

Abstract
Studies using cultured neurons have established the critical role of microtubule regulators in neuronal polarization. The c-Jun N-terminal kinase (JNK) pathway is one of the candidate signaling pathways driving microtubule regulation during neuronal polarization. However, the significance of the JNK pathway in axon formation, a fundamental step in neuronal polarization, in vivo, remains unclear. Here, we provide evidence supporting the notion that the JNK pathway contributes to axon formation, in vivo, by identifying the genetic interactions between mouse JNK1 and dual leucine zipper kinase (DLK). Double mutants exhibited severe defects in axon formation in the cerebral neocortex. Moreover, RNA interference rescue experiments, in vitro, showed that DLK and JNK1 function in a common pathway to support neuronal polarization by promoting short-neurite and axon formation. Defects in axon formation caused by perturbations of the DLK–JNK pathway were significantly improved by Taxol. However, defects in short-neurite formation caused by perturbations of the DLK–JNK pathway were enhanced by Taxol. Together, these in vivo and in vitro observations indicate that the DLK–JNK pathway facilitates axon formation in neocortical neurons via stage-specific regulation of microtubule stability.
Introduction
Neurons are highly polarized cells with a single long axon and multiple shorter dendrites that emerge from discrete regions of the cell body. The molecular mechanisms underlying neuronal polarization have been studied intensively, in vitro, using primary cultures of hippocampal or cortical neurons (Dotti et al., 1988). These studies revealed several signaling pathways involved in neuronal polarization in vitro, including the Akt–GSK3β–CRMP pathway (Jiang et al., 2005; Yoshimura et al., 2005) and the PKA–LKB1/STRAD–SAD pathway (Barnes et al., 2007; Shelly et al., 2007). However, the functional significance of these pathways, in vivo, has not yet been fully elucidated. In contrast, in vivo studies using genetically engineered mice revealed several molecules involved in axon formation (Wang et al., 2002; Tissir et al., 2005; Deuel et al., 2006; Koizumi et al., 2006). However, the signaling pathways regulating axon formation, in vivo, have not been fully established.
Microtubules are prominent components of the cytoskeleton that provide structural support for the axon and direct the transport of organelles and proteins through the axoplasm (Brady and Black, 1986; Mitchison and Kirschner, 1988). Therefore, microtubule stability appears to be critical to the progression of neuronal polarization (Zhou et al., 2004; Witte et al., 2008). Microtubule regulators involved in axon formation, both in vivo and in vitro, include doublecortin (DCX), microtubule-associated protein 2 (MAP2), and superior cervical ganglion 10 (SCG10). These have been shown to be phosphorylated and to have their activity modulated by c-Jun N-terminal kinase (JNK) (Chang et al., 2003; Gdalyahu et al., 2004; Grenningloh et al., 2004; Tararuk et al., 2006). Therefore, the JNK pathway is a candidate signaling pathway that drives these microtubule regulators.
The proposal that JNK activity contributes to axon formation, in vitro, was based on the observation that a pharmaceutical inhibitor of JNK abrogates axon formation in cultured hippocampal neurons (Oliva et al., 2006). More recently, the JNK-related genes, JNK1, JNK2, and JNK3, were also shown to contribute to axon regeneration in cultured dorsal root ganglion (DRG) neurons (Barnat et al., 2010). Evaluation of the direct contribution of JNK to axon formation, in vivo, has been hampered by the presence of these three JNK-related genes and their critical functions at early stages of embryogenesis (Kuan et al., 1999).
Dual leucine zipper-bearing kinase (DLK) is a mitogen-activated protein kinase kinase kinase (MAP3K) that activates the JNK pathway (Gallo and Johnson, 2002). Genetic knock-out of DLK results in a small but distinct defect in radial migration and axon formation in mouse neocortical neurons (Hirai et al., 2006; Bloom et al., 2007). An intriguing possibility is that the DLK–JNK pathway is a critical signaling pathway for axon formation in vivo. In this study, we evaluate the functional significance of the DLK–JNK pathway in axon formation through in vivo and in vitro approaches. We provide evidence supporting the notion that the DLK–JNK pathway plays a critical role in axon formation by regulating microtubule stability.
Materials and Methods
Mice.
Jnk1 homozygous mutant mice (Dong et al., 2000) were supplied by The Jackson Laboratory (B6.129S1-Mapk8tm1Flv; stock number 004319). Jnk1 mutant mice on a C57BL/6J background (N6) were crossed at least five times with C57BL/6N female and male mice. Jnk1 heterozygous mutant mice were then crossed with Dlk heterozygous mutant mice (Hirai et al., 2006) on a C57BL/6N background (N8). Mice used in these experiments were maintained according to protocols approved by the Institutional Animal Care and Use Committee at Yokohama City University School of Medicine.
Tissue sections.
Whole mouse embryos or brains harvested at embryonic day 18 (E18) to E19 were fixed overnight in 4% (w/v) paraformaldehyde (PFA) at 4°C and embedded in paraffin wax. Rehydrated paraffin sections (6 μm thick) were processed for Nissl staining [0.5% (w/v) cresyl violet; Sigma-Aldrich] and immunohistochemical staining following a standard protocol. Briefly, sections were heated for 20 min at 120°C in 10 mm sodium citrate, pH 6.0, and treated with 10% (v/v) goat serum in Tris-buffered saline with Tween 20, pH 8.0 (TBST), for 30 min at room temperature (RT). Sections were first incubated with either an affinity-purified rabbit anti-DLK primary antibody raised against the C-terminal part of DLK (dilution, 1:500) (Hirai et al., 2006), a rabbit anti-neurofilament M antibody (dilution, 1:5000; Millipore Bioscience Research Reagents), a mouse anti-vimentin antibody (dilution, 1:1000; Sigma-Aldrich), a rabbit anti-calbindin antibody (dilution, 1:3000; Millipore Bioscience Research Reagents), a rabbit anti-calretinin antibody (dilution, 1:2000; Millipore Bioscience Research Reagents), or an anti-active caspase-3 antibody (dilution, 1:1000; Promega) overnight at 4°C, and then with alkaline phosphatase-conjugated anti-rabbit IgG (dilution, 1:3000; BioSource International) for 2 h at RT. Alkaline phosphatase activity was detected with BM purple (Roche). Cyanine-3-conjugated anti-mouse IgM (Jackson ImmunoResearch), cyanine-3-conjugated anti-mouse IgG (GE Healthcare), and Alexa Fluor 488-conjugated anti-rabbit IgG (Invitrogen) were used as secondary antibodies in experiments involving fluorescence microscopy.
Construction of constitutively active JNK1, T7-JNK1-MKK7.
The coding region of mouse MKK7 cDNA was fused in-frame to the C-terminal end of T7-tagged human JNK1β1 cDNA, from which 30 nt encoding 9 aa and a termination codon were removed using linker oligonucleotides encoding a nuclear export signal derived from Net (Ducret et al., 1999) and five repeats of glutamate-glycine (Otto et al., 2000). A kinase-deficient mutant, T7-JNK1/KN-MKK7/KN, was constructed by replacing conserved lysine residues in the ATP binding site of JNK1β1 and MKK7 with aspartate.
Short hairpin RNA expression vectors.
