar (D) regions revealed cisplatin-induced ototoxicity. Scale bar: (in B) A, B, 50 μm. *p < 0.005.
Continued hair cell death after cisplatin treatment
To determine the impact of cisplatin ototoxicity on hair cell regeneration, we next compared hair cell numbers in cultured utricles at 1 versus 7 d after cisplatin treatment. As controls, we also quantified hair cells in utricles that were maintained in parallel under two different culture conditions. One set of controls consisted of specimens that received no drug treatment and were cultured for a total of 2 or 8 d. The second set of controls was comprised of utricles that were treated for 24 h with 1 mm streptomycin and then allowed to recover in vitro for 1 or 7 d. Previous studies have shown that culture in 1 mm streptomycin creates an extensive hair cell lesion and that significant hair cell regeneration is apparent after 7 d of recovery (Matsui et al., 2000; Warchol and Richardson, 2009). In the untreated utricles, we observed an ∼20% reduction in hair cell numbers over the 8 d culture period (Fig. 3A,D). As expected, hair cell numbers in utricles examined 1 d after streptomycin treatment were reduced by ∼90%, while hair cells in the cisplatin-treated utricles were reduced by ∼40% (Fig. 3D). After 7 d, we observed a moderate recovery of hair cells in the streptomycin-treated specimens, presumably due to spontaneous regeneration (Fig. 3E). Notably, however, we found no evidence for hair cell recovery in the cisplatin-treated utricles. Instead, hair cell numbers in those specimens had continued to decrease to ∼10% of control values (Fig. 3E).

Continued hair cell loss following 24 h of cisplatin treatment. A–C, To further characterize cisplatin ototoxicity and to assess the potential for hair cell recovery, three groups of utricles were treated for 24 h in control medium (A), 1 mm streptomycin (B), or 10 μm cisplatin (C). Specimens were then rinsed and cultured for an additional 7 d in drug-free medium. Images show hair cell survival and/or recovery after 7 d. D, E, Plots show the numbers of hair cells that were present in utricles after 1 (D) and 7 d (E). Cis, Cisplatin; Strep, streptomycin. Scale bar: (in C) A–C, 100 μm. *p < 0.005 (F(2,116) = 128.5) compared to untreated group; **p < 0.005 (F(2,107) = 557.8) compared to untreated and streptomycin-treated groups.
Cisplatin treatment inhibits supporting cell proliferation
The previous result indicated that hair cell regeneration had occurred after streptomycin injury, but not after cisplatin ototoxicity. Prior studies of regeneration in the avian vestibular organs have shown that most replacement hair cells are produced by the division of supporting cells (e.g., Weisleder and Rubel, 1993; Matsui et al., 2000). Since cisplatin is known to target mitotic cells, we next examined cell proliferation after cisplatin treatment. Cultured utricles received 1 mm streptomycin, 10 μm cisplatin, or no treatment (controls) for 24 h. They were then thoroughly rinsed and maintained in culture for an additional 7 d. The mitotic tracer BrdU was present in the culture medium for the entire 7 d period to label every cell that entered S phase during this recovery time (Fig. 4A–C). Quantification of BrdU-labeled cells revealed moderate proliferation in control cultures and high proliferation levels in the streptomycin-treated cultures (Fig. 4E). However, very few BrdU-labeled cells were observed in the cisplatin-treated specimens. We also colabeled these specimens with calretinin to determine the relative numbers of dividing cells that differentiated as hair cells. Consistent with previous studies (e.g., Matsui et al., 2000), many BrdU-labeled hair cells were observed in the streptomycin-treated utricles (Fig. 4D), but we observed almost no BrdU-positive hair cells in the cisplatin-treated specimens (Fig. 4F).

Cisplatin treatment inhibits supporting cell proliferation. A–D, Utricles were cultured for 24 h in 10 μm cisplatin, 1 mm streptomycin, or normal medium (controls). Following thorough rinsing, they were then maintained in vitro for 7 d in ototoxin-free medium that also contained BrdU. Images show proliferating cells (green, BrdU) in untreated (A), streptomycin-treated (B), and cisplatin-treated utricles (C). Some specimens were also immunolabeled for calretinin (D, red), and BrdU-labeled hair cells were observed in the control and streptomycin-treated specimens (D, arrow). E, F, Quantification of the numbers of BrdU-labeled cells suggests that cisplatin (Cis) treatment nearly eliminates regenerative proliferation (E). In addition, very few BrdU-labeled hair cells were present after recovery from cisplatin treatment (F). Strep, Streptomycin. Scale bars: (in C) A–C, 100 μm; D, 10 μm. *p < 0.005 compared to untreated group; **p < 0.005 (F(2,107) = 314.9) to untreated and streptomycin-treated groups; ***p < 0.005 (F(2,106) = 103.1) compared to streptomycin-treated group only.
