

Abstract
Some cases of autism spectrum disorder have mutations in the lipid phosphatase, phosphatase and tensin homolog on chromosome 10 (Pten). Tissue specific deletion of Pten in the hippocampus and cortex of mice causes anatomical and behavioral abnormalities similar to human autism. However, the impact of reductions in Pten on synaptic and circuit function remains unexplored. We used in vivo stereotaxic injections of lentivirus expressing a short hairpin RNA to knock down Pten in mouse neonatal and young adult dentate granule cells. We then assessed the morphology and synaptic physiology between 2 weeks and 4 months later. Confocal imaging of the hippocampus revealed a marked increase in granule cell size and an increase in dendritic spine density. The onset of morphological changes occurred earlier in neonatal mice than in young adults. We used whole-cell recordings from granule cells in acute slices to assess synaptic function after Pten knockdown. Consistent with the increase in dendritic spines, the frequency of excitatory miniature and spontaneous postsynaptic currents increased. However, there was little or no effect on IPSCs. Thus, Pten knockdown results in an imbalance between excitatory and inhibitory synaptic activity. Because reductions in Pten affected mature granule cells as well as developing granule cells, we suggest that the disruption of circuit function by Pten hypofunction may be ongoing well beyond early development.
Introduction
The phosphatidylinositol 3,4,5-triphosphate (PIP3), phosphatase and tensin homolog on chromosome 10 (Pten), catalyzes the reverse reaction of phosphoinositide 3-kinase (PI3K). Pten and PI3K are highly conserved throughout evolution and are expressed in all mammalian cell types. Although Pten-null mice die during embryogenesis (Di Cristofano et al., 1998), conditional knock-out mice have provided insights into the role of Pten in the CNS. Depending on the targeted cell type, cells in Pten conditional knock-out mice display cellular mislocation, hypertrophy, and increased survival (Groszer et al., 2001; Fraser et al., 2004). Conditional deletion of Pten in CNS neurons results in neuronal hypertrophy, macrocephaly, and abnormal social interaction (Kwon et al., 2006). The macrocephaly that occurs in 20% of patients with autism spectrum disorder (ASD) has been attributed to Pten mutations (Goffin et al., 2001; Butler et al., 2005; Buxbaum et al., 2007; Herman et al., 2007). Although multiple genes have been linked with ASD (Bourgeron, 2009), Pten conditional knock-out mice are the first animal model of ASD that incorporate the morphological and behavioral abnormalities seen in human cases (Greer and Wynshaw-Boris, 2006). The morphological deficits in neurons of Pten conditional mutant mice suggest a pattern of enhanced cellular growth, in keeping with the macroscopic increase in brain size seen in some cases of ASD.
Pten hypofunction may represent a prototype for examining perturbations of synaptic circuitry that are often considered as the fundamental abnormality in autism. Thus, to examine the cellular and synaptic phenotype of Pten deletion, we used a viral-based strategy that allows for precise temporal and spatial knockdown of Pten in vivo. After intrahippocampal injections of lentiviral particles, dentate granule cells targeted for Pten knockdown were identified by expression of a fluorescent marker. Analysis by immunohistochemistry, Western blots, and real-time PCR indicated a significant knockdown of Pten. Pten knockdown in neonatal mice [postnatal day 7 (P7)] increased soma size and dendritic spine density by 2 weeks after injection. In young adult mice (6–8 weeks), Pten knockdown resulted in a similar morphological phenotype that evolved by 4 months after injection. The frequency of miniature excitatory synaptic currents in Pten knockdown granule cells also increased. Our results suggest that Pten knockdown increases the net excitatory drive onto granule neurons in vivo.
Materials and Methods
Animals/stereotaxic injections.
C57BL/6 male mice were anesthetized using an isoflurane gas system (Veterinary Anaesthesia Systems). For intrahippocampal injections, mice were placed in a Kopf stereotaxic frame fitted to the anesthesia system, a skin incision was made, and holes were drilled at x, 1.1 mm, and y, −1.9 mm, from bregma. Using a 10 μl Hamilton syringe fitted with a 30 gauge needle and the Quintessential Stereotaxic Injector (Stoelting), 1 μl of virus was delivered at 0.25 μl/min at z-depths of 2.5 and 2.3 mm. The syringe was left in place for 1 min after each injection and then was slowly withdrawn. The skin above the injection site was closed using veterinary glue. Animals received postoperative lidocaine and drinking water containing Children's Tylenol. The experiments were approved by Institutional Biosafety and Institutional Animal Care and Use Committee review boards.
Cloning: FUGW-shPten and FURW-shPten.
The FUGW parent vector contains the HIV-1 Flap element, the human ubiquitin-c promoter, green fluorescent protein (GFP), and the woodchuck hepatitis virus posttranscriptional regulatory element (WRE) (Lois et al., 2002). A Pten short hairpin RNA (shRNA) lentivirus was previously published (Xin et al., 2005). To make the Pten shRNA compatible with multiple viral vectors, a PacI linker was ligated into the NheI site of FUGW. The Pten shRNA target sequence is as follows: AGGTGAAGATATATTCCTCCAA.
The H1 promoter and Pten shRNA were cloned into PacI/BstBI site of FUGW. mCherry was excised from pRSETB (Shaner et al., 2004) using the BamHI/EcoRI sites. The mCherry fragment was ligated into FUGW to generate red fluorescent FURW.
Viral packaging.
Lentiviral packaging was performed by calcium phosphate-mediated transfection of pCMVΔ8.9, pVSV-g, and the FUGW-based shuttle vector into HEK293 FT cells (Invitrogen) (Naldini et al., 1996). Cells are maintained in Iscove's modified Dulbecco's medium (IMDM) (high glucose) (Invitrogen; 12440-053), 10% fetal bovine serum (FBS), 0.1 mm MEM nonessential amino acids, 2 mm l-glutamine, 1% pen/strep, and 500 μg/ml Geneticin (only use during growth). One day before transfection, the HEK293 cells were split to a density of 2.5–3.0 × 106 cells per 10 cm dish (no Geneticin). Each vector is maintained at 1 mg/ml in TE. In one polystyrene tube, 20 μl of transfer vector, 13 μl of pCMVΔ8.9, 9 μl of pVSV-g, 860 μl of water, and 100 μl of 2.5 m CaCl2 are combined. In another polystyrene tube, add 1000 μl of 2× HBS (281 mm NaCl, 50 mm HEPES, 1.5 mm Na2HPO4 heptahydrate monobasic, pH to exactly 7.0). The contents of the first tube is then added slowly as single drops to the contents of the second tube while gently tapping the tube. The combined contents are incubated at room temperature in the dark for 30 min. One milliliter of this mixture is then added to each plate of HEK293 cells one drop at a time. At 24 h after transfection, the IMDM is replaced with low serum IMDM (2% FBS). The media is then harvested at 48 and 72 h after transfection, spun at 3000 rpm, and the supernatant filtered through a 0.45 μm PVDF (polyvinylidene difluoride) syringe tip filter. Viral particles are concentrated by ultracentrifugation at 20,000 rpm for 2 h and resuspended in PBS.