Two sets of the small interfering RNA (siRNA) sequences targeting mouse DLK, SCG10, DCX, and MAP2 were designed using the siDirect online design site (RNAi, Co., Ltd.). Double-stranded oligonucleotides encoding the siRNA sequences were cloned into a pSuper.gfp/neo vector (OligoEngine). The siRNA sequences used were as follows: DLK#1, CCTGTACATGGAACTGAAT; DLK#2, GGAACGTGCCACAGAAACT; SCG10#1, CGCGCAACATCAACATCTA; SCG10#2, GATCATGCGATATCAGTAT; DCX#1, GGGAGTGCGCTACATTTAT; DCX#2, CAAGGCTATTGGTGCTTAA; MAP2#1, CCGACGAGCGGAAAGATGA; MAP2#2, GATACAATCGGACAATTAT. For rescue experiments with tandem affinity purification (TAP)-DLK, two silent mutations were introduced into TAP-tagged (Stratagene) rat DLK at the corresponding target site of the DLK#1 siRNA. One silent base mismatch is present in the wild-type rat DLK sequence, meaning that three base mismatches in total were present in the TAP-DLK sequence that was cloned into a short hairpin RNA (shRNA) vector with an EF1α promoter. A lysine residue in the ATP-binding site of DLK was replaced with isoleucine in the kinase-deficient mutant, TAP-DLK/KN. For rescue experiments with constitutively active JNK1, the gfp/neo cassette of pSuper.gfp/neo vector was replaced with an EF1α promoter-driven expression cassette for green fluorescent protein (GFP) alone, or for both T7-JNK1-MKK7 and GFP. The open reading frames for T7-JNK1-MKK7 and GFP were connected with internal ribosome entry sites derived from pIRES (Invitrogen).
Primary culture of cortical neurons.
Whole brains were harvested from 10 E16 mouse embryos (ICR), and the meninges were removed. Neocortical regions were dissected from the cerebrum in ice-cold DMEM and pooled in a tube containing 5 ml of 0.05% (w/v) trypsin/EDTA (Invitrogen). For preparation of cortical neurons from a mutant embryo, the right and left cortices obtained from one embryo were placed in a microtube containing 0.5 ml of the trypsin solution. After 30 min incubation at 37°C, the tissue was triturated gently with a fire-polished Pasteur pipette. Viscous material was removed and cell aggregate was left to sediment for 5 min at RT. The cell suspension was then transferred to a new tube and the number of cells was counted using a hemocytometer chamber. Typically, ∼5 × 107 cells were obtained from the cortices of 10 embryos. Cells were transfected with shRNA vectors using the Amaxa Nucleofector system (VPI-1003; program O-005). Transfected cells were pooled in 3 ml of 10% (v/v) fetal bovine serum/DMEM, and 5 × 104 cells were seeded onto 14 mm round coverslips coated with poly-dl-ornithine (50 μg/ml; Sigma-Aldrich) or 24-well culture plates. Cells were cultured at 37°C in the presence of 5% CO2, but the culture medium was changed to B27/Neurobasal medium (without antibiotics) at 1 d in vitro (DIV). Taxol (5 nm paclitaxel; Sigma-Aldrich), anthra[1,9-cd]pyrazol-6(2H)-one (SP600125) (3 or 10 μm JNK inhibitor II; Calbiochem), LiCl (2 mm; Sigma-Aldrich), 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)1H-pyrrole-2,5-dione (SB216763) (5 μm; Tocris), 3-[(3-chloro-4-hydroxyphenyl)amino]-4-(2-nitrophenyl)-1H-pyrrole-2,5-dione (SB415286) (25 μm; BIOMOL), brain-derived neurotrophic factor (BDNF) (50 ng/ml; PeproTech), or vehicle (DMSO or Neurobasal medium) was added at 1 DIV. At 3 DIV, cells were fixed with 3% (w/v) PFA for 15 min at RT for immunostaining. For Western blotting, 3 × 106 cells were seeded onto 6 cm culture dishes coated with poly-dl-ornithine and then lysed with SDS-PAGE sample buffer at 3 DIV. Other culture conditions were identical with those used to maintain cells seeded on coverslips.
Immunocytochemistry.
Fixed cells were permeabilized with 0.1% (v/v) Triton X-100 in PBS for 10 min at RT, and any free aldehyde groups remaining in cells were blocked with 0.1 m glycine in PBS (10 min, RT). For immunostaining, cells were pretreated with 10% (v/v) calf serum in TBST (30 min, RT) and then incubated with primary antibodies (overnight, 4°C) and secondary antibodies (2 h, RT). The primary antibodies used in this study included a chicken anti-GFP antibody (dilution, 1:2000; Aves Lab), a mouse anti-class III β-tubulin antibody (Tuj1) (dilution, 1:20,000; BAbCO), an affinity-purified rabbit anti-DLK primary antibody raised against the C-terminal part of DLK (dilution, 1:500) (Hirai et al., 2006), a mouse anti-T7 tag antibody (dilution, 1:2000; Santa Cruz Biotechnology), a mouse anti-acetylated tubulin antibody (dilution, 1:2000; Sigma-Aldrich), a mouse anti-Tau-1 antibody (dilution, 1:5000; Millipore Bioscience Research Reagents), a mouse anti-MAP2 antibody (dilution 1:2000; Sigma-Aldrich), and a rat anti-tyrosinated α-tubulin antibody (dilution, 1:2000; Harlan Sera-Lab). Secondary antibodies used in this study included cyanine-3-conjugated anti-mouse or rabbit IgG (GE Healthcare), Alexa Fluor 488-conjugated anti-chicken or rat IgG (Invitrogen), cyanine-5-conjugated anti-mouse IgG (GE Healthcare), and Alexa Fluor 647-conjugated anti-rat IgG (Invitrogen). For nuclear staining, 0.5 μm 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) was added to the buffer used in the final wash (TBST). Stained cells were examined using a fluorescence microscope.
Western blotting.
Cell lysate protein extracts were separated by SDS-PAGE and analyzed by Western blotting, according to standard protocols, using the following antibodies: an anti-DLK antibody (dilution, 1:300), a mouse anti-T7 tag antibody (dilution, 1:2000; Santa Cruz), a mouse anti-acetylated tubulin antibody (dilution, 1:3000; Sigma-Aldrich), a rat anti-tyrosinated α-tubulin antibody (dilution, 1:3000; Harlan Sera-Lab), a mouse anti-SCG10 antibody (dilution, 1:1000; Millipore), a goat anti-DCX antibody (dilution, 1:1000; Santa Cruz), a mouse anti-MAP2 (dilution, 1:200; Sigma-Aldrich), a mouse anti-JNK1 antibody (dilution, 1:1000; BD Biosciences Pharmingen), a rabbit anti-active JNK antibody (dilution, 1:2000; Cell Signaling Technology), and a rabbit anti-active extracellular signal-regulated kinase (ERK) antibody (dilution, 1:2000; Promega). The secondary antibodies used in this study were horseradish peroxidase-conjugated anti-rabbit (dilution, 1:3000; GE Healthcare) IgG, anti-mouse (dilution, 1:3000; GE Healthcare) IgG, anti-goat (dilution, 1:2000; Santa Cruz) IgG, and anti-rat (dilution, 1:2000; ICN) IgG. Peroxidase activity was detected using the ECL or ECL-Plus system (GE Healthcare), and chemiluminescence was quantified using a FUJI LAS-1000 plus Luminescent Image Analyzer.
Neurite length measurement.