Supporting cells cannot transdifferentiate after cisplatin ototoxicity
Hair cell regeneration can occur either through the renewed proliferation of supporting cells or by the nonmitotic transdifferentiation of those cells into new hair cells (Baird et al., 1996; Adler et al., 1997; Roberson et al., 2004). In addition, hair cell differentiation is regulated by Notch pathway signaling and can be enhanced by pharmacological inhibition of γ-secretase (Woods et al., 2004). Treatment with the γ-secretase inhibitor DAPT has been shown to promote transdifferentiation and hair cell recovery in the chick cochlea after aminoglycoside ototoxicity (Daudet et al., 2009). To determine whether such transdifferentiation could also occur after cisplatin treatment, we treated cultured utricles for 24 h with 10 μm cisplatin and then allowed them to recover in vitro for 7 d in either 10 μm (n = 10) or 25 μm (n = 10) DAPT. Equal numbers of control specimens were maintained in parallel but did not receive DAPT. Following fixation, hair cells in all specimens were immunolabeled with HCS-1. No increase in hair cell numbers was seen in DAPT-treated utricles (7.6 ± 2.7 HCs/10,000 μm2) compared to controls (8.2 ± 2.4 HCs/10,000 μm2) (Fig. 5). In fact, utricles that received 25 μm DAPT contained fewer hair cells (1.2 ± 1.18 HCs/10,000 μm2) than controls (7.7 ± 3.1 HC/10,000 μm2). Finally, as positive controls we also examined the effects of DAPT during regeneration from streptomycin ototoxicity. Utricles (n = 16) were placed in culture and treated for 24 h with 1 mm streptomycin. The specimens were then thoroughly rinsed and maintained in vitro for 7 d. During this recovery time, half of the utricles were also treated with 10 μm DAPT. Following fixation, hair cells were labeled with the HCS-1 antibody and quantified from randomly selected portions of the extrastriolar regions of each utricle (n = 22 samples/treatment group). Untreated utricles contained 63.2 ± 23.6 hair cells/10,000 μm2, while those treated with DAPT contained 295.6 ± 80.4 hair cells/10,000 μm2 (p < 0.0001; t test). This nearly fivefold increase in the recovery of vestibular hair cells in response to DAPT treatment is consistent with results obtained from studies of regeneration in the chick cochlea (Daudet et al., 2009).

Inhibition of γ-secretase does not augment direct transdifferentiation after cisplatin damage. A, B, Utricles were treated for 24 h with 10 μm cisplatin and then allowed to recover for 7 d in either normal medium (A) or medium supplemented with 10 μm DAPT (B). Hair cells were labeled for HCS-1 (green). Similar numbers of hair cells were observed in both groups of specimens, suggesting that cisplatin ototoxicity blocks the ability of supporting cells to transdifferentiate into hair cells. C, D, To verify the activity of DAPT, we also cultured utricles for 24 h in 1 mm streptomycin (Strep) followed by 7 d in either control medium (C) or DAPT-containing medium (D). After streptomycin injury, treatment with DAPT resulted in a large increase in hair cell recovery. Scale bar: (in D) A–D, 50 μm.