Quantitative PCR.
Quantitative PCR (QPCR) analysis of Pten knockdown was performed in both HEK293 cells and hippocampal primary neuronal cultures. Hippocampal neurons were cultured from P0 animals as previously published (Tovar and Westbrook, 1999). Both hippocampal neurons and HEK293 cells were infected on the day of plating. After 7 d in culture, RNA was isolated from hippocampal neurons or HEK293 cells using Trizol (Invitrogen) and the manufacturer's recommended procedures. The samples were DNase treated using the DNA-free kit (Ambion) followed by an additional ethanol precipitation. The RNA concentration was determined and 200 ng of RNA was used to generate cDNAs with random hexamer primers of the First-Strand cDNA Synthesis kit (Fermentas). One sample was serially diluted to provide a standard curve for the real-time reaction. All other samples were diluted 1:3. The real-time reaction was performed in duplicate using FastStart SYBR Green Master (Roche). PCR was performed on an Opticon OP346 (MJ Research).
QPCR primers were as follows: Pten, sense (s), 5′-CTG GTG TAA TGA TAT GTG CA-3′; antisense (as), 5′-AAC GGC TGA GGG AAC TC-3′; GAPDH, s, 5′-GGA GAT TGT TGC CAT CAA CGA-3′; as, 5′-TGA AGA CAC CAG TAG ACT CCA CGA C-3′; PHF20, s, 5′-GTT TCT TCC TGG CAT GGT GT-3′; as, 5′-AGG CTG TAT GGG CTC TGC TA-3′; ENGASE, s, 5′-TGT TGA ACG CAG AAA CAA GC-3′; as, 5′-TGA GAA GCA AAC GAG GAG GT-3′; DDX60, s, 5′-CCC AGG GTC CAG GAT TTT AT-3′; as, 5′-GAA CAG TTG CTG CCA CTT GA-3′.
Western blots.
HEK293 cells were infected with Pten shRNA lentivirus at 10 multiplicities of infection on the day of plating in six-well plates. On day 7, protein was isolated by homogenizing in 650 μl of 100 nm Tris-Cl, pH 6.8, 200 mm DTT, 4% SDS, 0.2% bromophenol blue, and 20% glycerol. Protein concentrations were determined using the BCA kit (Pierce). Twenty-five micrograms of protein were loaded onto 12% SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were then Ponceau S stained, photographed, and probed with anti-Pten (1:250; Millipore) overnight. The blots were then developed using the ChemiGlow West reagent (Alpha Innotech), films photographed, and densitometry performed using the measure intensity function of ImageJ. The densitometry values were normalized to the Ponceau S staining intensity and quantified using ImageJ.
Electrophysiology.
Hippocampi from adult mice were perfused and cut in an ice-cold solution containing the following (in mm): 110 choline-Cl, 7 MgCl2, 2.5 KCl, 1.25 NaH2PO4 · 2H2O, 0.5 CaCl2, 1.3 Na-ascorbate, 25 NaHCO3 bubbled with 95% O2–5% CO2. Slices were stored and recordings performed in a solution containing the following (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, and 25 d-glucose, bubbled with 95% O2–5% CO2. For EPSC recordings, we used a Cs-gluconate internal (in mm: 100 gluconic acid, 0.2 EGTA, 5 HEPES, 2 Mg-ATP, 0.3 Li-GTP, pH 7.2, 295 mOsm, pH to 7.2 using 50% CsOH for a final concentration of 120 Cs-gluconate). SR95531 (4-[6-imino-3-(4-methoxyphenyl)pyridazin-1-yl]butanoic acid hydrobromide) (10 μm) was added to block GABAA-mediated IPSCs. For all recordings, cells were held at −70 mV in the presence of magnesium (1 mm); thus, the measured spontaneous EPSCs (sEPSCs) and miniature EPSCs (mEPSCs) were AMPA receptor mediated. For IPSC recordings, NBQX (10 μm) was added to block AMPA receptor-mediated EPSCs, and we used a CsCl internal solution (in mm: 125 CsCl, 10 HEPES, 10 EGTA, 2 Mg-ATP, 0.3 Na3-GTP, 0.5 CaCl2, pH 7.2 and 295 mOsm). Series resistance (5–20 MΩ) was monitored, and experiments were discarded if increases >10 MΩ occurred. Currents were filtered at 2 kHz and sampled at 20 kHz (Axopatch 200B or MultiClamp 700A; Molecular Devices). Synaptic responses were evoked with a tungsten bipolar electrode in the outer molecular layer of the dentate gyrus. Paired-pulse ratios were calculated as the ratio of peak amplitudes of 10 averaged episodes. The interstimulus interval was 50 ms. Spontaneous currents were detected using template matching in the Neuromatic plug-in in Igor Pro. Miniature synaptic events were detected using the same template matching function, but with Axograph software. The template rise time was 1 ms; decay time, 6 ms (8 ms for IPSCs); baseline, 10 ms; length, 30 ms; and a threshold of 3. These values represent an optimal template and threshold value to detect all PSCs (Clements and Bekkers, 1997). Captured events were manually reviewed to exclude false-positive events. For the miniature synaptic events, the investigator was blinded to the cellular genotype. Our template function detected events with rise times between 0.5 and 3.2 ms. Rise times were defined as the time between 10 and 90% of the maximal amplitude. The decay of PSCs was determined by fitting averaged events to a single exponential (AMPA) or a double exponential (GABA). One-way ANOVA statistics were performed in Igor Pro, paired t tests in Microsoft Excel, and Kolmogorov–Smirnov test or the Kruskal–Wallis test in BrightStat (Stricker, 2008).