Cortical neurons transfected with shRNA vectors were identified through the expression of GFP encoded by the same plasmid vector. To detect low-level GFP expression, the fluorescent signal was enhanced by application of an anti-GFP antibody. Images of cells were taken with an epifluorescence microscope (BX50; Olympus Optical) equipped with a 10× objective lens and a CCD camera (SenSys KAF1401E G2; Photometrics). Sample names were masked until all photographs had been taken. The length of the longest neurite in each neuron was measured by manual tracing using imaging software (IPLab; Scanalytics). Neurons were identified by anti-class III β-tubulin (Tuj1) immunostaining. Cells undergoing apoptosis, identified by nuclear staining with DAPI, were omitted from counts. Neurite lengths in 100 cells were measured for each transfection assay, and the results were analyzed using different parameters as indicated in the figure legends. For cortical neurons from a mutant embryo, neurite lengths in 150 cells were measured after staining with α-tubulin antibody. Sample names were masked until all measurements had been made.
Statistical analysis.
All transfection experiments were repeated three to seven times on different days, and the effect of each treatment was assessed using two-tailed Student's t tests. A value of p < 0.05 was considered statistically significant. Neurite length of cortical neurons from mutant embryos was compared using Welch's t tests.
Results
Genetic interaction of DLK and JNK1
Disruption of the JNK1 gene had only a subtle effect on axon formation in the mouse embryonic neocortex (Chang et al., 2003) (Fig. 1A, JNK1−/− panels). However, it markedly exacerbated the defective axon formation caused by disruption of the DLK gene (Fig. 1A, compare DLK−/− panels, DLK−/−JNK1−/− panels). Brain size and neocortical thickness just before birth were comparable among DLK/JNK1 double mutants, DLK or JNK1 single mutants, and wild-type embryos, except that the lateral ventricles and third ventricle were enlarged in the double mutant (Fig. 1A, asterisks). Staining with the neurofilament antibody revealed that axon density in the neocortex (Fig. 1A, arrows) and corona radiata, a structure consisting of pyramidal tract axons (Fig. 1A, arrowheads), was greatly reduced in the double mutant. The corpus callosum and anterior commissure were also smaller or absent in double-mutant embryos (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). At higher magnification, a reduction in the number of neurofilament-labeled axon tracts arranged in parallel to the ventricular surface (Fig. 1B, arrows) was evident in DLK/JNK1 double-mutant embryos. However, staining of the radially oriented neurofilaments was not reduced and occasionally appeared to form thick aggregates (Fig. 1B, arrowheads). The marked reduction in axon density in the cerebral cortex was also shown by Tau-1 staining and differential interference contrast microscopy (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). These axonal defects seen in DLK/JNK1 double-mutant embryos were also observed in four nonlittermates.

Axon formation is severely impaired in the cerebral cortex of DLK/JNK1 double mutants. A, Coronal sections of E19 brain, around the bregma (top panels) and 600 μm more caudal (bottom panels), were stained with a neurofilament antibody. Axon tracts emerging from neocortical regions (arrows) and forming the corona radiata (arrowheads) were reduced in DLK/JNK1 double mutants. Enlargement of the lateral ventricles and the third ventricle was also prominent in the double mutants (asterisks). Scale bar, 1 mm. B, Coronal sections around the bregma of E18 embryos were treated with a neurofilament antibody to label axon tracts (green), a vimentin antibody to label cells in the ventricular zone and radial glial fibers (red), and DAPI to label nuclei (blue). The massive axon tracts that run in parallel to the ventricular zone (arrows) were hardly seen in the cortex of double mutants. Radially aligned neurofilaments frequently formed thick aggregates in double mutants (arrowheads). VZ, Ventricular zone. Scale bar, 100 μm. C, Coronal sections taken through the hippocampus of E19 embryos were stained with cresyl violet (Nissl) or a neurofilament antibody. Axon tracts seen in the alveus and in the molecular layer (arrows) were diminished in the double mutants. CA1 and CA3, Hippocampal regions; DG, dentate gyrus; Sm, molecular layer; al, alveus; fi, fimbria. Scale bar, 500 μm.
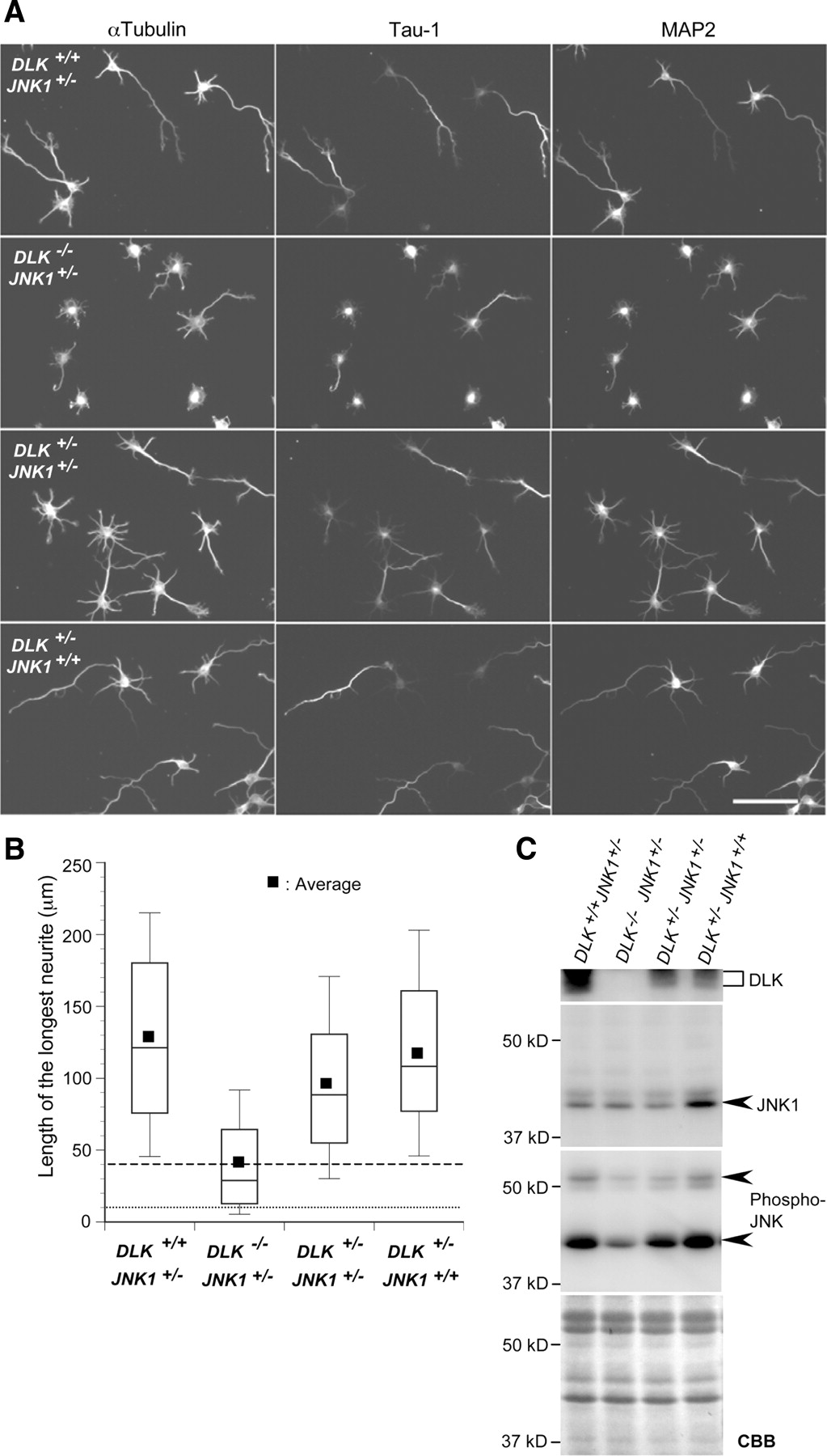
Tau-1 staining revealed that axon formation in E16 cortical neurons, in vitro, was also influenced by genotype (Fig. 2A). The length of the longest neurite, presumably the axon, in cortical neurons prepared from a DLK+/−/JNK1+/− embryo, was significantly shorter than that in cortical neurons prepared from a DLK+/+/JNK1+/− or a DLK+/−/JNK1+/+ embryo (Fig. 2B). Cortical neurons prepared from a DLK−/−/JNK1+/− embryo exhibited more severe defects in neurite formation (Fig. 2B). JNK activity, monitored with a phosphospecific JNK antibody, was also affected by DLK and JNK1 gene dosage (Fig. 2C).