Cisplatin treatment causes a unique pattern of hair cell injury in the avian cochlea
Having established that cisplatin is toxic to hair cells in the chick utricle and that it also blocks the regeneration of utricular hair cells, we next examined the effects of cisplatin on the chick cochlea. In preliminary experiments, we observed that hair cells in the chick cochlea were slightly less sensitive to cisplatin toxicity than were their utricular counterparts (data not shown). As a result, our experiments with cultured cochleae used a slightly higher dose of cisplatin. We first cultured cochleae for 24 h in 20 μm cisplatin and then labeled hair cell stereocilium bundles with phalloidin (Fig. 6). Marked disruption and loss of stereocilia bundles were observed at the distal end (low-frequency region) of the cochlea. Surprisingly, however, hair cell injury appeared less severe in the middle and proximal regions of the cochlea (Fig. 6B). We then quantified the time course of cisplatin-induced injury to cochlear hair cells. Cochleae were treated with cisplatin for 24 h and then maintained in culture for an additional 0, 24, or 72 h. After fixation, hair cell strereocilia were counted from 10,000 μm2 regions spaced at 250 μm intervals, beginning at the distal tip of the sensory epithelium. Cisplatin treatment caused a progressive loss of stereocilia, which began in the distal region and spread proximally (Fig. 7). To further confirm the distal-to-proximal pattern of cisplatin ototoxicity, we also examined the spatial distribution of apoptosis after cisplatin treatment. Cochleae (n = 9) were cultured for 24 h with 20 μm cisplatin and then fixed and processed for immunohistochemical labeling with HCS-1 (to identify hair cells) and an antibody against activated caspase-3 (to identify apoptotic cells). An equal number of cochleae were cultured in parallel but did not receive cisplatin; these specimens served as controls. The numbers of apoptotic cells in both cisplatin-treated and control cochleae were quantified from 10,000 μm2 regions located at 500, 1000, and 1500 μm from the distal tip. We observed a pronounced increase in immunoreactivity for activated caspase-3 in cochleae that were treated with cisplatin (Fig. 8). Notably, the greatest number of apoptotic cells was observed near the distal tip (cisplatin-treated utricles, 27.95 ± 8.48 cells/10,000 μm2 at 500 μm from the distal end; 19.65 ± 8.49 at 1000 μm; 6.35 ± 5.56 at 1500 μm) (Fig. 8C).

Cisplatin preferentially damages hair cells in the distal (low frequency) region of the chick cochlea. A, B, Cochleae were cultured for 24 h in either normal medium (A) or medium that contained 20 μm cisplatin (B). Following fixation, specimens were labeled with phalloidin (green), to reveal stereocilium bundles. Low magnification images of the distal, middle, and proximal regions showed that the initial hair cell damage occurred at the distal end of the cochlea (B, left). In contrast, little damage was noted in the proximal region (B, right). Scale bar: (in B) A, B, 100 μm.

Spatial progression of cisplatin damage in the chick cochlea. Cultured cochleae were treated for 24 h in 20 μm cisplatin and then allowed to recover in vitro for 0, 24, or 72 h. Following fixation, stereocilium bundles were labeled with phalloidin and quantified at 250 μm intervals along the length of the cochlea, beginning at the distal tip and proceeding proximally. Surviving stereocilium bundles at each location are expressed as percentage of bundles present in control cochlea (i.e., cultured for identical times, but without cisplatin treatment).

Cisplatin induces apoptosis in the chick cochlea. A, B, Cochleae were cultured for 24 h in either normal medium (A) or medium supplemented with 20 μm cisplatin (B). Specimens were immunolabeled for HCS-1 (green, hair cells) and activated caspase-3 (red, apoptotic cells). Following cisplatin treatment, we observed numerous cells with immunoreactivity for activated caspase-3 (B, red). Such apoptotic cells were quantified from 10,000 μm2 regions that were located 500, 1000, and 1500 μm from the distal tip. C, Plot shows quantitative data for untreated and cisplatin-treated cultures. Scale bar (in B) A, B, 50 μm. *p < 0.05.
Additional experiments examined the proliferation of cochlear supporting cells after cisplatin ototoxicity. Cochleae were treated for 24 h in 20 μm cisplatin and then maintained for an additional 4 d in cisplatin-free medium that also contained BrdU. One set of control cochleae was cultured for an identical time period but was first treated for 24 h with 1 mm streptomycin, while additional control cochleae received no drug treatments. After fixation and immunohistochemical processing, BrdU-labeled cells were counted in 10,000 μm2 regions that were located 500, 1000, and 1500 μm from the distal tip of the sensory epithelium. The effects of cisplatin treatment on cochlear supporting cells were very similar to those observed in the utricle. Numerous BrdU-labeled cells were present in the streptomycin-treated cochleae, and a moderate number of proliferative cells were observed in untreated controls (Fig. 9C). In contrast, we observed almost no BrdU incorporation in the specimens that had been treated with cisplatin.