Histology.
For vibratome sectioning, mice were anesthetized using 2% Avertin injected intraperitoneally and were intracardially perfused with ice-cold PBS followed by 4% (w/v) paraformaldehyde (PFA) in PBS. The brain was postfixed in 4% PFA overnight and embedded in 2.5% agarose. For immunohistochemistry, 50-μm-thick coronal sections were cut and incubated with rabbit anti-Pten (1:100; Cell Signaling; 9559) and mouse anti-NeuN (1:500; Millipore Bioscience Research Reagents; MAB377). Primary antibodies were detected using secondary antibodies conjugated with Cy3 (Jackson ImmunoResearch) or Alexa Fluor 488 (Invitrogen) followed by counterstaining with DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories). An additional antigen retrieval procedure was used for Pten staining in the neonatal-injected animals (see Fig. 1B). Sections were incubated at 80°C for 10 min in 10 mm tri-sodium citrate, pH 8.5, in water. The sections were then cooled to room temperature and stained using standard immunohistochemistry methods.
Morphology.
For all morphological analyses, image z-stacks were obtained using a Zeiss LSM 510 laser-scanning confocal microscope. An investigator blinded to the genotype of the animal imaged and analyzed granule neurons from anatomically equivalent sections of the suprapyramidal blade of the dorsal portion of dentate gyrus. Soma images were taken using a 40× c-Apochromat water lens (Zeiss) at 1024 × 1024 and a 1.3 μm virtual section thickness with a z-step of 2 μm. Stacks of three images were compressed. The circumference of the neuron was manually traced using ImageJ. Based on this circumference, the cross-sectional area was recorded. For dendritic spine images, the brightest neurons located in the suprapyramidal blade of the dentate gyrus were imaged. For the adult injected group and neonate injections analyzed at 14 d after injection, the investigator was blinded during image acquisition and analysis. For the neonatal group at 4 months after injection, the investigator was not blinded during image acquisition but the images were scrambled and renamed to blind the analysis. Dendritic spine density was estimated from image stacks captured within 100 μm of the dendritic tips for the outer molecular layer, and within 50 μm of the granule cell layer for the inner molecular layer. To exclude qualitative judgment from quantitative measurements, no distinction was made between spines and filopodia. All protrusions were considered spines. Images were captured using a 63× Plan-Apochromat oil lens (Zeiss) at 512 × 512 and an electronic zoom of 3×. The virtual section thickness was 0.8 μm with a step size of 0.5 μm and a scan speed of 9. Using ImageJ, a dendritic section was traced by hand and each spine on that dendritic segment marked. From these measurements, dendritic spine density was estimated. No correction was made for spines obscured in the z-plane. To measure the dendrite diameter in the inner molecular layer, z-stacks were collapsed as maximal projections and four lines drawn across the diameter of the dendrite at random intervals perpendicular to the axis of the dendrite. We recorded the diameter of a single dendrite as the average length of these four lines. For mossy fiber analysis, images were captured using the same parameters as for dendritic spines. We focused on the region where the axons emerged from the hilus and the CA3 cell layer began. The circumference of the mossy fiber boutons was traced to measure their cross-sectional area and four lines drawn across random axonal sections to determine their average diameter.
Fluorescence-activated cell sorting.
For fluorescence-activated cell sorting (FACS) sort of cells from injected animals, hippocampi were isolated using a fluorescence dissection microscope in D1 media (137 mm NaCl, 5.3 mm KCl, 10 mm HEPES, 33 mm d-glucose, 44 mm sucrose, 0.167 mm NaHPO4 · 7H2O, 0.220 mm KH2PO4, 0.067 mm phenol red, 1% pen/strep). Isolated hippocampi were digested with papain for 40 min [10 ml of D1, 2 mg of cysteine, 150 μl of calcium solution (100 μm), 100 μl of EDTA solution (50 mm), 200 U of papain]. Digestion was terminated by adding 10 ml of heat-inactivated fetal calf serum, 25 mg of BSA, and 25 mg of trypsin inhibitor. The cells were then mechanically dissociated using glass pipettes with progressively smaller tips. Cells were sorted in dissociation media containing 200 U of RNase inhibitor/5 ml media. The flow cytometer (FACSVantage Diva; BD Biosciences) was equipped with an argon ion laser (488 nm, 200 mW) operated at 10 psi and 20,000 drops/s with a 130 μm tip. Enhanced green fluorescent protein (EGFP) fluorescence was detected using a 530/30 nm bandpass filter. EGFP+ cells were sorted into culture tubes containing 1 ml of tissue culture media at a rate of 2000–4000 cells/s.
Results
Viral-mediated Pten knockdown in vivo
To examine Pten hypofunction at the single-cell level, we developed a strategy to knock down Pten in vivo using lentivirus. An effective shRNA sequence targeting Pten driven by the H1 promoter has been previously published (Xin et al., 2005). We subsequently constructed lentiviral vectors containing this shRNA, and a downstream fluorescent reporter (GFP or mCherry) driven by the ubiquitin promoter (Lois et al., 2002). The fluorescent reporters allowed unambiguous identification of cells expressing the Pten shRNA. We first injected a virus expressing Pten shRNA and mCherry into young adult mice (6–8 weeks of age). In vivo injection resulted in robust infectivity as indicated by mCherry expression in granule neurons at 14 d after injection as well as decreased anti-Pten immunoreactivity in virus-injected, mCherry+ cells (Fig. 1A). Using the virus expressing the Pten shRNA and EGFP, Pten knockdown was also apparent in neonatal (P7) injected mice at 14 d after injection, but not in granule cells infected with an EGFP control virus (Fig. 1B).