DLK and JNK1 mutations affect axon formation in cortical neurons in vitro. A, Cortical neurons prepared from E16 embryos were cultured for 3 d in vitro and stained with antibodies against α-tubulin (left panels), Tau-1 (middle panels), and MAP2 (right panels). Three embryos (DLK+/+JNK+/−, DLK+/−JNK+/−, DLK+/−JNK+/+) were littermates, whereas one (DLK−/−JNK+/−) was not. Scale bar, 100 μm. B, The length of the longest neurite was measured in 150 neurons for each genotype and displayed in a box plot. The dotted line and the broken line mark the transition between stage 1 and stage 2 neurons and stage 2 and stage 3 neurons, respectively, using the staging criteria indicated in Figure 7. The lengths of the longest neurites were compared using Welch's t tests. The average neurite length in DLK+/−JNK+/− and DLK−/−JNK+/− neurons was significantly lower than that in DLK+/+JNK+/− neurons (p = 8E-6 and 7E-31), whereas the difference was not significant between DLK+/+JNK+/− neurons and DLK+/−JNK+/+ neurons (p = 0.15). The difference between DLK+/−JNK+/− neurons and DLK−/−JNK+/− neurons was significant (p = 3E-21). C, JNK activity in primary cortical neurons was assessed by Western blotting using antibodies against phosphorylated (active) JNK. DLK and JNK1 protein levels were also estimated using the appropriate antibodies. A Coomassie Brilliant Blue (CBB)-stained membrane is shown as a loading control.
These results clearly establish the genetic interactions between DLK and JNK in axon formation in neocortical neurons.
The effect of DLK/JNK1 mutations in different nervous systems
The effects of the DLK/JNK1 double mutation varied between different areas of the cerebrum. Calretinin staining showed that layer I of the cortex was preserved but that the internal capsule, which was partially reduced in DLK-deficient embryos, was mostly absent in double-mutant embryos (supplemental Fig. S2C, top panels, available at www.jneurosci.org as supplemental material). In contrast, calbindin staining showed that the caudoputamen, induseum griseum, and stria terminalis, a structure that carries axons from the amygdala, were preserved (supplemental Fig. S2C, bottom panels, available at www.jneurosci.org as supplemental material). The hippocampus, especially the anterior end, was severely affected in double-mutant embryos, whereas it appeared normal in DLK and JNK1 single-mutant embryos (Fig. 1C). Specifically, neurofilament staining in the alveus and molecular layer of the hippocampus was diminished in the double mutant, rendering these layers indistinguishable (Fig. 1C, arrows). Nevertheless, the fimbria and ventral hippocampal commissure were well preserved (Fig. 1C; supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
The laminar arrangement of the cortical plate in the E19 cerebrum was similarly less defined in DLK/JNK1 double-mutant embryos (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). However, the radially oriented glial fibers in the neocortex appeared to be unaffected in the double mutant (Fig. 1B, vimentin staining). Whereas the laminar structure of the olfactory bulb was preserved in the double mutant, the thickness of the internal and external plexiform layers and the glomerular layer was markedly reduced, and the accessory olfactory bulb was barely detectable (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). The peripheral nervous system appeared to be unaffected in the double mutant (Fig. 3A).

Effects of a DLK/JNK1 double mutation on axon formation in the peripheral nervous system and apoptosis in the neocortex. A, Sagittal sections of E19 mouse brain stained with a neurofilament antibody. Ganglia and axon tracts located in the upper trunk (top panels) and abdomen (bottom panels) were not significantly affected. DRG, Dorsal root ganglia; SG, stellate ganglion; In, intestine; Bd, bladder. Scale bars, 500 μm. B, Quantification of apoptotic cells in the E19 neocortex. Apoptotic cells were detected by immunostaining using an anti-active caspase-3 antibody (left panels). Scale bar, 10 μm. For quantification, five serial sections (6 μm thick) were taken from eight different sites along the rostrocaudal axis in two different embryos for each of the four genotypes. The average number of apoptotic cells in each region of the neocortex was calculated from the five serial sections. An overall average and SD was then calculated from these eight different regions and depicted graphically (right panels, *p < 0.05).
Apoptotic cells in the neocortex, detected with an active caspase-3 antibody, were slightly increased by the JNK1 gene disruption. However, additional disruption of the DLK gene did not significantly change the number of apoptotic cells present in the tissue (Fig. 3B). Cell density in the neocortex was also relatively unaffected by the DLK and JNK1 gene disruptions (Fig. 1B, bottom panels). Together, these observations suggest that the functional significance of DLK and JNK1 lies mainly in axon formation in neocortical neurons during embryogenesis.
DLK kinase activity is required for axon formation in vitro
As forced expression of DLK has been shown to induce JNK activity in several cell lines (Gallo and Johnson, 2002), it is conceivable that DLK-dependent JNK activation in neuronal cells is the basis of the genetic interaction between DLK and JNK1. However, the DLK-related, JNK-activating MAP3K enzyme, MLK3, has been reported to regulate the activity of the small G-protein, Rho, in a kinase activity-independent manner (Swenson-Fields et al., 2008). Therefore, DLK may influence axon formation either through the JNK-dependent signaling pathway or through a JNK-independent signaling pathway.
To test whether the protein kinase activity of DLK is essential for axon formation, we performed in vitro RNA interference (RNAi) rescue experiments using primary cultures of mouse embryonic cortical neurons. Most cortical neurons cultured for 3 DIV grew a single long neurite, which was presumably an axon. The length of the longest neurite varied among neurons, averaging ∼100 μm in control vector-transfected cells (Fig. 4A; supplemental Fig. S4C, available at www.jneurosci.org as supplemental material). Cell transfection with an expression vector for shRNA targeting DLK mRNA (DLK#1) resulted in a decrease in DLK protein levels (Fig. 4B,C) and perturbed axon formation (Fig. 4A). The median length of the longest neurite in DLK RNAi cells was 32.8% of that in control cells (Fig. 4D). Neurons were identified by class III β-tubulin staining, and neurite length was measured in neurons expressing GFP, which was encoded by the same plasmid vector designed for shRNA expression (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material). Expression of shRNA targeting a different part of the DLK mRNA (DLK#2) resulted in comparable levels of neurite growth suppression (see Fig. 6B). The inhibitory effect of DLK shRNA (DLK#1) on axon formation was nearly completely offset by the expression of exogenous DLK bearing three silent base substitutions at the RNAi target site, whereas its kinase-deficient counterpart, DLK/KN, was unable to reestablish normal neurite growth (Fig. 4; supplemental Fig. S4B,C, available at www.jneurosci.org as supplemental material). These results indicate that the ability of DLK to facilitate axon formation depends on its kinase activity.