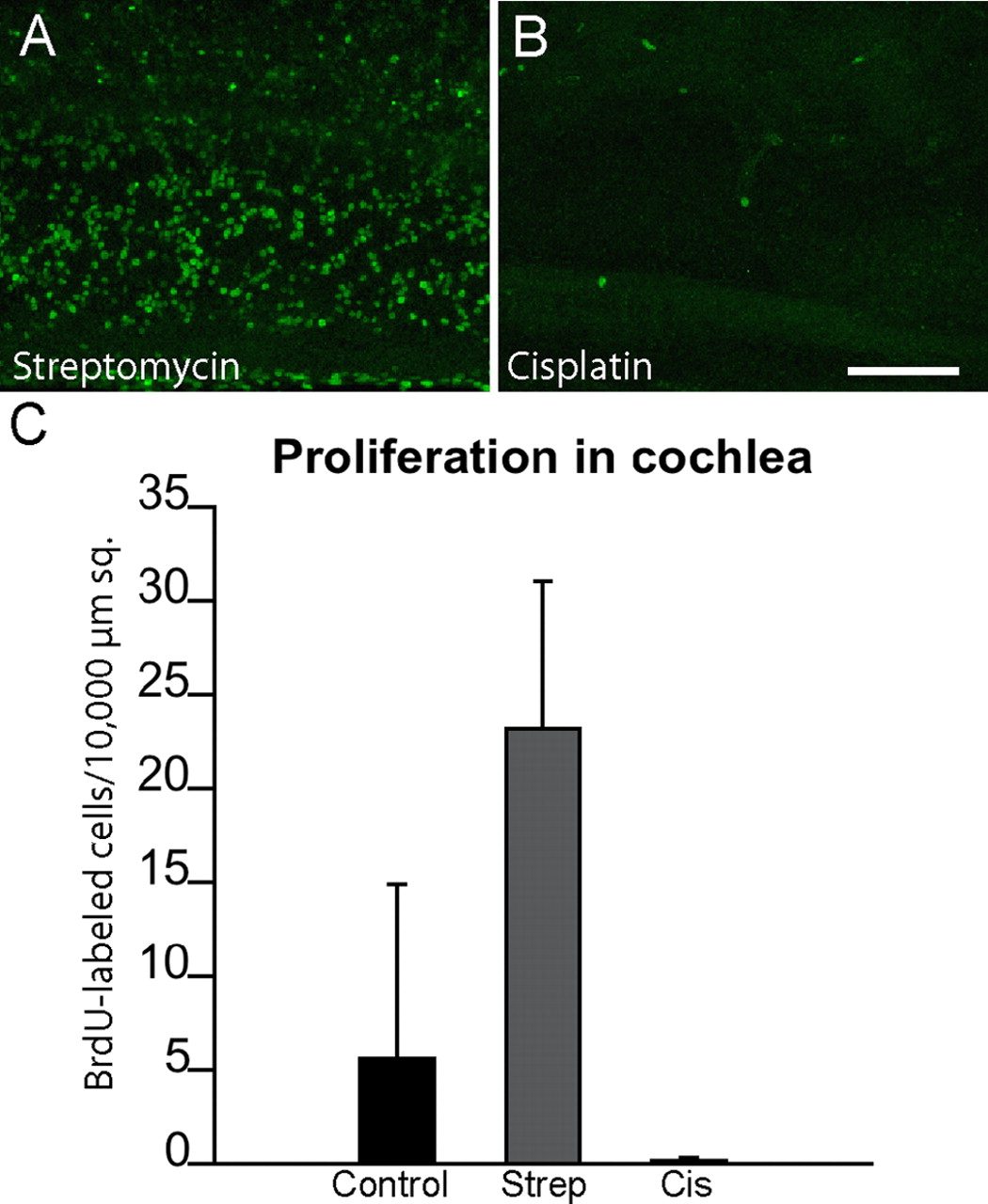
Cisplatin exposure reduces cell proliferation in the chick cochlea. Cultured cochleae were treated for 24 h in either 1 mm streptomycin (A) or 20 μm cisplatin (B). Specimens were then rinsed and maintained for 96 h in medium that contained BrdU. Numerous BrdU-positive nuclei were present in the streptomycin-treated (Strep) specimens (A, C), but very few such cells were observed in the cisplatin-treated (Cis) cultures (B, C) [p < 0.0001, F(2,16) = 23.2].. Scale bar (in B) A, B, 100 μm.