Viral-mediated knockdown of Pten expression in vivo. Lentiviral particles driving expression of an shRNA directed against Pten and the GFP or mCherry protein were injected in vivo into neonatal (P7) and young adult (6- to 8-week-old) mice. A, Immunohistochemical staining for Pten revealed that mCherry expression correlated with loss of Pten immunoreactivity in young adult mice at 14 d after injection. Infected granule cells in the cell body layer were labeled with mCherry, but were not labeled with anti-Pten antibody as shown in the “Pten shRNA,” “anti-Pten,” and “merged” panels. B, Neonatal (P7) animals were injected with a Pten shRNA plus GFP virus or a virus expressing GFP alone. There was a marked decrease in Pten immunoreactivity (anti-Pten) in cells infected with the GFP-expressing Pten shRNA virus compared with neurons infected with the GFP-only control. C, We isolated sparsely infected HEK293 cells using fluorescence activated cell sorting to examine the degree of Pten knockdown in cells with a multiplicity of infection of 1. There was a 77.5 ± 10.2% reduction in Pten transcript, but no change in GAPDH, PHF20, ENGASE, or DDX60 controls (*p < 0.05, t test; error bars are SD). D, In Western blots from infected HEK293 cells, Pten expression was reduced 73.7 ± 1.8% by the Pten shRNA. We also did not detect a change in the intensity of nonspecific bands revealed by the Ponceau S staining of total protein (D, red bands). E, The Pten ShRNA also reduced Pten transcripts in infected primary hippocampal cultures. There was a 79.2 ± 0.3% reduction in Pten expression compared with uninfected or GFP-only infected neurons (*p < 0.05, t test; error bars are SEM). This reduction was not observed for the GAPDH control (data not shown).
The shRNA target sequence is identical in the mouse and human genome, so we first used HEK293 cells to quantify virally mediated Pten knockdown. Given the relatively low infectivity seen in vivo, it is likely that only a single viral particle infects a neuron. To mimic such a situation in vitro, we infected HEK293 cells with dilutions of virus, achieving infection rates of 24.6 and 13.8% in independent cultures. The sparsely infected cells likely represent infection by a single viral particle per cell. At 1 week after infection, we isolated the infected cells using FACS. By real-time PCR, there was a 77.5 ± 10.2% reduction in the Pten transcript (Fig. 1C) (mean ± SEM; p < 0.03, t test; n = 3 uninfected control and 2 Pten shRNA infected cultures). To examine possible off-target effects, we used the Primer BLAST specificity-checking database to determine whether the Pten shRNA sequence was conserved in other genes. There were at least four basepair mismatches between the shRNA sequence and the closest potential off-target gene. We performed QPCR for Pten, GAPDH, and the potential off-target genes PHF20 (4 nt mismatches), ENGASE (5 nt mismatches), and DDX60 (5 nt mismatches). We found no significant difference in the expression of these control genes (Fig. 1C), consistent with specific knockdown of Pten.
We next examined Pten protein levels by infecting HEK293 cells at 10 viral particles per cell achieving 95% infectivity. After 7 d of viral expression, we assayed for endogenous Pten expression by Western blot (Fig. 1D). Densitometry of the Pten band normalized to Ponceau S stain indicated a 73.7 ± 1.8% reduction in Pten (GFP, mCherry, and uninfected cultures were grouped as control and compared with Pten shRNA-expressing cultures; p < 0.001, t test; n = 3 control and 2 Pten shRNA cultures). Thus, the decrease in protein levels closely paralleled the knockdown of the Pten transcript. We also observed a similar degree of Pten knockdown in primary hippocampal cultures. At 10 viral particles per neuron, 95% of the neurons were infected with a resulting 79.2 ± 0.3% reduction in Pten transcripts as measured by real-time PCR (Fig. 1E) (mean ± SEM; p < 0.05; n = 2 cultures per condition).
To examine the degree of Pten knockdown in vivo, we isolated cells infected with the Pten shRNA virus using fluorescence-activated cell sorting and measured the Pten transcript with QPCR. We injected the Pten shRNA-EGFP virus into six animals at P7. At 10 d after injection, we noted prominent EGFP labeling of the dentate gyrus in 10 of the 12 hippocampi. After dissociation, we isolated 25,000 EGFP+ cells and two sets of 25,000 EGFP− control cells. There was a 51.6% reduction of Pten in the EGFP+ sample compared with the EGFP− sample. This is likely an underestimate of Pten knockdown in infected cells because EGFP transcript in the “EGFP−” sample was detected at 17.6% of the level in the EGFP+ sample (i.e., 17.6% false negatives). Adjusting for the Pten shRNA-EGFP cells in the control sample suggests that in vivo knock-out was very similar to our measurements of knockdown in vitro. These experiments corroborate that viral-mediated Pten shRNA expression results in specific and efficient, but not complete, knockdown of Pten in vivo.
Pten knockdown results in increased neuronal size and dendritic spine density
To determine the effects of viral-mediated Pten knockdown on single neurons in vivo, we infected granule cells in wild-type animals by injecting viruses driving expression of EGFP with or without Pten shRNA. We examined the impact of Pten knockdown in neonatal (P7) and young adult (6- to 8-week-old) mice. We selected P7 to determine the effects of developmental Pten knockdown because it is the peak of granule cell generation (Bayer and Altman, 1974). After injections at P7, we detected a distinctive morphological phenotype at P21 (14 d after injection). The cross-sectional area of infected granule cells increased from 92.3 ± 1.2 μm2 in GFP control to 113.4 ± 2.3 μm2 after Pten knockdown (Fig. 2A) (p < 0.001, t test; n = 63 control, n = 52 Pten knockdown neurons; two animals per group). In addition, Pten knockdown neurons had thicker dendrites with more filopodial-like protrusions and aberrant protrusions extruding from existing spines in Pten knockdown granule cells (Fig. 2B). The outer two-thirds of the molecular layer zone receive the major excitatory input from entorhinal cortex. We found an increase in the density of spines in this dendritic zone as quantified in the outer molecular layer within 100 μm of the dendritic tip (Fig. 2B) (GFP control, 2.16 ± 0.10 spines/μm; Pten shRNA, 2.52 ± 0.09 spines/μm; p < 0.01, t test; n = 25 and 22 neurons from two animals per group, respectively). Four months after injections at P7, Pten knockdown neurons were clearly larger. The somatic area was 79.4 ± 1.8 μm2 for control neurons and 99.9 ± 3.1 μm2 for Pten knockdown neurons (Fig. 2A, graph) (p < 0.0001, t test; n = 66 and 62 neurons from two animals per group, respectively). Dendritic spine density in the outer molecular layer remained elevated in knockdown neurons (Fig. 2B, graph) (control, 2.10 ± 0.05 spines/μm; Pten shRNA, 2.73 ± 0.13 spines/μm; p < 0.001, t test; n = 15 and 18 neurons from two animals per group, respectively).