DLK kinase activity is required for the polarization of cortical neurons in vitro. A, Polarization of cortical neurons at 3 DIV. Enzymatically dispersed E16 cortical neurons were transfected with a plasmid vector carrying expression cassettes for GFP and for nonsilencing shRNA (NC), DLK shRNA (DLK#1), DLK shRNA and RNAi-resistant DLK (DLK#1+DLK), or DLK shRNA and an RNAi-resistant DLK kinase-deficient mutant (DLK#1+DLK/KN). Scale bar, 100 μm. B, DLK knockdown in cortical neurons was monitored by Western blotting. shRNA vector-transfected cells were lysed at 3 DIV and DLK protein levels were analyzed with the DLK antibody. Considerable amounts of DLK were detected in the DLK shRNA vector-transfected sample. However, it should be noted that ∼30% of cells expressed GFP when examined under a fluorescence microscope; thereby DLK protein levels were likely very low in GFP-positive cells. The expression of exogenous DLK (TAP-tagged DLK), or its kinase-deficient mutant, was not enough to return DLK expression levels to control levels, and the broad Western band, which includes endogenous DLK, appeared to be slightly intensified. The expression of exogenous DLK and its kinase-deficient mutant was confirmed using Neuro 2a cells (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material). C, DLK expression in GFP-labeled neurons (arrows) was tested by staining with a DLK antibody. In control cells (NC) and DLK RNAi-rescue cells, DLK protein was detected in the soma (arrows) and in a GFP-labeled long neurite (arrowhead). Note that DLK expression decreased to background levels in DLK RNAi cells (DLK#1). Scale bar, 50 μm. D, The median length of the longest neurite (n = 100) at 3 DIV is shown graphically. The bars represent the average of values obtained from four independent experiments. Error bars represent the SD. *p < 0.0001. The average of values obtained from control neurons was expressed as 100. The transfected plasmid vectors are shown on the x-axis.
Active JNK rescues the DLK RNAi phenotype
To test whether JNK activation is an essential part of DLK function in axon formation in neocortical neurons, we performed DLK RNAi rescue experiments using active JNK1, a fusion construct of JNK1 and MKK7 (T7-JNK1-MKK7) (supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material). The defects in axon formation seen in DLK RNAi-treated neocortical neurons were almost completely eliminated by the expression of active JNK1 (Fig. 5B,C; supplemental Fig. S5C, available at www.jneurosci.org as supplemental material). Conversely, the fusion construct of kinase-deficient JNK1 and kinase-deficient MKK7 (T7-JNK1/KN-MKK7/KN) was unable to offset the axon formation defects seen in DLK RNAi cells, even though expression levels were comparable with those of the active construct (Fig. 5A,B). These results indicate that DLK and JNK function in a common pathway to support axon formation. Together with the results of the genetic analysis in mice, these in vitro data reveal the critical function of “the DLK–JNK pathway” in axon formation in neocortical neurons.

Constitutively active JNK1 suppressed the polarization defects seen in DLK RNAi neurons. A, DLK protein levels were monitored by Western blotting using a DLK antibody (top panel). The expression of active JNK1 and its kinase-deficient mutant was confirmed by Western blotting using an anti-T7-tag antibody (middle panel). A CBB-stained membrane is shown as a loading control (bottom panel). B, The expression of DLK, T7-JNK1-MKK7, and T7-JNK1/KN-MKK7/KN in GFP-labeled E16 cortical neurons at 3 DIV (arrows) was examined by staining with a DLK antibody and a T7-tag antibody. Scale bar, 50 μm. C, The median length of the longest neurite (n = 100) at 3 DIV is shown graphically. The bars represent the average of values obtained from four independent experiments. Error bars represent the SD. *p < 0.0001. The average of values obtained from control neurons was expressed as 100. A plasmid vector carrying expression cassettes for GFP and for nonsilencing shRNA (NC), DLK shRNA (DLK#1), DLK#1 and T7-JNK1-MKK7, or DLK#1 and T7-JNK1/KN-MKK7/KN was transfected before seeding.
Forced stabilization of microtubules improves the polarization of cortical neurons following perturbation of the DLK–JNK pathway
Subsequently, we attempted to elucidate the molecular mechanisms underlying DLK–JNK pathway-dependent axon formation. Microtubule stabilization is critical for axon formation (Zhou et al., 2004; Witte et al., 2008). Acetylated tubulin, a marker of stable microtubules (Baas and Black, 1990), is enriched in growing axons (Witte et al., 2008) and reduced in the short neurites of DLK RNAi cells (supplemental Fig. S6A, available at www.jneurosci.org as supplemental material).
To test whether the DLK–JNK pathway facilitates axon formation by regulating microtubule stability, we evaluated the ability of low-dose Taxol treatment (Witte et al., 2008) to offset axon formation defects caused by suppression of the DLK–JNK pathway. Axon formation in DLK shRNA-treated cells was significantly improved with the administration of 5 nm Taxol (Fig. 6A,B). Administration of a JNK inhibitor (SP600125) (Bennett et al., 2001) disrupted axon formation in a dose-dependent manner, but this was also significantly improved by treatment with 5 nm Taxol (Fig. 6A,C). In cortical neurons, Taxol treatment did not affect JNK activity but led to an accumulation of acetylated tubulin and a reduction of tyrosinated tubulin, a marker of unstable microtubules or unpolymerized tubulin (Fig. 6D). These results indicate that the DLK–JNK pathway supports axon formation, at least in part, by stabilizing microtubules.

Taxol improves the polarization of cortical neurons impaired by perturbation of the DLK–JNK pathway. A, Polarization of E16 cortical neurons at 3 DIV in the presence or absence of 5 nm Taxol. Cells were transfected with a plasmid vector encoding nonsilencing shRNA (NC) or DLK shRNA (DLK#1 or DLK#2) before seeding. A JNK inhibitor (10 μm SP600125), Taxol (5 nm), or vehicle (DMSO) was added separately or in combination to the culture medium at 1 DIV. A plasmid vector encoding nonsilencing shRNA was transfected to monitor the effects of the JNK inhibitor. The arrows indicate stage 1 neurons. Scale bar, 100 μm. B, C, Quantitative analysis of the effects of Taxol on the polarization of (B) DLK RNAi or (C) JNK inhibitor-treated cells. The median length of the longest neurite (n = 100) at 3 DIV is shown graphically. The bars represent the average of values obtained from four (B) or three (C) independent experiments. Error bars represent the SD. *p < 0.0005; **p < 0.005. The average of values obtained from control neurons was expressed as 100. D, Western blot analysis monitoring the effects of Taxol on JNK activity and microtubule stability. Primary cultures of E16 cortical neurons were treated with 5 nm Taxol or vehicle at 1 DIV. Cell lysates were prepared at 3 DIV, and proteins were analyzed by Western blotting using antibodies against phosphorylated (active) JNK, acetylated tubulin, and tyrosinated tubulin. A CBB-stained membrane is shown as a loading control.