Impaired clearance of hair cell debris and reduction in resident macrophages following cisplatin treatment
After aminoglycoside ototoxicity, the remains of apoptotic hair cells are either extruded from the sensory epithelium or phagocytosed by surrounding supporting cells or tissue macrophages (Li et al., 1995; Warchol, 1997, 1999; Abrashkin et al., 2006). Examination of the sensory epithelium of cisplatin-treated utricles revealed the presence of large quantities of hair cell debris, suggesting that the clearance of apoptotic cells had been impaired (Fig. 10). In light of this observation, we next examined the activity of resident macrophages after cisplatin ototoxicity. Cultured utricles were treated for 24 h in 10 μm cisplatin and then maintained for an additional 0, 24, or 48 h in cisplatin-free medium. Control specimens were treated with 1 mm streptomycin and maintained for identical recovery times. After fixation, macrophages were labeled with the KUL01 antibody (Mast et al., 1998). As reported previously (Warchol, 1999), large numbers of macrophages were present in the aminoglycoside-treated specimens (Fig. 11, top). In contrast, very few macrophages were observed in the cisplatin-treated utricles (Fig. 11, bottom). After 48 h recovery, the density of immunolabeled macrophages from both streptomycin- and cisplatin-treated utricles was quantified. Streptomycin-treated utricles contained 22.5 ± 13.7 macrophages/50,000 μm2, while cisplatin-treated specimens contained 5.0 ± 4.2 macrophages/50,000 μm2 (n = 12 samples/group; p < 0.001).

Maintained presence of hair cell debris in the sensory epithelium of the cisplatin-treated utricle. Utricles were cultured for 24 h in 10 μm cisplatin and then allowed to recover in vitro for 1, 5, or 7 d. Frozen sections of these specimens were immunolabeled for HCS-1 (green, hair cells), Sox2 (red), and DAPI (blue). High levels of hair cell debris (arrows) were still present in the injured sensory epithelia, even after 7 d of recovery. Scale bar: (bottom) top–bottom, 50 μm.

Cisplatin depletes resident macrophages in the chick utricle. Chick utricles were cultured in either 1 mm streptomycin (top) or 10 μm cisplatin (bottom) and then allowed to recover for 0, 24, or 48 h. Macrophages (green) were labeled with the KUL01 antibody, and hair cells (red) were immunolabeled for calretinin. Numerous macrophages were present in the streptomycin-treated specimens, but very few macrophages were observed after cisplatin treatment. Scale bar, 60 μm.
Discussion
Sensory hair cells are responsible for the perception of sound and head movements. Injury to hair cells is a well known side effect of treatment with aminoglycoside antibiotics and with the chemotherapeutic agent cisplatin. Because the mammalian ear has a very limited capacity for regeneration, ototoxic injury often leads to permanent deficits in hearing and equilibrium. Notably, however, the ears of nonmammalian vertebrates are able to quickly regenerate hair cells after aminoglycoside injury (Brigande and Heller, 2009; Brignull et al., 2009). This is the first study to examine the regenerative ability of the nonmammalian ear after cisplatin ototoxicity. We found that exposure to cisplatin blocks regenerative proliferation of avian supporting cells and also prevents those cells from transdifferentiating into replacement hair cells. As a result, the avian ear appears to be incapable of regeneration after cisplatin injury. We conclude that cisplatin has direct toxic effects on both hair cells and supporting cells.
Mechanisms of cisplatin oototoxicity
Although the ototoxic effects of cisplatin are well known, the cellular mechanisms of cisplatin-induced hair cell death are poorly understood. In contrast, considerable research has focused on characterization of the antineoplastic effects of cisplatin. Following cellular uptake, aquated cisplatin becomes incorporated into nuclear DNA, leading to the formation of intrastrand cross-links. These “DNA adducts” are recognized by DNA nucleotide excision–repair mechanisms (particularly during the G1 to S phase transition), which then activate cellular apoptotic pathways (Wang and Lippard, 2005). Although this mechanism is thought to preferentially target proliferating cells, it is clear that cisplatin is also toxic to a number of permanently postmitotic cell phenotypes. For example, systemic treatment with cisplatin leads to the formation of DNA adducts in sensory neurons of mouse dorsal root ganglia, and the presence of such adducts is correlated with the extent of resulting peripheral neuropathy (Dzagnidze et al., 2007). Given the similarities between neurons and sensory hair cells, it is likely that the ototoxic effects of cisplatin involve similar mechanisms. Consistent with this suggestion, cisplatin treatment leads to immunoreactivity for platinated DNA in cochlear hair cells before hair cell death (van Ruijven et al., 2005).