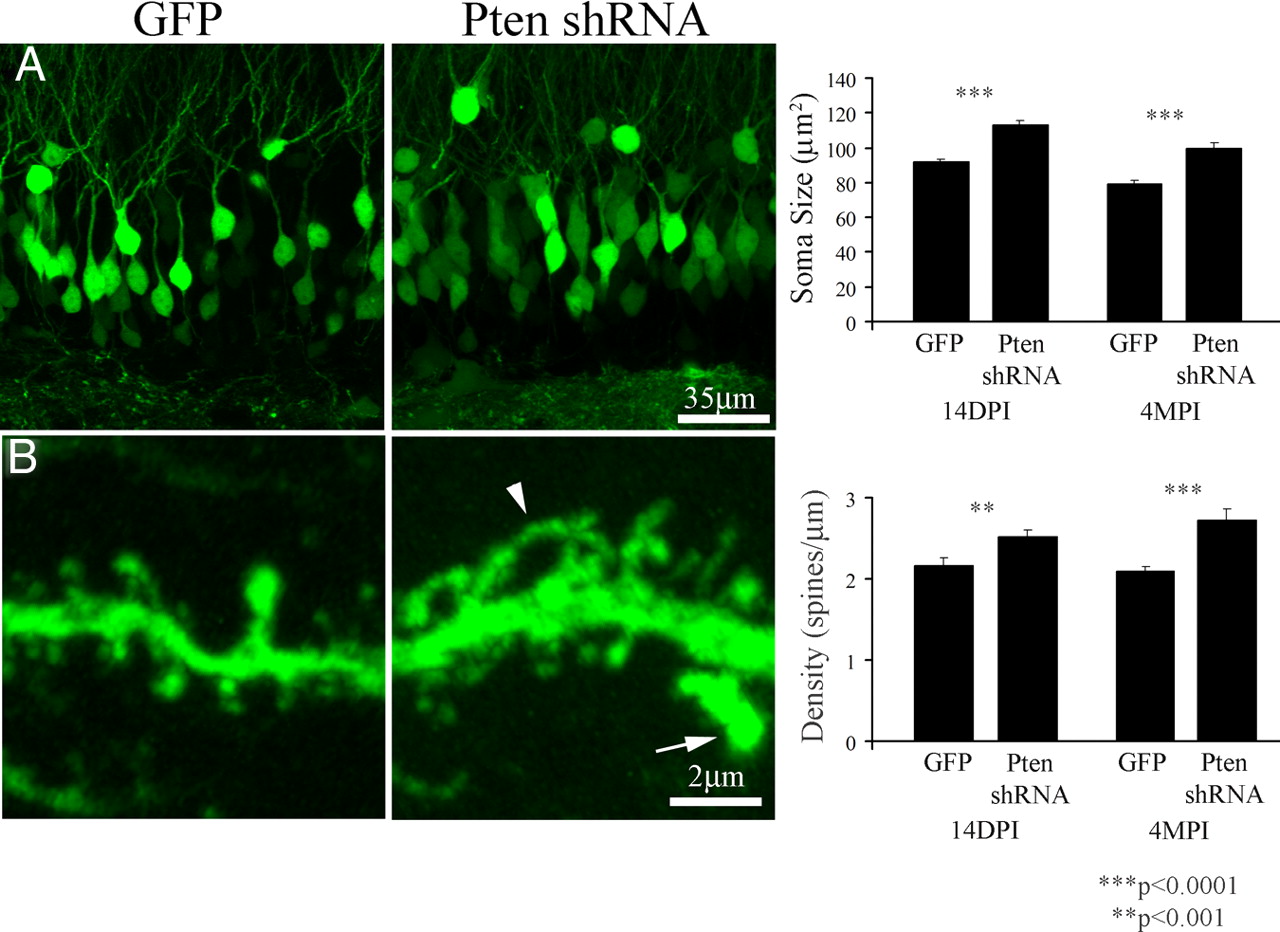
Pten knockdown in neonatal animals resulted in rapid onset of neuronal hypertrophy and an increase in dendritic spines. Neonatal (P7) mice were injected with the GFP control or a lentivirus expressing the Pten shRNA. By 14 d after injection, soma size and dendritic spine density increased in granule cells expressing the Pten shRNA. A, The images at left are 14 d after injection. There was an increase in soma size at both 14 d and 4 months after injection (right panel). B, Dendritic spines in distal dendrites of Pten knockdown cells were increased at 14 d after injection (images at left) including an increase in long, thin filopodial-like protrusions (B, arrowhead). We also noted aberrant spine-like protrusions that appeared to arise from other spines (B, arrow). The increase in dendritic spine density was significant 14 d and 4 months after injection (right panel).
To determine whether the neuronal hypertrophy was limited to a particular cellular compartment, we also examined dendrites in the inner molecular layer (commissural/associational inputs) and mossy fiber axons at 4 months after injection of P7 animals. All examined regions of the granule cell were increased in size. The diameter of the dendrites in the inner molecular layer (within 50 μm of the granule cell layer) increased from 0.738 ± 0.034 μm in control to 0.977 ± 0.059 μm in Pten knockdown cells (Fig. 3A) (p < 0.01, t test; n = 16 and 23 neurons from two animals per group). The dendritic spine density also increased in the inner molecular layer (Fig. 3A) (control, 1.56 ± 0.09 spines/μm; Pten shRNA, 2.39 ± 0.14 spines/μm; p < 0.0001, t test; n = 16 and 20 neurons from two animals per group). Mossy fiber axons in the CA3 region, examined at the point where they exit from the hilus, were thicker in Pten knockdown neurons than in the GFP-expressing control neurons (Fig. 3B) (control, 0.360 ± 0.010 μm; Pten shRNA, 0.450 ± 0.014 μm; p < 0.0001, t test; n = 39 and 37 axons from two animals per group). Furthermore, we examined the cross-sectional area of the mossy fiber boutons, which were larger in Pten knockdown neurons (Fig. 3B) (control, 6.45 ± 1.60 μm2; Pten shRNA, 9.92 ± 2.08 μm2; p < 0.01, t test; n = 24 and 22 boutons from two animals per group). From these morphological analyses, we conclude that Pten knockdown results in global hypertrophy of granule cells in vivo and that the density of dendritic spines increased in both of the two main inputs to granule cell dendrites in the molecular layer.