Glycogen synthase kinase 3β (GSK3β) is part of a major signaling pathway that regulates microtubule assembly in neurons undergoing polarization. Specifically, GSK3β inactivation leads to axon specification and growth in vitro (Zhou et al., 2004; Jiang et al., 2005; Yoshimura et al., 2005). However, none of the GSK3 inhibitors used in this study was able to rescue the DLK RNAi phenotype (supplemental Fig. S6B, available at www.jneurosci.org as supplemental material). Therefore, the DLK–JNK pathway may regulate microtubule stability through a GSK3β-independent mechanism.
BDNF is a well characterized cytokine that promotes axon growth in vitro and activates intracellular signaling pathways regulating microtubule stability (Yuan et al., 2003; Yoshimura et al., 2005; Shelly et al., 2007). We tested the possibility that the DLK–JNK pathway supports axon formation by stimulating the secretion of BDNF. The addition of BDNF to the culture medium led to acute and chronic activation of ERK in cortical neurons, but it did not reverse the defects in axon formation caused by DLK RNAi, nor did it affect JNK activity (supplemental Fig. S6C, available at www.jneurosci.org as supplemental material). Although it is still possible that the DLK–JNK pathway supports axon formation by inducing the secretion of a factor that cannot be replaced by BDNF, this result supports the notion that the DLK–JNK pathway facilitates axon growth by regulating microtubule stability via a cell-autonomous pathway.
The DLK–JNK pathway contributes to the fixation of cell polarity
In the early stages of neuronal polarization in vitro, nonpolar stage 1 neurons begin to grow multiple short neurites (stage 1/stage 2 transition). A multipolar stage 2 neuron will allow one of its neurites to rapidly grow so as to become an axon-forming stage 3 neuron (stage 2/stage 3 transition) (Dotti et al., 1988; de Anda et al., 2005; Jacobson et al., 2006). As shown in Figure 6, Taxol significantly reversed axonal defects caused by perturbations in the DLK–JNK pathway. However, it should be noted that Taxol could not counter defects in axon formation to match control levels of axon formation, and it often inhibited the transition of stage 1 neurons to stage 2 neurons during neuronal polarization (Fig. 6A, arrows). This dual effect of Taxol treatment on axonal defects caused by perturbation of the DLK–JNK pathway suggests that microtubule stabilization is not the sole function of the DLK–JNK pathway.
Accordingly, our next aim was to evaluate the effect of DLK–JNK pathway perturbations and Taxol treatment on the various stages of neuronal polarization. To this end, we compared the number of neurons present at different stages of polarization. Stages were defined by the length of the longest neurite (Fig. 7A). Most of the control cortical neurons seen at 3 DIV were stage 3 neurons (86.75%). A smaller number of stage 2 neurons were also observed (13%), whereas stage 1 neurons were rare (0.25%). DLK RNAi or JNK inhibitor treatment resulted in a significant decrease in the number of stage 3 neurons, suggesting that these treatments disrupt transition of stage 2 neurons to stage 3 neurons. Indeed, there was a concomitant increase in the number of stage 2 neurons (Fig. 7B,C). Moreover, DLK RNAi resulted in a significant increase in the number of stage 1 neurons. The nonsignificant increase in the number of stage 1 neurons after JNK inhibitor treatment became significant by advancing the timing of inhibitor administration from 1 DIV (Fig. 7C) to the time of cell seeding (supplemental Fig. S7A, available at www.jneurosci.org as supplemental material). This increase in the number of stage 1 neurons may reflect a perturbation in the transition of stage 1 neurons to stage 2 neurons during polarization. Both the reduction of stage 3 neurons and the accumulation of stage 2 neurons were significantly countered by Taxol treatment. Conversely, the increase in the number of stage 1 neurons was significantly enhanced by Taxol (Fig. 7B,C). Taxol treatment alone also resulted in a small, but significant, increase in the number of stage 1 neurons (Fig. 7B,C). Advanced administration of Taxol resulted in a marked increase in the number of stage 1 neurons (supplemental Fig. S7A, available at www.jneurosci.org as supplemental material). These observations suggest that the significance of microtubule stabilization and the function of the DLK–JNK pathway may vary at different stages of neuronal polarization.

DLK RNAi or JNK inhibition impairs the transition of a multipolar cell to an axon-forming cell. A, Staging criteria for polarizing cortical neurons at 3 DIV. Representative images are shown of GFP-labeled cells that differ with respect to the length of their longest neurite. Cells were defined by the length of the longest neurite: stage 1 (nonpolar), <10 μm; stage 2 (multipolar), <40 μm; stage 3 (axon-forming), >40 μm. Scale bar, 50 μm. B, C, The relative number of cortical neurons at each of the three different stages is depicted graphically. Neurite length data, shown in Figure 6, were used to count the number of cells at each stage. Error bars represent the SD of values obtained from four (B) or three (C) independent experiments. The statistical significance of the effect of DLK knockdown and JNK inhibitor treatment on the number of neurons at respective stages was evaluated using two-tailed Student's t tests (*p < 0.05). The statistical significance of Taxol treatment on the number of neurons at respective stages was also evaluated the same way (**p < 0.05). ns, Not significant. DLK knockdown or JNK inhibitor treatment significantly enhanced the marginal increase in the number of stage 1 neurons seen with Taxol treatment (#p < 0.01).
To evaluate the contribution of the DLK–JNK pathway to the transition of stage 1 neurons to stage 2 neurons, and stage 2 neurons to stage 3 neurons, we calculated the efficiency of stage transitions by using the data shown in Figure 7. The efficiency of stage 1/stage 2 transitions was defined by the following formula: (ST2 + ST3)/100, where ST2 and ST3 correspond to the number (as a percentage) of stage 2 and stage 3 neurons, respectively. Similarly, the efficiency of stage 2/stage 3 transitions was defined by the following formula: ST3/(ST2 + ST3) (Table 1). Stage 2/stage 3 transitions were markedly perturbed by DLK RNAi (48.8 ± 8.9% of control RNAi) and JNK inhibitor treatment (52.0 ± 4.2% of vehicle). Stage 1/stage 2 transitions were moderately affected by DLK RNAi (94.7 ± 2.2% of control RNAi) and JNK inhibitor treatment (98.0 ± 1.2% of vehicle). Stage 2/stage 3 transitions, perturbed by DLK RNAi or JNK inhibitor treatment (48.8 ± 8.9% of control RNAi or 52.0 ± 4.2% of vehicle), were significantly improved after Taxol treatment (87.4 ± 2.8% of control RNAi or 89.6 ± 6.1% of vehicle). However, the disruption in stage 1/stage 2 transitions by DLK RNAi or JNK inhibitor treatment (94.7 ± 2.2% of control RNAi or 98.0 ± 1.2% of vehicle) was exacerbated by Taxol (86.5 ± 2.6% of control RNAi or 85.0 ± 4.2% of vehicle). In fact, perturbations in stage 1/stage 2 transitions by Taxol was also observed in control neurons (96.6 ± 2.0% of vehicle) transfected with a nonsilencing control shRNA vector (NC). In contrast, ectopic expression of RNAi-resistant DLK or active JNK1 restored the progression of stage 2/stage 3 transitions without perturbing stage 1/stage 2 transitions (Table 1; supplemental Fig. S7B,C, available at www.jneurosci.org as supplemental material). These results indicate that the DLK–JNK pathway contributes to both stage 1/stage 2 transitions and stage 2/stage 3 transitions and that, although Taxol can essentially mimic the role of the DLK–JNK pathway in stage 2/stage 3 transitions, Taxol disrupts stage 1/stage 2 transitions during neuronal polarization.