Time course of cisplatin-induced hair cell death
Aminoglycoside antibiotics enter hair cells through mechanotransduction channels (Dai et al., 2006), leading to observable changes in morphology within 1 h (Richardson and Russell, 1991) and cell death within 24 h (Matsui et al., 2004). In contrast, the results presented here indicate that cisplatin ototoxicity may occur over a slower time course. Our data demonstrate that moderate concentrations of cisplatin (e.g., 10–20 μm for 24 h) can kill the majority of hair cells in the avian cochlea and utricle, but the apoptotic process requires ∼3–7 d (Fig. 2). Notably, some prior in vitro studies have observed hair cell death within 24 h of cisplatin application, but those studies typically employed much higher concentrations of cisplatin than those used here (Schmitt et al., 2009). One interpretation of these data is that there exists a dose–time relationship for cisplatin ototoxicity so that higher doses of cisplatin kill hair cells more quickly. Such a relationship between cisplatin concentration and the latency of injury has been demonstrated for lateral line hair cells in zebrafish (Oh et al., 2007). Quantitative measurements conducted in vivo suggest the cisplatin concentration in inner ear fluids peaks at ∼10 μm, following systemic injection (Hellberg et al., 2009). For this reason, we believe that our organotypic culture methods provide a reasonably accurate model of cisplatin ototoxicity. The long latency of cisplatin-induced hair cell death may also suggest possible differences in the cellular mechanisms that underlie aminoglycoside versus cisplatin ototoxicity. For example, it is not known how cisplatin enters hair cells, but the levels of cisplatin within tumor cells appear to be regulated by active uptake and efflux (e.g., Wang and Lippard, 2005). If similar transport mechanisms were to mediate the entry of cisplatin into hair cells, they may occur more slowly than passive entry through mechanotransduction channels. Also, the possible incorporation of cisplatin into hair cell DNA and subsequent recognition of DNA adducts by nucleotide excision repair mechanisms might require a longer time course than the intrinsic (i.e., mitochondrial) apoptotic pathway that is thought to underlie aminoglycoside-induced hair cell death (Matsui et al., 2004; Cheng et al., 2005).
Cisplatin treatment blocks regeneration in the avian ear
One of the key observations in the present study is that cisplatin blocks the proliferation of avian supporting cells. Since those cells act as progenitors during hair cell regeneration, we also found that the avian ear appears to be incapable of regeneration after cisplatin ototoxicity. Given the highly proliferative nature of avian supporting cells (Warchol 1995; Warchol, 2002), it is likely that cisplatin interacts with supporting cell DNA in a manner similar to that observed in tumor cells (Wang and Lippard, 2005). One limitation of the present work is that we examined cell proliferation for only 7 d after cisplatin treatment. As such, we cannot conclusively state whether cisplatin treatment leads to a permanent impairment in the proliferative ability of supporting cells or whether those cells possess the ability for repair after cisplatin exposure. It is conceivable, for example, that cisplatin uptake leads to the formation of DNA adducts in supporting cells, but such adducts may be removed (via nucleotide excision repair mechanisms) over a time course of several weeks, leading to restoration of proliferative ability. In this regard, it is notable that cisplatin is incorporated into nuclear DNA of peripheral glial cells after systemic injection, but those cells can repair much of the resulting DNA injury after 14 d of recovery (Dzagnidze et al., 2007). Also, our results suggest that the ototoxic effects of cisplatin occur over a relatively long time course (3–7 d), and this may cause a corresponding delay in the onset of regeneration. Nevertheless, we did observe the loss of ∼30% of utricular hair cells within 24 h of cisplatin treatment (Fig. 3D). A comparable injury from aminoglycosides would evoke a proliferative response within 48 h (Matsui et al., 2000), yet we observed essentially no regenerative proliferation during the following 7 d (Fig. 4E). This finding further underscores the differences between regenerative proliferation after cisplatin versus aminoglycoside ototoxicity. Finally, it is possible that only a subpopulation of avian supporting cells are capable of regenerative proliferation following hair cell injury (see review by Brignull et al., 2009) and that cisplatin selectively targets those proliferative cells. A more complete understanding of cisplatin ototoxicity in the nonmammalian ear may reveal differences in supporting cell phenotypes that are not apparent from morphological analysis.