Pten knockdown results in hypertrophic axons and proximal dendrites. We examined axons and proximal dendrites at 4 months after injection of neonatal (P7) animals. A, Expression of the Pten shRNA resulted in dendrites with increased diameter and spine density in the inner molecular layer (A, right panel) when compared with granule cells expressing GFP alone (A, left panel). B, Mossy fiber axons arising from Pten shRNA-infected granule cells were thicker and the cross-sectional area of mossy fiber boutons was increased (B, right panel) when compared with granule cells expressing GFP alone (B, left panel).
In mice injected as young adults, the onset of the morphological phenotype appeared to be somewhat slower. At 14 d after injection, we did not see a significant increase in cell size or dendritic spine density (images not shown). The somas of control granule cells had cross-sectional areas of 108.1 ± 1.2 μm2 compared with 111.2 ± 1.6 μm2 for neurons expressing Pten shRNA (p > 0.1, t test; n = 121 cells from three animals per group). Cells expressing Pten shRNA had 2.18 ± 0.07 spines/μm compared with 2.15 ± 0.06 spines/μm in control animals (p > 0.5, t test; n = 55 dendritic segments from six animals for control and n = 63 dendritic segments from seven animals for Pten shRNA). By 4 months after injection, the hippocampus appeared grossly normal (Fig. 4A); however, the soma size and the density of dendritic spines were markedly increased. The cross-sectional soma area was 164.7 ± 3.5 μm2 in the Pten knockdown compared with 114.0 ± 2.6 μm2 in the control (p < 0.0005, t test, n = 35 cells from two animals for control and n = 33 cells from two animals for Pten shRNA) (Fig. 4B). In the outer molecular layer, there was a 43.9 ± 15.6% increase in dendritic spine density (3.3 ± 0.17 in the Pten knockdown compared with 2.3 ± 0.19 spines/μm in the control; p < 0.005; n = 20 dendritic segments from two animals for GFP control and n = 20 dendritic segments from two animals for Pten knockdown) (Fig. 4C).

Pten knockdown in young adult mice results in neuronal hypertrophy and increased dendritic spine density. Young adult animals were injected with either GFP control or Pten shRNA lentivirus. At 14 d after injection, we did not detect significant morphological changes (see Results). By 4 months after injection, there was an increase in cell size as well as dendritic spine density. A, Low-magnification images show the dentate gyrus infected with the control GFP (left) and Pten shRNA virus (right). B, Higher magnification images of the granule cell layer revealed an increase in soma size (164.7 ± 3.5 μm2 in the Pten knockdown compared with 114.0 ± 2.6 μm2 in the control). C, There was also an increase in dendritic spine density of Pten shRNA-infected neurons (3.3 ± 0.17 spines/μm in the Pten knockdown compared with 2.3 ± 0.19 spines/μm in the control; quantitated in the outer molecular layer).
Consistent with the increased size of infected granule cells, Pten knockdown significantly increased the total cell capacitance and decreased the input resistance in both neonate and young adult injected groups at 4 months after injection (Fig. 5A). For the P7 injection, the values obtained in whole-cell recordings were 11.7 ± 1.6 pF and 204.7 ± 15.6 MΩ in control cells, and 16.4 ± 1.4 pF and 128.7 ± 13.3 MΩ for Pten shRNA cells. For young adult injections, the values were 12.8 ± 0.9 pF and 181.3 ± 15.6 MΩ for control cells, and 19.0 ± 1.3 pF and 92.6 ± 9.3 MΩ for Pten shRNA cells. Interestingly, the soma fluorescence intensity correlated with the cross-sectional area of Pten shRNA-infected neurons (r = 0.39; p < 0.01) (Fig. 5B), suggesting that increased viral expression results in higher levels of the Pten shRNA and a more pronounced morphological phenotype. This correlation was not present in cells expressing only EGFP (r = 0.09; p < 0.5, for control), indicating that variation in fluorescence intensity did not distort measurements of cell size. In sections from 62 Pten shRNA-injected animals, we did not observe fluorescent labeling of mossy fibers in the inner molecular layer or dendrites extending into the hilus. However, mossy fiber sprouting and ectopic hilar dendrites have been observed in conditional Pten knock-out mice (Kwon et al., 2006) and in animals with spontaneous seizures (Jessberger et al., 2007; Walter et al., 2007).

Neuronal hypertrophy correlated with the passive membrane properties and GFP expression levels. A, At 4 months after injection, increases in cell size were accompanied by changes in cell capacitance (see text) and input resistance as measured in whole-cell voltage-clamp recording. A 4 mV, 50 ms test pulse (holding potential, −70 mV) from representative recording shows a larger current response for Pten knockdown neurons (red trace) than for control neurons (black trace), indicative of a reduced input resistance. The input resistance was decreased in Pten knockdown neurons in both neonate and adult injections (A, histogram) (***p < 0.001, t test; n in parentheses). B, In images of infected neurons, fluorescence intensity correlated with soma size in the Pten knockdown (right, red dots), but not in control (left, black dots).
Pten knockdown increases the frequency of EPSCs
Although deletion of Pten enhances brain and neuronal growth (Kwon et al., 2001, 2006), the effects on single-cell synaptic physiology have not been explored. To assess synaptic activity after Pten knockdown, we first examined granule cell spontaneous synaptic currents under four conditions: cells from uninjected mice, uninfected cells in injected mice, cells infected with Pten shRNA-mCherry, and cells infected only with mCherry (Fig. 6A). At 2–3 weeks after injection of young adult animals, Pten knockdown markedly increased the sEPSC frequency (Fig. 6B,C) without changing the amplitude, rise time, or decay of the sEPSC (data not shown). There was no significant difference of the paired-pulse ratio of EPSCs evoked by perforant path stimulation (1.13 ± 0.12 uninfected control; 1.39 ± 0.18 Pten shRNA; n = 6 and 4 neurons, respectively; p > 0.1, paired t test), suggesting that the increase in sEPSC frequency was not attributable to an increase in the probability of glutamate release. In contrast to the increase in sEPSCs, Pten had no apparent effect on sIPSC frequency (Fig. 6D).