Effects of DLK–JNK pathway inactivation and Taxol treatment on the different stages of neuronal polarization
A gross accumulation of stage 2 neurons was also observed after treatment with shRNA against the microtubule modulators, SCG10, DCX, and MAP2 (Fig. 8A–C), which are thought to be physiological substrates for JNK (Chang et al., 2003; Gdalyahu et al., 2004; Grenningloh et al., 2004; Tararuk et al., 2006). As with DLK RNAi and JNK inhibitor treatment, RNAi directed against the microtubule-stabilizing proteins, DCX or MAP2, disrupted stage 2/stage 3 transitions of neuronal polarization, whereas its effect on stage 1/stage 2 transitions was relatively moderate (supplemental Table S1, available at www.jneurosci.org as supplemental material). In contrast, RNAi directed against SCG10, a microtubule-destabilizing protein, strongly perturbed stage 1/stage 2 transitions (supplemental Table S1, available at www.jneurosci.org as supplemental material). Furthermore, SCG10 RNAi enhanced Taxol-induced accumulation of nonpolar cells, whereas DCX RNAi and MAP2 RNAi exhibited no such effect (Fig. 8C; supplemental Fig. S7D,E, available at www.jneurosci.org as supplemental material).
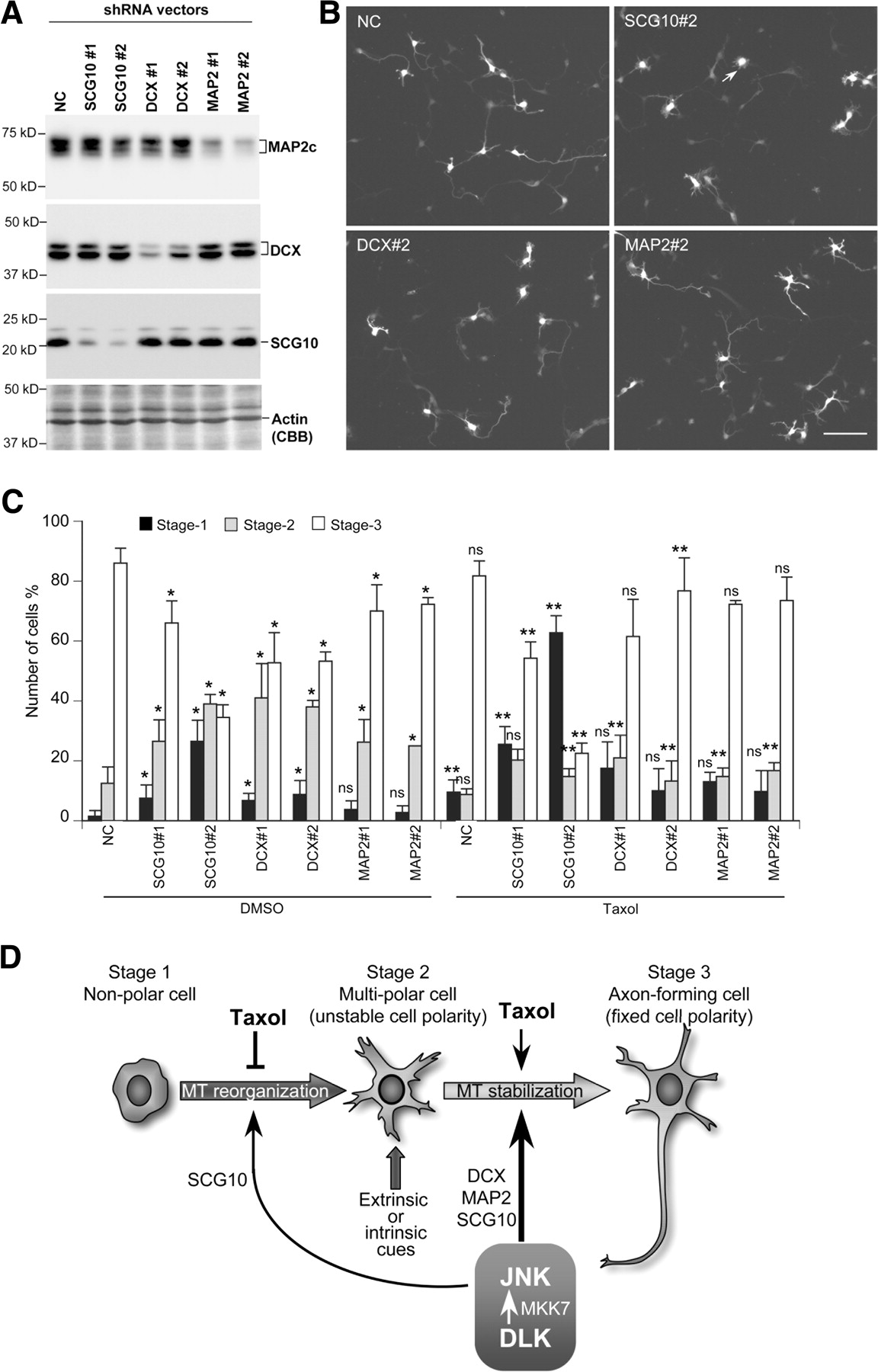
Knockdown of microtubule modulators that serve as JNK substrates affects the polarization of cortical neurons. A, E16 cortical neurons were transfected with shRNA expression vectors for MAP2, DCX, and SCG10, and protein levels of each of these microtubule modulator were examined by Western blotting. B, Polarization of neurons transfected with shRNA vectors at 3 DIV. Scale bar, 100 μm. C, The relative number of cortical neurons at each of the three different stages is shown graphically. The relative number of stage 2 neurons was significantly increased by introducing shRNA vectors for SCG10, DCX, or MAP2. Error bars represent the SD of values obtained from four independent experiments. The statistical significance of the effect of DLK knockdown and JNK inhibitor treatment on the number of neurons at respective stages was evaluated by two-tailed Student's t tests (*p < 0.05). The statistical significance of Taxol treatment on the number of neurons at respective stages was evaluated the same way (**p < 0.05). ns, Not significant. SCG10 knockdown significantly enhanced the marginal increase in the number of stage 1 neurons seen with Taxol treatment (#p < 0.005). D, Stage-specific stabilization of microtubules by the DLK–JNK pathway. In vivo or in vitro polarization of cortical neurons begins with the formation of multiple unstable neurites (Dotti et al., 1988; Tabata and Nakajima, 2003; Noctor et al., 2004). This step is likely to be highly dependent on the reorganization of microtubules, a process supported by SCG10. Extrinsic or intrinsic cues allow one of these neurites to become stable and to progressively elongate (Hatanaka and Murakami, 2002; de Anda et al., 2005; Jiang et al., 2005; Shelly et al., 2007). The DLK–JNK pathway likely supports microtubule reorganization and stabilization through different sets of microtubule-binding proteins, including DCX, MAP2, and SCG10.
Together, these data suggest that dynamic and stable microtubules are essential in stage 1/stage 2 and stage 2/stage 3 transitions, respectively, and that the DLK–JNK pathway allows for appropriate stage-specific regulation of microtubule stability by activating different sets of microtubule regulators (Fig. 8D).
Discussion
In vivo and in vitro experiments performed in this study have demonstrated the functional significance of the DLK–JNK pathway in axon formation in mouse neocortical neurons. During polarization of cortical neurons in vitro, the DLK–JNK pathway plays a significant role in the transition of stage 1 neurons to stage 3 neurons, the time when neuronal polarity is fixed by the growing axon. This role of the DLK–JNK pathway depends, at least in part, on timely regulation of microtubule stability.