In addition to creating replacement hair cells though renewed proliferation, supporting cells in the nonmammalian ear can also produce new hair cells via direct transdifferentiation (Baird et al., 1996; Adler et al., 1997; Stone and Rubel, 2000; Roberson et al., 2004). Moreover, this phenotypic change can be greatly enhanced by blocking Notch pathway signaling via treatment with the γ-secretase inhibitor DAPT (Daudet et al., 2009). Our results suggest that exposure to cisplatin interferes with the ability of supporting cells to transdifferentiate into hair cells, both spontaneously and after Notch inhibition. These findings are further evidence for a direct effect of cisplatin on supporting cells. Additional research has shown that γ-secretase inhibitors can enhance the toxicity of cisplatin toward certain types of tumor cells (Aleksic and Feller, 2008). Such results may have relevance for the development of techniques to induce mammalian inner ear regeneration. Specifically, the promotion of supporting cell transdifferentiation—via retroviral transfection or by treatment with γ-secretase inhibitors—is emerging as a promising strategy for inducing sensory regeneration in the mammalian ear (for review, see Brigande and Heller, 2009). However, if cisplatin were to also block the ability of mammalian cells to transdifferentiate, it may not be possible to use such methods for the treatment of cisplatin ototoxicity.
Atypical pattern of cisplatin-induced hair cell death in the avian cochlea
One unexpected finding of the present study is that cisplatin-induced hair cell damage in the chick cochlea first occurs in the distal (low-frequency) region of the sensory epithelium and then progresses proximally over a time course of several days. This topographic pattern of injury is strikingly different from that observed in mammals, where cisplatin damage first occurs in the high-frequency (basal) region and then moves apically (Cardinaal et al., 2000). A similar distal-to-apical injury pattern is also observed in the cochleae of both mammals (for review, see Rizzi and Hirose, 2007) and birds (Cheng et al., 2003) after aminoglycoside treatment. These conflicting observations are difficult to explain, given our current knowledge of ototoxic signaling. It is possible that cisplatin enters proximal and distal hair cells at similar rates but that the resulting apoptotic mechanisms (e.g., formation of DNA adducts and recognition by DNA repair mechanisms) occur more quickly in distally located hair cells. On the other hand, hair cell uptake of cisplatin may occur more quickly in distal versus proximal hair cells. Both of these suggestions are speculative and will require further study. A more complete understanding of these opposing tonotopic patterns of hair cell death may lead to fundamental insights into the molecular mechanisms of cisplatin ototoxicity.
Impaired phagocytosis and reduction in tissue macrophages after cisplatin ototoxicity
Finally, examination of sensory epithelia after cisplatin treatment revealed the presence of considerable hair cell debris, even after 7 d of recovery (Fig. 10). Following aminoglycoside injury, apoptotic hair cells are either extruded from the sensory epithelium or phagocytosed by adjoining cells (Li et al., 1995). In addition, the avian inner ear contains a resident population of macrophages that may also participate in the removal of hair cell debris (Warchol, 1997; Bhave et al., 1998; Warchol, 1999). Notably, we found that cisplatin treatment resulted in a significant reduction in the numbers of tissue macrophages within the chick utricle. It should be emphasized that the specific roles of macrophages in the processes of ototoxicity and sensory regeneration are not known. They could function strictly as scavengers, removing cellular debris to promote epithelial repair. However, recruitment of macrophages has been shown to play a stimulatory role in many forms of neural regeneration (e.g., Cui et al., 2009), and it is conceivable that the toxic action of cisplatin on tissue macrophages further contributes to impairment of regeneration after cisplatin ototoxicity.
Footnotes
-
This work was supported by Grants R01 DC006283 (M.E.W.), T32 DC000022 (J. G. Neely), and P30 DC04665 (R. A. Chole) from the National Institutes of Health–National Institute on Deafness and Other Communication Disorders and by the National Organization for Hearing Research. We thank Angie Schrader for assistance with confocal imaging.
- Correspondence should be addressed to Mark E. Warchol, Department of Otolaryngology, Box 8115, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110. warcholm{at}ent.wustl.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}