Pten knockdown in young adult animals increased the frequency of spontaneous EPSCs. Whole-cell recordings of dentate granule cells were performed on acute slices at 2–3 weeks after injection of 6- to 8-week-old animals. A, Biocytin fills (blue) with the patch pipette during whole-cell recording indicate the morphology of granule cells that were targeted for analysis. Infected cells expressed mCherry (red). We recorded from granule cells in uninjected animals (blue, left panel), uninfected granule cells from injected animals (blue, middle panel), and infected granule cells (purple, right panel). B, Sample traces of spontaneous EPSCs are shown from the uninjected, uninfected, and infected conditions. C, There was a significant increase in the frequency of sEPSC in granule cells infected with the Pten shRNA-mCherry lentivirus (p < 0.01, one-way ANOVA and Tukey's post hoc test). The number of cells is indicated in parentheses in the histogram. D, In the presence of NBQX to block EPSCs, there was no significant change in the frequency of sIPSCs compared with uninfected control granule cells (n in parentheses).
To determine the effect of Pten knockdown on unitary excitatory synaptic transmission, we recorded miniature synaptic events in the presence of tetrodotoxin. In control recordings, there was no difference between infected cells expressing only the fluorophore (mCherry or GFP) compared with uninfected cells for sEPSCs (Fig. 6C), sIPSCs at 14 d after injection of adult injected animals (uninfected, 6.52 ± 0.69 Hz; fluorophore, 6.66 ± 1.16 Hz; p > 0.9; n = 9 and 5, respectively), or mEPSCs at 4 months after injection of adult animals (uninfected, 0.06 ± 0.01 Hz; fluorophore, 0.06 ± 0.02 Hz; p > 0.8; n = 15 and 8 neurons, respectively). Because infection of granule cells with the fluorophore-only virus did not alter the frequency of synaptic events, we compared Pten shRNA-expressing cells to neighboring uninfected cells in the same slice in the analysis of miniatures (Figs. 7, 8). As expected, the miniature EPSC frequency in dentate granule cells was lower than spontaneous EPSCs. Pten knockdown increased mEPSC frequency 14 d after neonatal injection (Fig. 7A) (control, 0.12 ± 0.04 Hz; Pten shRNA, 0.33 ± 0.05 Hz; p < 0.001, t test; n = 14 and 17 neurons, respectively). In the adult, the increase did not reach statistical significance at 14 d after injection (Fig. 7A) (control, 0.05 ± 0.01 Hz; shRNA, 0.07 ± 0.01 Hz; p = 0.09; n = 8 and 7 neurons, respectively). However, by 4 months after injection, mEPSC frequency increased twofold to threefold in both neonate and adult injected mice (Fig. 7A) (neonatal injection: 0.06 ± 0.01 Hz, control, and 0.15 ± 0.03 Hz, Pten shRNA; adult injection: 0.08 ± 0.01 Hz, control, and 0.14 ± 0.02 Hz, Pten shRNA). This pattern is consistent with the increases in dendritic spine density, which had an earlier onset in mice injected at P7. We detected a slight increase in mEPSC amplitude in adult injected mice but not in the neonatal group (Fig. 7B).

Pten knockdown increased miniature EPSCs. We performed an electrophysiological analysis of both neonate (P7) and adult (6–8 weeks) injected animals (see Results). The figure shows results obtained 4 months after injection of both neonate and adult injected animals. A, Sample traces from control (black) and Pten knockdown neurons (red) at 4 months after injection revealed an increase in the frequency of mEPSCs (graph at right; p < 0.05, t test; n in parentheses). B, There was a small increase in the amplitude of mEPSCs in adult Pten knockdown neurons (red trace) compared with uninfected control neurons (black trace), but this difference was not detected in mice injected at P7 (graph at right; *p < 0.05, adult injection, and ns, p > 0.40, neonate injection, t test; n in parentheses).

Pten knockdown did not alter mIPSCs. Miniature IPSCs were measured at 4 months after injection in both neonate (P7) and adult (6–8 weeks) injected animals. A, Sample traces show mIPSCs in control (black) and Pten knockdown granule cells (red). There was no significant change in mIPSC frequency in either the neonate or adult injected groups (p > 0.25, t test; n in parentheses). B, The amplitude of mIPSCs were comparable between Pten knockdown granule cells (red trace) compared with uninfected control granule cells (black trace) in adult and neonate groups (right panel; p < 0.36, t test; n in parentheses).
We analyzed miniature IPSCs in the presence of NBQX to block AMPA receptor-mediated EPSCs. Similar to the analysis of sIPSC, we did not detect a statistically significant difference in mIPSC frequency or amplitude in either neonatal or young adult injected mice (Fig. 8). Four months after injection in neonates, the IPSC amplitude and frequency was 32.6 ± 3.5 pA and 0.48 ± 0.09 Hz in control cells compared with 37.1± 4.5 pA and 0.68 ± 0.18 Hz in Pten shRNA-expressing cells (Fig. 8A). For adult injections at 4 months after injection, the IPSC amplitude and frequency was 37.7 ± 3.2 pA and 1.33 ± 0.14 Hz in control cells compared with 35.5 ± 1.8 pA and 1.77 ± 0.37 Hz in Pten shRNA-expressing cells (Fig. 8B).
Discussion
Our studies demonstrate the effects of Pten knockdown on single neuronal synaptic function. We used a shRNA-expressing lentivirus to efficiently knockdown Pten expression in vivo. Pten knockdown results in a distinctive morphological phenotype in both neonatal mice and in young adults consisting of neuronal hypertrophy and increases in dendritic spine density. The onset of these changes occurred more rapidly in neonatal mice but reached similar levels by 4 months after injection in both young adult and neonatal mice. Acute hippocampal slices taken from injected animals revealed a synaptic phenotype consisting of a disproportionate increase in excitatory synaptic transmission compared with inhibition, which may reflect the circuit defect in autistic patients that have Pten mutations.