We have shown that mutations in the JNK1 gene markedly exacerbate the defective axon formation seen in DLK mutant mice. In neocortical regions of DLK/JNK1 double-mutant mice, the axon tracts forming the anterior commissure, corpus callosum, and corona radiata, which connect different areas of the brain and spinal cord, were mostly abolished. The synergistic effects of the DLK and JNK1 mutations is likely explained by a massive reduction in total JNK activity in cortical neurons, which becomes lower than the activity required for axon tract formation. The functional link between DLK and JNK in axon formation was further supported by the following in vitro observations: (1) DLK kinase activity was essential to its function in axon formation, supporting the notion that DLK functions as a MAP3K. (2) A constitutively active JNK1 construct mitigated the axonal defects seen in DLK RNAi cells, indicating that JNK is a downstream target of DLK. Together, these in vivo and in vitro observations strongly suggest that the DLK–JNK pathway plays an essential role in axon formation.
Quantification of neurons at different stages of polarization allowed us to clarify the role of the DLK–JNK pathway at different stages of neuronal polarization. Suppression of DLK expression by shRNA or suppression of JNK activity by a pharmaceutical inhibitor impaired both stage 1/stage 2 and stage 2/stage 3 transitions during neuronal polarization. Taxol significantly improved stage 2/stage 3 transitions impaired by perturbation of the DLK–JNK pathway, whereas it exacerbated the suppression of stage 1/stage 2 transitions by the perturbation of DLK–JNK pathway. These observations indicate that microtubule stabilization is essential for stage 2/stage 3 transitions in cortical neurons, as has been reported in hippocampal neurons (Witte et al., 2008) and that the DLK–JNK pathway contributes to these stage transitions by stabilizing microtubules. Conversely, dynamic rather than stable microtubules may be essential for stage 1/stage 2 transitions, where the DLK–JNK pathway plays a distinct role in microtubule regulation (Fig. 8D).
Targets of the DLK–JNK pathway in axon formation may comprise microtubule modulators that have been identified as JNK substrates. These include the microtubule-stabilizing proteins, DCX and MAP2 (Horesh et al., 1999; Dehmelt et al., 2003), and the microtubule-destabilizing protein, SCG10 (Grenningloh et al., 2004), which all play significant roles in axon formation in vivo and in vitro (Teng et al., 2001; Chang et al., 2003; Gdalyahu et al., 2004; Deuel et al., 2006; Koizumi et al., 2006; Tararuk et al., 2006). In our experiments, the suppression of DCX or MAP2 expression in cultured cortical neurons treated with shRNA resulted in impaired stage 2/stage 3 transitions, as was observed after suppression of the DLK–JNK pathway. Moreover, it has been suggested that the phosphorylation of DCX or MAP2 by JNK enhances their ability to support axonal growth (Gdalyahu et al., 2004) and to bind microtubules (Chang et al., 2003). Therefore, the DLK–JNK pathway may contribute to the stabilization of microtubules during stage 2/stage 3 transitions, at least in part, through the modulation of these microtubule-stabilizing proteins.
In contrast, the suppression of SCG10 expression impairs not only stage 2/stage 3 transitions, but also stage 1/stage 2 transitions. The requirement for SCG10 in stage 1/stage 2 transitions is consistent with the negative effects of Taxol treatment on the transition of stage 1 neurons to stage 2 neurons. The requirement for SCG10 in stage 2/stage 3 transitions might be explained by its role in the growth cones of developing neurites/axons, where it binds to microtubules to keep them unstable (Grenningloh et al., 2004; Morii et al., 2006). The DLK–JNK pathway may be involved in stage 2/stage 3 transitions via a phosphorylation-dependent reduction in SCG10 activity (Antonsson et al., 1998; Grenningloh et al., 2004) in certain areas of the cell, such as the shaft of the presumptive axon. In any case, these observations do not rule out the possibility that other targets of the DLK–JNK pathway, such as transcriptional regulation via c-Jun, also support stage transitions during neuronal polarization (Raivich et al., 2004).
Whereas most of the thick axon bundles in DLK/JNK1 double-mutant mice were abolished, neurofilament staining for radially oriented fibers in the cortical wall remained unaffected. This may reflect the limited function of the DLK–JNK pathway in the early stages of axon formation in vivo. Polarization of cortical neurons, in vivo, begins with the formation of multiple short neurites, shortly after the loss of epithelial cell polarity on neuronal differentiation (Tabata and Nakajima, 2003; Noctor et al., 2004). Early multipolar neurons migrate toward their final destination, assuming a bipolar morphology and leaving behind a trailing process, the primary axon (Hatanaka and Murakami, 2002; Noctor et al., 2004). This step depends on the presence of specific cues (Yi et al., 2010), which may not be available to neurons cultured in vitro. The axon grows continuously during and after neural migration, directed by several guidance systems, until it reaches its target (Jones et al., 2002; Wang et al., 2002; Tissir et al., 2005; Canty and Murphy, 2008). The DLK–JNK pathway is likely to play a major role in this process by stabilizing microtubules within axonal shafts. Additional study is required to clarify the interplay between axonal cues, guidance molecule-dependent signaling, and the DLK–JNK pathway. This would help elucidate the essential molecular systems involved in establishing the projection of cortical neurons.
The effects of the DLK/JNK1 double mutation on axon formation were mostly confined to the cerebral cortex. The DLK protein is widely distributed in embryonic neural tissue (Hirai et al., 2005, 2006), and JNK1 is expressed in almost all embryonic tissues (Kuan et al., 1999). Therefore, this specificity may be indicative of an environment that supports axon growth (Jones et al., 2002; Wang et al., 2002; Tissir et al., 2005; Canty and Murphy, 2008), a specific differentiation mode for cortical neurons (Hatanaka and Murakami, 2002; Noctor et al., 2004), or an influence of redundant signaling pathways. This regional specificity of axonal defects, however, does not mean that the DLK–JNK pathway is dispensable for neurons in unaffected regions, including the peripheral nervous system. In Caenorhabditis elegans and Drosophila, the effect of a dlk-1/wallenda mutation is significant in injured axons and in rpm-1/highwire mutants, although mutation of dlk-1/wallenda alone does not cause a significant defect in axon or synapse formation during ontogeny (Nakata et al., 2005; Collins et al., 2006; Hammarlund et al., 2009; Miller et al., 2009; Yan et al., 2009). This suggests that the mammalian DLK–JNK pathway also functions in axon regeneration/degeneration and synapse plasticity in a wide variety of neural tissues. In fact, it has been reported that Wallerian degeneration in mouse DRG neurons, in vitro, depends on DLK and JNK activity (Miller et al., 2009). This study has uncovered a specific and remarkable function of the DLK–JNK pathway in the development of cortical neurons, which provides a novel perspective to understanding the molecular basis of mental retardation related to developmental disorders.
Footnotes
-
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank E. Nishida for the MKK7 cDNA and C. Kusaka for her technical assistance with mouse genotype analysis.
- Correspondence should be addressed to Dr. Syu-ichi Hirai, Department of Molecular Biology, Yokohama City University Graduate School of Medicine, 3-9 Fukuura, Kanazawa-ku, Yokohama 236-0004, Japan. sh3312{at}med.yokohama-cu.ac.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}