The synaptic phenotype
Our results demonstrate that Pten hypofunction leads to a preferential increase in excitatory synaptic function. The increase in EPSC frequency we observed was accompanied by increases in dendritic spine density throughout the dendritic arbor of both neonates and young adults at the 4 month after injection time point. This result suggests an accompanying increase in the number of excitatory synapses on cells in which Pten was knocked down. Furthermore, spine density was increased in the outer portions of the molecular layer, the site of perforant path innervation from uninfected pyramidal neurons of the entorhinal cortex. Thus, postsynaptic deletion of Pten is sufficient to increase synapse formation with wild-type presynaptic neurons.
Although our data are entirely consistent with a postsynaptic locus for the increase in mEPSCs, we also considered possible presynaptic mechanisms. The possibilities include either a transynaptic effect between Pten knockdown dendrites and uninfected nerve terminals, or an increase in release probability (or number of release sites) of any dentate hilar cells infected by the Pten shRNA. We have no evidence for the first possibility. However, in some animals, there were scattered infected cells in the dentate hilus, either inhibitory interneurons or excitatory mossy cells. Substantial numbers of shRNA-expressing excitatory mossy cell input from the inner molecular layer could contribute to increases in EPSC frequency. However, in sparsely infected animals in which there were no infected hilar neurons, sEPSC and mEPSC frequency in infected dentate granule cells were still increased. Thus, the increased spine density in the outer molecular layer, increased EPSC frequency in sparsely infected slices, and increased mEPSC frequency support a cell-autonomous effect in granule cells. The lack of a presynaptic effect on transmitter release from hilar neurons also is consistent with results in which conditional knock-out of Pten in nigral dopaminergic neurons did not alter striatal dopamine release (Diaz-Ruiz et al., 2009). Although the increase in mEPSC frequency appeared to result from an increase in the number of synapses, our results do not preclude other effects of Pten knockdown on axons or nerve terminals (Christie et al., 2010; Drinjakovic et al., 2010). In fact, we observe that Pten knockdown results in increased axon diameters and mossy fiber bouton size.
It is interesting to consider why Pten knock-out causes a specific increase of excitatory synaptic activity with little or no effect on inhibitory synaptic activity. There are at least two possibilities. As the volume of a neuron increases with reduced Pten, the increase in surface area will be greater in distal dendrites than in the soma and proximal dendrites. This growth pattern in dentate granule cells could favor formation of excitatory synapses on more distal dendrites compared with proximal inhibitory synapses from basket and axoaxonic cells. Alternatively, the synapse specificity could result from a Pten dependence of excitatory synapse and dendritic spine formation. PI3K signaling is necessary for axonal filopodial motility (Gallo and Letourneau, 1998; Ketschek and Gallo, 2010). Furthermore, Pten knockdown enhances the number and motility of dendritic filopodia (Luikart et al., 2008). If these structures participate exclusively in the formation of excitatory synapses (Wierenga et al., 2008), one might expect manipulation of Pten to specifically increase excitatory synapse number.
Pten and autism
Germ line heterozygous mutations in Pten have been identified in human subjects with ASD (Goffin et al., 2001; Butler et al., 2005; Buxbaum et al., 2007; Herman et al., 2007). These mutations are likely loss-of-function because conditional knock-out mice display behavioral deficits analogous to those seen in humans. However, it is unlikely that mutations identified in human patients result in the complete loss of Pten function because Pten-null animals are not viable (Di Cristofano et al., 1998). Using in vitro approaches to carefully quantify Pten levels, we obtained reasonable estimates of the degree of knockdown in our in vivo experiments. These measurements suggest knockdown in the range of 70–80% and were qualitatively confirmed in vivo using immunohistochemistry. It is interesting to compare our results with the morphological phenotype of conditional Pten deletion mice, which show progressive increases in brain size and neuronal hypertrophy (Backman et al., 2001; Kwon et al., 2001, 2006). By 8 months of age, conditional knock-outs show massive changes in morphology including a doubling of the thickness of the molecular layer of the dentate gyrus and a 40% increase in the density of dendritic spines in granule cells (Kwon et al., 2006). The knockdown of Pten in our experiments caused a nearly identical 40% increase in neuronal size and spine density by 4 months after injection. However, the macroscopic laminar organization of the dentate gyrus was not affected by Pten knockdown. The absence of cellular mislocalization is consistent with a proposed cell-non-autonomous mechanism whereby glial Pten knockdown results in the disruption of cerebellar organization (Yue et al., 2005). It may also reflect a difference between Pten knock-out and knockdown, or the timing and distribution of the Pten ablation. The width of the molecular layer containing the granule cell dendrites appeared somewhat thicker in animals expressing the Pten shRNA, but this increase was less obvious than in conditional knock-out animals. It will be interesting to determine the degree of Pten loss of function in individuals with varying degrees of ASD severity.
Although ASD can be triggered by different molecular and cellular mechanisms, abnormal circuit function may be the common link. It has been postulated that an increase in the ratio of excitation to inhibition is a hallmark of ASD (Rubenstein and Merzenich, 2003; Bourgeron, 2009). However, in the neuroligin-3 mouse model of autism, this ratio is shifted in the opposite direction (Tabuchi et al., 2007), suggesting that changes in the balance of excitation and inhibition in either direction cause circuit instability and a resulting behavioral phenotype (Cline, 2005). Although our results indicate specific increases in excitatory synapse function, these data do not address an important issue—the appropriateness of the newly formed excitatory synapses. It is possible that the additional synapses form between cells that would normally not synapse on one another. Furthermore, the similar phenotype in neonatal and young-adult animals suggests that the synaptic remodeling is ongoing. The formation of such “inappropriate” synapses could contribute to the disruption of information processing in the brain. This idea could be consistent with the inappropriate response to sensory stimuli that is a prominent feature of ASD. It will be important to examine neural circuit function in models of autism, particularly as treatments to reverse circuit abnormalities become available (Zhou et al., 2009).
Footnotes
This work was supported by National Institutes of Health Grants T32NS007381, F32MH079548 (B.W.L.), and MH46613 (G.L.W.). We thank Cooper French, Stephen Magill, Mike Walogorsky, and Karin Müllendorff for their contributions to this work.
- Correspondence should be addressed to Bryan W. Luikart, Department of Physiology and Neurobiology, Dartmouth Medical School, 1 Medical Center Drive, Lebanon, NH 03756-0001. bryan.w.luikart{at}dartmouth.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}